

Abstract
Structuring over many length scales is a design strategy widely used in Nature to create materials with unique functional properties. We here present a comprehensive analysis of an adult sea urchin spine, and in revealing a complex, hierarchical structure, show how Nature fabricates a material which diffracts as a single crystal of calcite and yet fractures as a glassy material. Each spine comprises a highly oriented array of Mg-calcite nanocrystals in which amorphous regions and macromolecules are embedded. It is postulated that this mesocrystalline structure forms via the crystallization of a dense array of amorphous calcium carbonate (ACC) precursor particles. A residual surface layer of ACC and/or macromolecules remains around the nanoparticle units which creates the mesocrystal structure and contributes to the conchoidal fracture behavior. Nature’s demonstration of how crystallization of an amorphous precursor phase can create a crystalline material with remarkable properties therefore provides inspiration for a novel approach to the design and synthesis of synthetic composite materials.
Keywords: calcium carbonate biomineralization, echinoderm skeleton, hierarchical structuring, mesocrystal, skeletal elements
Some of the most remarkable, and best-studied biominerals, are sea urchin (SU) skeletal elements, which are materials that have challenged scientists for decades. Despite exhibiting sponge-like morphologies and curved surfaces, which are features normally associated with amorphous materials, each element behaves as a single crystal of calcite as judged by techniques such as polarized light microscopy (1), X-ray Diffraction (XRD) (2), and electron backscatter diffraction (3). Prior to recent studies which demonstrated that morphologically complex calcite single crystals can be produced synthetically using templating methods (4–6), such crystals were believed to be restricted to the biological world. Intriguingly, this single crystal structure has been disputed on many occasions, which has led to the suggestion that SU spines and plates may actually comprise a highly oriented array of crystalline nanoparticles rather than a true single crystal (7). The fracture behavior of SU skeletal elements is also unusual and contradictory. While calcite single crystals cleave along the low-energy {104} planes (8), SU spines cleave with conchoidal fracture surfaces (7, 9, 10). This behavior has been attributed in part to the presence of intracrystalline macromolecules (11–15), which are typically present in concentrations of < 1 wt%.
Considering the ultrastructure of SU spines in more detail, Towe suggested over 40 years ago that SU skeletal plates may comprise a single crystal interior and a polycrystalline exterior (8). He postulated that this structural organization could derive from the initial formation of an oriented polycrystalline mineral which subsequently underwent fusion and recrystallization to give a single crystal product. In such a scheme, the resulting material would invariably contain a nonnegligible fraction of disordered regions, which could be attributed to organic and/or amorphous inclusions. Depending on the quantity and distribution of defects, the sea urchin spine can therefore be seen either as a composite material in which disordered inclusions are embedded in a single crystal matrix, or as an assembly of crystalline nanoparticle domains which are separated by disordered layers and which exhibit a high degree of crystallographic registration. The latter structure is described as “mesocrystalline,” (16, 17) where a mesocrystal ideally comprises a 3D array of isooriented single crystal particles of size 1–1,000 nm (mesoscale dimensions). The highly oriented subunits therefore distinguish a mesocrystal from a randomly oriented polycrystal, and the identifiable nano-sized building units distinguish it from a single crystal containing impurities (18). It is emphasized that the term mesocrystal defines the structure of a material rather than its mechanism of formation. Therefore, while oriented aggregation of crystalline nanoparticles can give rise to either a single crystal or mesocrystal product, in analysis of the structure of sea urchin spines we here present data which suggests that a mesocrystal can also form when a dense array of amorphous nanoparticles crystallizes to give a highly cooriented end-product material. Other authors have argued that sea urchin spines cannot be mesocrystals as they are space-filling, as demonstrated by the low surface area measured by Brunauer-Emmett-Teller (BET) surface area analysis (19). This result, however, does not exclude its mesocrystallinity as many biominerals form through secondary growth stages, enabling mesocrystalline domains to exist in the absence of porosity. Recent experimental evidence has added weight to the proposition that many biominerals exhibit mesocrystalline structures. High resolution transmission electron microscopy (HRTEM) has revealed nanoparticle building units within sea urchin spines (20, 21), while synchrotron XRD studies of a range of single crystal calcareous elements have identified mosaic structures where the calcitic elements exhibited coherence lengths of 50–200 nm (18, 22–24). Further, microscopic studies have suggested that the SU spine is an extended mesocrystal composed of nanoparticles (25–27). There is also growing evidence that mesocrystals may be present in related organisms. Mesocrystalline regions have been observed in the ossicles of a sea star (7), in the skeletal units of a crinoid echinoderm (28), and in the aragonite platelets of nacre (29). According to Sethmann and coworkers (30), similar mesocrystals have been found in sponge spicules, which are distantly related to echinoderms. In an attempt to finally clarify the structure of sea urchin skeletal elements, and to reconcile conflicting historical analyses, this paper describes a uniquely detailed structural examination of adult SU spines which employs a wide range of analytical techniques including scanning and transmission electron microscopy, X-ray scattering, solid-state NMR (31). The complex ultrastructure revealed succeeds in uniting existing contrasting viewpoints which consider the spine as either single crystalline or polycrystalline and also provides the basis for suggesting a unique growth mechanism.
Results and Discussion
Looking first at the mechanical properties of this unique material, the chonchoidal fracture behavior of SU spines is well documented (9, 32, 33). Our results in Fig. 1 are in good agreement with these earlier investigations with one important exception. In addition to the characteristic curved, chonchoidal surfaces, we frequently observe that the fracture surfaces exhibit areas that are facetted like a calcite single crystal and that planar surfaces are exposed (8). Review of the literature shows that similar fracture surfaces have been observed with a number of SU species (9, 32) (Fig. S1), but that these features were not discussed (8). The sponge-like morphology of the SU spine does not itself contribute to this mixed fracture behavior as was confirmed by fracturing synthetic calcite single crystals with morphologies identical to urchin skeletal plates. These crystals, which were produced in the absence of organic additives by a templating method (5), showed no conchoidal fracture surfaces, but cleaved with the planes expected for geological calcite (Fig. S2) (8).
Fig. 1.

Fracture surfaces of a sea urchin spine shown at different magnifications under SEM (A)–(D). The spine was fractured perpendicular to its growth direction which corresponds to the crystallographic c-axis. Note: (C) is area bounded by white box in (B) and (D) is area bounded by white box in (C)
Examination of the chonchoidal fracture surfaces at higher magnifications using SEM (scanning electron microscopy) provided further insight into the spine structure (Fig. 1 A–C) and revealed the presence of substructures with sizes 10–40 nm [mean diameter 16.5 nm ± 3.6 (SD)] (Fig. 1C). This suggests that the SU spine is composed of nanoparticulate building blocks, a finding which is in agreement with recent studies which proposed that SU skeletal elements are mesocrystalline in structure (25, 26). The observed nanoparticle size also agrees with the 20–40 nm found from SEM and TEM (transmission electron microscopy) examination of other echinoderm species including Echinometra mathaei, Anthocidaris crassispina, and Heterocentrotus mammillatus (25, 26). Although this size is smaller than the 200 nm coherence length measured in the c-axis direction using synchrotron diffraction (24), it agrees reasonably well with the 50 nm coherence length determined in the a-direction. Despite this apparent particulate structure, single crystal diffraction spots confirm that each spine is a calcite single crystal with space group R-3c, a result which is consistent with the literature (24). This data supports a mesocrystal model of a SU spine which combines a polycrystalline architecture with single crystalline scattering behavior. A similar organization has been determined in several SU species (25, 26).
TEM examination of thin sections cut through SU spines provided further insight into their ultrastructures (Fig. 2). Ultramicrotomed sections, which had been prepared by floating onto water, showed regions with a granular substructure comprising a heterogeneous distribution of elongated nanoparticles with lengths of 100–300 nm [mean diameter 118 nm ± 44 nm (SD), 30 measurements] and widths of approximately 50 nm [mean diameter 43 nm ± 14 nm (SD), 30 measurements]. Electron diffraction patterns of these sectioned spine samples (Fig. 2A, Inset) indicate isoorientation of the constituent particles, which is consistent with an average coherence length of 50 nm in the a-direction (24). Significantly, regions with lower electron density were clearly visible between the nanoparticle units; these regions were no longer observed if sections were floated onto ethylene glycol, in which CaCO3 is not soluble (Fig. 2B) (34). This strongly suggests that the calcite nanoparticles are coated with a layer of amorphous calcium carbonate (ACC), as was observed for the aragonite single crystal tablets in nacre (35). ACC has a marked solubility in water even at neutral pH values (36), while crystalline calcium carbonate etches only slowly under the same conditions, leading to preferential dissolution at the interface between the nanoparticles. This technique also enabled the preparation of more complete fragments of the spine in which the coexistence of microscale regions of mesotextured (Fig. 2C) and single crystalline material (Fig. 2D) could be observed. Attempts were also made to prepare spine sections using focussed ion-beam (FIB) techniques, but these proved unsuccessful due to the beam sensitivity of the samples (Fig. S3). That the mesocrystalline ultrastructure observed is a true reflection of the material structure was confirmed using AFM measurements which showed a granular structure in the phase contrast image consistent with the TEM images, and which was lost after calcination of the spine (Fig. S4). Surprisingly, wide angle X-ray diffraction (WAXS) of the sample showed only sharp peaks corresponding to calcite (Fig. S5). The failure to observe an amorphous halo corresponding to ACC can be attributed to the small fraction of ACC present in the spine.
Fig. 2.

(A) TEM image of a thin section of a sea urchin spine (floated onto water). Inset: SAED pattern indicating the section was cut perpendicular to the [001] direction of calcite. (B) TEM image of a thin section of a sea urchin spine (floated onto ethylene glycol). (C), (D) Images of fractured sea urchin spine etched in water overnight.
That the sea urchin spine dissolves nonuniformly in water was confirmed by SEM examination of a ground SU spine which had been stored in water overnight at room temperature (Fig. 2 C and D). The etched regions between the nanoparticles became clearly visible after this treatment, while the heterogeneous size distribution of the nanoparticles was consistent with the TEM analysis [mean lengths 179 nm ± 53 (SD), and mean widths 62 nm ± 13 nm (SD)] (Fig. 2 A and B). These SEM images also showed that the etched samples comprised domains of both etched and nonetched material, where the nonetched domains were several micrometers in size (Fig. 2D, Left). This result therefore supports the structural picture developed from analysis of the fracture surfaces, where single crystalline domains (corresponding to the nonetched regions) are seen to coexist with etched regions.
HRTEM of the crystalline nanoparticle building units provided further insight into their ultrastructures and large defect-free regions are observed which show lattice planes with 0.304 nm fringes (Fig. 3B), which correspond to {104} lattice planes. However, some defects can be identified which differ from the kinking of fringes between mosaic domains observed by Peacor and coworkers 20 y ago (28). Fig. 3A shows several areas up to 4 nm in size where the continuity of the lattice planes is lost. SU spines occlude a small amount of intracrystalline organic matrix (0.02 wt%) (18) and we believe that these defect sites represent the location of these macromolecules and ACC. The area of these defects greatly exceeds the organic matrix fraction, which demonstrates that it must also correspond to ACC.
Fig. 3.

HRTEM of the microstructure of the sea urchin spine (A) A microtomed section of sea urchin spine imaged by HRTEM (The white arrows show the defect regions around the lattice fringes) (B) Higher magnification zoom showing the lattice fringes (white arrows depict the spacing between the start and end of fringe)
FTIR analysis of the spines was also carried out to investigate the atomic-scale ordering of the structure. The data indicated a high level of local distortion around the calcium ions, as shown by the higher ν2/ν4 peak intensity ratio (5.4) as compared with geological calcite (3.0) (37, 38) (See Fig. S6). This distortion can be attributed to a number of factors including the existence of ACC, the presence of magnesium (4 atom-%) and the incorporation of macromolecules (0.02 wt%) (18). The Mg content was determined using ICP analysis which yielded values of 0.93 (0.9) wt.-% Mg2+ (corresponding to a Mg2+ content of 4.0 atom-%) and 36.7 (35.5) wt.-% Ca2+. These values agree well with those obtained from X-ray single crystal structure determination (Table S1, Fig. S7). The ν2 peak does not show broadening, although broadening occurs in ν4 due to the Mg incorporation in the calcite lattice (39) and the existence of ACC. WAXS examination of the spines failed to show any significant peak shifts associated with inclusion of the Mg2+ ions in the calcite lattice. However, Falini and coworkers (39) showed that the presence of up to 5.7 atom-% Mg2+ in Mg-calcite only results in very small peak shifts in the XRD spectra. Single crystal X-ray analysis (Table S1) did, however, show a contraction due to 4 atom-% Mg2+ incorporation, giving lattice constants of 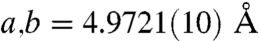 and
and 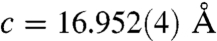 for the urchin Mg-calcite as compared with
for the urchin Mg-calcite as compared with 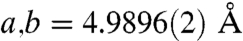 and
and 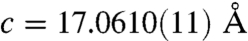 in pure calcite. This corresponds to a c-axis contraction of 0.6%.
in pure calcite. This corresponds to a c-axis contraction of 0.6%.
In order to confirm the mesocrystal structure of the spine, small-angle X-ray scattering (SAXS) and nano-XRD experiments were carried out, yielding data averaged over large volumes and providing information complementary to the SEM and TEM studies. In particular, due to a difference in electron density, ACC should be distinguishable from calcite using SAXS. Measurements using a laboratory instrument and a synchrotron pinhole/Bonse-Hart camera revealed two distinct contributions to the SAXS signal, as evidenced from the azimuthally integrated data (Fig. S8). This immediately suggests the presence of scattering objects with two different size distributions. The analysis of the region at higher angles (higher q-values) on the radial profile using a method applied to other biominerals (40) provided an estimated correlation length of 1.4 ± 0.3 nm (Fig. S9A). The fact that the slope in the intermediate region is approximately -2 tends to indicate that these inclusions are elongated in shape (41). This points to the presence of occluded biomacromolecules with a diameter of 1.4 nm. In order to improve the resolution at lower q-values, additional ultra-SAXS (USAXS) measurements were made to study larger scattering structures. These showed that the second correlation length exceeded 100 nm, which is therefore in good agreement with a structure in which calcite nanoparticles are surrounded by ACC, as observed using TEM (Fig. 2) and reported in the literature (29, 31). It was not, however, possible to precisely determine the ACC layer thickness, most probably due to large variations in the size distribution. The SAXS analysis therefore provides confirmation of the structural model of the SU spine developed from the microscopic analysis. From recent nano-XRD measurements, the angular spread of the nanocrystalline constituents was determined to be less than < 1°, validating the highly single crystalline behavior in the spine (Fig. S9 B–D).
The SU spines were also studied using solid-state 13C NMR. The spectra showed three lines after signal deconvolution which are referred to as “narrow,” “broad,” and “weak” (Table 1) (42). The relative 13C content is listed in the last column, taking into account a required T1 intensity correction factor of 1.84 for the narrow line at 168.55 ppm because the repetition delays of 60 min and 120 min used in the experiments were comparable with the T1 relaxation time of 102 min of this component. The CP line (“weak”, Table 1) has the broadest line width and as the 13C spin is cross-polarized from 1H, it is assigned as ACC (about 8 atom-%). This small amount of ACC is consistent with only ≈2 wt% water, as is determined with thermogravimetric analysis (Fig. S10), and may derive from water present in the structure (CaCO3·H2O). The two other lines (“narrow” and “broad”) show no CP effect. The 13C chemical shift of the narrow component shows that this derives from calcite while the “broad” component is shifted by only 0.2 ppm and is therefore “calcite-like.” Both signals can be explained by the presence of magnesium in the calcite lattice, where each Ca2+ ion and each Mg2+ ion are coordinated by six carbonate groups (43). The 13C chemical shifts of these carbonates are expected to be slightly different from those coordinated to calcium due to the different ionic radii of these ions, which will cause small displacements of atoms in the lattice. As the 13C content of the “broad” line is about 24%, it can be deduced that 24% of carbonate units have a single Mg2+ ion in their coordination shell, which corresponds to a magnesium content of 4 atom % (24 divided by 6). This is in very good agreement with the chemical analysis (4 mol %). The “narrow” 13C line therefore derives from the carbonate groups in the ideal calcite lattice, while the “broad” line corresponds to carbonate groups adjacent to magnesium-substituted sites.
Table 1.
Results of the line shape analysis. Left: Positions and 13C shifts for the three lines: narrow, broad (both from the single pulse excitation (SP) spectra), and “weak” from the cross polarization (CP) spectrum (Fig. S13). Right: Quantitative data for the relative 13C spin content of the three lines (right column: data after T1 correction for the narrow component and subsequent normalization to 100%. The numbers correspond to atom %
| Fit of difference line shapes |
Fit of 13C NMR spectrum; Area (%) |
|||
| Line |
Isotropic shift (ppm) |
Gaussian width (ppm) |
without |
with T1 correction for the narrow line |
| Narrow | 168.55 | 0,45 | 53 | 68 +/− 10* |
| Broad | 168.74 | 0.95 | 35 | 24 +/− 4* |
| “weak” | 168.82 | 1.21 | 12 | 8 +/− 1* |
*Numbers rounded.
Further insight into the structure of the SU spine was also obtained by annealing samples in air at 450 °C for 4 h. Examination of samples that had been fractured after annealing revealed the presence of nonspherical holes which ranged from tens, to several hundreds of nanometres in size (Fig. S11). The size of the pores decreases from the center to the edge of the fractured specimens. As the pores are generated by the decomposition of occluded macromolecules and crystallization of ACC, this may indicate a higher concentration of occlusions near the center. Similar effects were observed from AFM analysis of native and similarly annealed spine samples (Fig. S4). Image analysis shows that the pores occupy 13.5 area %, which corresponds to 3.7 vol % if a spherical pore shape is assumed. Crystallization of calcite from ACC results in a 40% loss in the total volume, as estimated from the densities of these two phases (DACC = 1.62 g/cm3, DCalcite = 2.75 g/cm3) (44). In terms of a molar volume change from CaCO3·H2O to calcite, the change is 49%. In a similar fashion, at higher annealing temperatures (approximately 800 °C) the transformation of calcite to CaO would constitute in a 55% loss in total volume, creating macropores in the material (45). If the pores are considered to result from the volume shrinkage accompanying ACC recrystallization during heating, an ACC content in the native urchin spine of 9.3 vol % (4.9 atom-%) can be calculated. This value is lower than the 8 atom % value derived from the NMR studies and can be attributed to the assumption that the pores are spherical and the minimum threshold size of pores (≈5 nm) that can be discriminated in the image analysis. As both macromolecules and ACC are entrapped within the dense mineral phase, it is also possible that complete decomposition and loss of side-products does not occur on annealing, leading to reduced pore formation. In summary, the ACC content is calculated to be about 8 atom % by using NMR analysis and to be about 4.9 atom % by calculating the area of the pores in the annealed spine. FTIR also demonstrated the presence of ACC, while both the WAXS and HRTEM provide indirect proof for the existence of ACC in the sea urchin spine (Fig. S14).
Our demonstration that the adult urchin spine contains ACC distributed throughout the biomineral is highly significant and provides insight into the formation mechanism of the spine. With the total quantity of ACC in the spine estimated at 8 atom %, our calculations of a 5 atom % ACC layer coating each 50 × 50 × 150 nm calcite nanoparticle would correspond to approximately 2 nm thick layer. The remaining 3 atom % of ACC is occluded within the calcite nanoparticles as domains < 5 nm in diameter (Fig. 3). The dimensions of these regions are in the size range of typical biomacromolecules with a diameter of 1.4 + /-0.3 nm (SAXS). This implies that macromolecules occluded within the developing calcite nanocrystals may locally hinder crystallization, leading to ACC inclusions. Such a mechanism is analogous to poisoning of a calcite growth front which has been suggested to occur during the crystallization of ACC (35). Macromolecules active in stabilizing ACC are expelled from the newly forming crystal lattice, which leads to an increased concentration in the residual ACC. This eventually results in its permanent stabilization as the observed ACC layer (35). The fracture properties of the spine are also consistent with this structural model. The two-phase structure of the spine is a mechanically optimized composite, where a filler (here, soft ACC and macromolecules) modulates the mechanical properties of the hard, but brittle calcite. The nanoparticulate structure also enhances the mechanical properties in that fracture is driven to follow a tortuous path, resulting in the observed chonchoidal fracture surface. The larger, facetted, micron-sized regions observed on the fracture surface correspond to larger single crystal particles within the spine, which can arise due to the nonuniform distribution of macromolecules in the ACC precursor phase.
A model for the formation mechanism of the adult spine can now be developed based both on the structural data presented here and on prior literature (Fig. 4). SU spines and spicules form via a transient ACC precursor phase (37, 46) and domains of 40–120 nm have been observed in the early stages of development of the spicule (Fig. S12B) (47, 49). Biogenic ACC contains occluded macromolecules and Mg2+ ions which stabilize it against rapid crystallization (9, 14, 18, 43), a process which is essential if nucleation is to be initiated at a single site only. Once nucleation of calcite has occurred within the ACC phase, the calcite crystal grows at the expense of the ACC phase according to a mechanism of secondary nucleation, as described by Killian et al. (48), partly expelling and partly occluding macromolecules from the newly formed crystal lattice. The occluded macromolecules are retained as small, amorphous regions within the calcite nanocrystal (Fig. 3 and Fig. S12B). Growth of the calcite nanocrystal is terminated when a critical macromolecule concentration is reached in the growth layer which inhibits further crystallization and results in the formation of an ACC layer on the nanoparticle surface (35). As the concentration of macromolecules in the ACC is low (ca. 0.02 wt%) (9, 14, 18), their distribution is likely to be inhomogeneous, resulting in some regions which are not stabilized against crystallization. Such regions can continue to grow through the ACC layer until they impinge upon adjacent ACC particles (which contain a lower concentration of occluded macromolecules). The presence of the calcite crystals then triggers crystallization of the ACC nanoparticles, which proceeds by an identical mechanism (Fig. S12C). This process leads to the formation of mineral bridges between neighboring nanoparticles as observed by Oaki and Imai (25) and Killian et al. (48), and explains their perfect mutual crystallographic orientation, as the lattice structure is in fact continuous. As the formation of connections is random, the crystallization path is tortuous (47). Crystallization terminates when the initial ACC phase has been consumed, and the structure has been transformed into a magnesium calcite mesocrystal. Residual ACC is present as a surface layer on the calcite nanoparticles, and as inclusions within the nanocrystallites in approximately equal quantities (Fig. S12D). It is emphasized that mineral bridges are not required for perfect nanocrystal orientation as recent molecular dynamics simulations have shown that silver nuclei in an amorphous phase can undergo rotation and realignment to bring them into register (50).
Fig. 4.

Schematic representation of structural hierarchy in the sea urchin spine. At the cm-mm scale, as visualized and rendered from microtomography measurements, the spine is shown to be a complex, porous material [scale bar = 5 μm]. When examined at the μm scale, the microstructure of the mineral becomes apparent, and rhombohedral as well as chonchoidal fracture surfaces are typically observed. Here, light areas represent macroporosity while dark purple areas represent the mineralized sea urchin spine. At the nanoscale, a mesocrystalline organization is apparent where the ordered nanocrystal building units are shown in blue, while ACC inclusions and surface layers are shown as pink dots and lines, respectively.
Conclusion
We show a structural model of the adult sea urchin spine which both resolves the dichotomies present in existing data and explains the observed mechanical properties. This ultrastructural study conclusively demonstrates that the sea urchin spine has a mesocrystalline structure and provides the foundation for a unique growth mechanism based on the concerted crystallization of a 3D array of amorphous nanoparticles. Formation of a mesostructured material from an amorphous precursor phase clearly provides an organism with many advantages, as it combines the ability to rapidly form a material with complex morphology with ease of control over the composition, ultrastructure, and material properties. It would be highly surprising if more biominerals are not subsequently shown to form via similar mechanisms. Recent work utilizing the X-PEEM technique on the highly mineralized sea urchin tooth have found that each crystalline component possesses highly cooriented crystallographic axes and undergoes a secondary nucleation mechanism, further supporting the formation mechanism shown in the spine (48, 51, 52). Nature demonstrates how this crystal growth mechanism can be used to form composite materials with remarkable and highly controllable structures and properties; now provides enormous potential for the translation of similar design strategies to the fabrication of synthetic functional materials.
Materials and Methods
FTIR.
A sea urchin spine from the sea urchin Authoeidaris Erassispina was ground in a mortar with a pestle and infrared spectra were recorded using a FTIR (NICOLET 380, Thermo Scientific) spectrometer at 4 cm-1 resolution. KBr pellets were prepared for the FTIR samples.
Solid-State NMR.
All NMR experiments were carried out using a BRUKER AVANCE 600 spectrometer using double resonant NMR probes with 7 mm zirconia rotors spun at 6.5 kHz under the magic angle (Magic Angle Sample Spinning, MAS).
SAXS/USAXS/Nano-XRD.
SAXS data was collected using a Nanostar microdiffractometer setup (Bruker AXS Gmbh) with a copper anode generator operated at 40 kV and 35 mA. USAXS data was collected at the ID02 beamline of the European Synchrotron Radiation Facility (ESRF). For nanocrystallography measurements, a X-ray beam at beamline ID13 (ESRF) was monochromatized to an energy of 15.26(5) keV (λ = 0.812(2) Å) was used.
Electron Microscopy.
TEM/HRTEM.
Sample sections were floated onto either water or ethylene glycol and collected onto 3 mm copper mesh grids. TEM/HRTEM analyses were performed using a JEOL JEM 1200 EX (JEOL GmbH) and a JEOL JEM-2100F-UHR (JEOL GmbH), respectively.
Supplementary Material
ACKNOWLEDGMENTS.
Rona Pitschke (MPI) is acknowledged for help with microtoming and Ingrid Zenke (MPI) for WAXS and laboratory SAXS measurements. Jon Jones and Sean Lenegan (Bristol) are acknowledged for their help on EM studies. We thank the Max Planck Society and the German Research Foundation (DFG, SPP 1117) for financial support of this work. Y.Y.K. thanks the EPSRC for funding (EP/E037364/2). J.S. acknowledges support from EU Marie Curie EST Fellowship on Biomimetic Systems, MEST-CT-2004-504465. The European Synchrotron Radiation Facility (ESRF) is acknowledged for provision of beamtime.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. F.W. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1109243109/-/DCSupplemental.
References
- 1.Raup D. Physiology of Echinodermata. New York/London: Wiley; 1966. pp. 379–395. [Google Scholar]
- 2.Donnay G, Pawson D. X-ray diffraction studies of echinoderm plates. Science. 1969;166:1147–1150. doi: 10.1126/science.166.3909.1147. [DOI] [PubMed] [Google Scholar]
- 3.Moureaux C, et al. Structure, composition and mechanical relations to function in sea urchin spine. J Struct Biol. 2010;170:41–49. doi: 10.1016/j.jsb.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Aizenberg J, Muller D, Grazul J, Hamann D. Direct fabrication of large micropatterned single crystals. Science. 2003;299:1205–1208. doi: 10.1126/science.1079204. [DOI] [PubMed] [Google Scholar]
- 5.Park R, Meldrum F. Shape-constraint as a route to calcite single crystals with complex morphologies. J Mat Chem. 2004;14:2291–2296. [Google Scholar]
- 6.Li C, Qi L. Bioinspired fabrication of 3D ordered macroporous single crystals of calcite from a transient amorphous phase. Angew Chem Int Edit. 2008;47:2388–2393. doi: 10.1002/anie.200705403. [DOI] [PubMed] [Google Scholar]
- 7.O’Neill P. Polycrystalline echinoderm calcite and its fracture mechanics. Science. 1981;213:646–648. doi: 10.1126/science.213.4508.646. [DOI] [PubMed] [Google Scholar]
- 8.Towe K. Echinoderm calcite: Single crystal or polycrystalline aggregate. Science. 1967;157:1048–1050. doi: 10.1126/science.157.3792.1048. [DOI] [PubMed] [Google Scholar]
- 9.Weiner S, Addadi L. Design strategies in mineralized biological materials. J Mat Chem. 1997;7:689–702. [Google Scholar]
- 10.Emlet R. Echinoderm calcite: A mechanical analysis from larval spicules. Biol Bull. 1982;163:264–275. [Google Scholar]
- 11.Beniash E, Addadi L, Weiner S. Cellular control over spicule formation in sea urchin embryos: A structural approach. J Struct Bio. 1999;125:50–62. doi: 10.1006/jsbi.1998.4081. [DOI] [PubMed] [Google Scholar]
- 12.Wilt F. Matrix and mineral in the sea urchin larval skeleton. J Struct Bio. 1999;126:216–226. doi: 10.1006/jsbi.1999.4105. [DOI] [PubMed] [Google Scholar]
- 13.Addadi L, Weiner S. Interaction between acidic proteins and crystals: Stereochemical requirements in biomineralization. Proc Natl Acad Sci USA. 1985;82:4110–4114. doi: 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albeck S, Aizenberg J, Addadi L, Weiner S. Interactions of various skeletal intracrystalline components with calcite crystals. J Am Chem Soc. 1993;115:11691–11697. [Google Scholar]
- 15.Berman A, Addadi L, Weiner S. Interactions of sea-urchin skeleton macromolecules with growing calcite crystals—A study of intracrystalline proteins. Nature. 1988;331:546–548. [Google Scholar]
- 16.Cölfen H, Antonietti M. Mesocrystals: Inorganic Superstructures Made by Highly Parallel Crystallization and Controlled Alignment. Angew Chem Int Edit. 2005;44(35):5576–5591. doi: 10.1002/anie.200500496. [DOI] [PubMed] [Google Scholar]
- 17.Cölfen H, Antonietti M. Mesocrystals and Nonclassical Crystallization. Chichester: Wiley; 2008. p. 288. [Google Scholar]
- 18.Aizenberg J, Hanson J, Koetzle T, Weiner S, Addadi L. Control of macromolecule distribution within synthetic and biogenic single calcite crystals. J Am Chem Soc. 1997;119:881–886. [Google Scholar]
- 19.Gilbert P, Yang L, Killian C, Kunz M, Tamura N. Biomineral nanoparticles are space-filling. Nanoscale. 2011;3:603–609. doi: 10.1039/c0nr00697a. [DOI] [PubMed] [Google Scholar]
- 20.Tsipursky S, Buseck P. Structure of magnesium calcite from sea urchins. Am Mineral. 1993;78:775–781. [Google Scholar]
- 21.Travis D, editor. The Comparative Ultrastructure and Organization of Five Calcified Tissues. New York: Appleton-Century-Crofts; 1970. pp. 203–311. [Google Scholar]
- 22.Berman A, et al. Intercalation of sea-urchin proteins in calcite—Study of a cyrstalline composite-material. Science. 1990;250:664–667. doi: 10.1126/science.250.4981.664. [DOI] [PubMed] [Google Scholar]
- 23.Berman A, et al. Biological control of crystal texture: A widespread strategy for adapting crystal properties to function. Science. 1993;259:776–779. doi: 10.1126/science.259.5096.776. [DOI] [PubMed] [Google Scholar]
- 24.Aizenberg J, et al. Biologically induced reduction in symmetry—A study of crystal texture of calcitic sponge spicules. Chem-Eur J. 1995;1(7):414–422. [Google Scholar]
- 25.Oaki Y, Kotachi A, Miura T, Imai H. Bridged nanocrystals in biominerals and their biomimetics: Classical yet modern crystal growth on the nanoscale. Adv Funct Mater. 2006;2:1633–1639. [Google Scholar]
- 26.Oaki Y, Imai H. Nanoengineering in echinoderms: The emergence of morphology from nanobricks. Small. 2006;2:66–70. doi: 10.1002/smll.200500246. [DOI] [PubMed] [Google Scholar]
- 27.Sethmann I, Putnis A, Grassmann O, Löbmann P. Observation of nano-clustered calcite growth via a transient phase mediated by organic polyanions: A close match for biomineralization. Am Mineral. 2005;90:1213–1217. [Google Scholar]
- 28.Blake D, Peacor D, Allard L. Ultrastructural and microanalytical results from echinoderm calcite: Implications for biomineralization and diagenesis of skeletal material. Micron Microsc Acta. 1984;15(2):85–90. [Google Scholar]
- 29.Li X, Huang Z. Unveiling the formation mechanism of pseudo-single-crystal aragonite platelets in nacre. Phys Rev Lett. 2009;102:075502-1–075502-4. doi: 10.1103/PhysRevLett.102.075502. [DOI] [PubMed] [Google Scholar]
- 30.Sethmann I, Wörheide G. Structure and composition of calcareous sponge spicules: A review and comparison to structurally related biominerals. Micron. 2008;39:209–228. doi: 10.1016/j.micron.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Jäger C, Cölfen H. Fine structure of nacre revealed by solid state 13C and 1H NMR. Cryst Eng Comm. 2007;9:1237–1244. [Google Scholar]
- 32.Lowenstam H, Weiner S. On Biomineralization. New York: Oxford University Press; 1989. p. 323. [Google Scholar]
- 33.Simkiss K, Wilbur K. Cell Biology and Mineral Deposition. San Diego: Academic; 1989. Biomineralization. [Google Scholar]
- 34.Pingitore N, et al. Dissolution Kinetics of CaCO3 in Common Laboratory Solvents. J Sediment Petrol. 1993;63(4):641–645. [Google Scholar]
- 35.Nassif N, et al. Amorphous layer around aragonite platelets in nacre. Proc Natl Acad Sci USA. 2005;102:12653–12655. doi: 10.1073/pnas.0502577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aizenberg J, Lambert G, Addadi L, Weiner S. Stabilization of amorphous calcium carbonate by specialized macromolecules in biological and synthetic precipitates. Adv Mater. 1996;8(3):222–226. [Google Scholar]
- 37.Beniash E, Aizenberg J, Addadi L, Weiner S. Amorphous calcium carbonate transforms into calcite during sea urchin larval spicule growth. P Roy Soc Lond B Bio. 1997;264(1380):461–465. [Google Scholar]
- 38.Gueta R, et al. Local atomic order and infrared spectra of biogenic calcite. Angew Chem-Int Edit. 2007;46(1-2):291–294. doi: 10.1002/anie.200603327. [DOI] [PubMed] [Google Scholar]
- 39.Falini G, Fermani S, Gazzano M, Ripamonti A. Structure and morphology of synthetic magnesium calcite. J Mat Chem. 1998;8:1061–1065. [Google Scholar]
- 40.Gourrier A, et al. Scanning X-ray imaging with small-angle scattering contrast. J Appl Crystallogr. 2007;40:s78–s82. [Google Scholar]
- 41.Glatter O, Kratky O, editors. Small-Angle X-ray Scattering. New York: Academic; 1982. [DOI] [PubMed] [Google Scholar]
- 42.Papenguth H, Kirkpatrick R, Montez B, Sandberg P. 13 C MAS NMR spectroscopy of inorganic and biogenic carbonates. Am Mineral. 1989;74:1152–1158. [Google Scholar]
- 43.Addadi L, Raz S, Weiner S. Taking advantage of disorder: Amorphous calcium carbonate and its roles in biomineralization. Adv Mater. 2003;15(12):959–970. [Google Scholar]
- 44.Bolze J, et al. Formation and Growth of Amorphous Colloidal CaCO3 Precursor Particles as detected by time-resovled SAXS. Langmuir. 2002;18:8364–8369. [Google Scholar]
- 45.Rodriguez-Navarro C, Ruiz-Agudo E, Luque A, Rodriguez-Navaro A, Ortega-Huertas M. Thermal decomposition of calcite: Mechanisms of formation and textural evolution of CaO nanocrystals. Am Mineral. 2009;94:578–593. [Google Scholar]
- 46.Politi Y, Arad T, Klein E, Weiner S, Addadi L. Sea urchin spine calcite forms via a transient amorphous calcium carbonate phase. Science. 2004;306:1161–1164. doi: 10.1126/science.1102289. [DOI] [PubMed] [Google Scholar]
- 47.Politi Y, et al. Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule. Proc Natl Acad Sci USA. 2008;105(45):17362–17366. doi: 10.1073/pnas.0806604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Killian C, et al. Mechanism of Calcite Co-Orientation in the Sea Urchin Tooth. J Am Chem Soc. 2009;131:18404–18409. doi: 10.1021/ja907063z. [DOI] [PubMed] [Google Scholar]
- 49.Gibbins J, Tilney L, Porter K. Microtubules in formation and development of primary mesenchyme in arbacia punctulata. i. Distribution of microtubules. J Cell Biol. 1969;41(1):201–226. doi: 10.1083/jcb.41.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang J, You H, Kong P, Ding B, Song X. Size-dependent structure transformation from amorphous phase to crystal. Appl Phys Lett. 2008;92(14):143111–143114. [Google Scholar]
- 51.Killian C, Wilt F. Characterization of the proteins Comprising the Integral Matrix of Strongylocentrotus purpuratus embryonic spicules. J Biol Chem. 1996;271(15):9150–9159. doi: 10.1074/jbc.271.15.9150. [DOI] [PubMed] [Google Scholar]
- 52.Gilbert P, Young A, Coppersmith S. Measurement of c-axis angular orientation in calcite (CaCO3) nanocrystals using X-ray absorption spectroscopy. Proc Natl Acad Soc USA. 2011;108(28):11350–11355. doi: 10.1073/pnas.1107917108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.