
Figure 1.
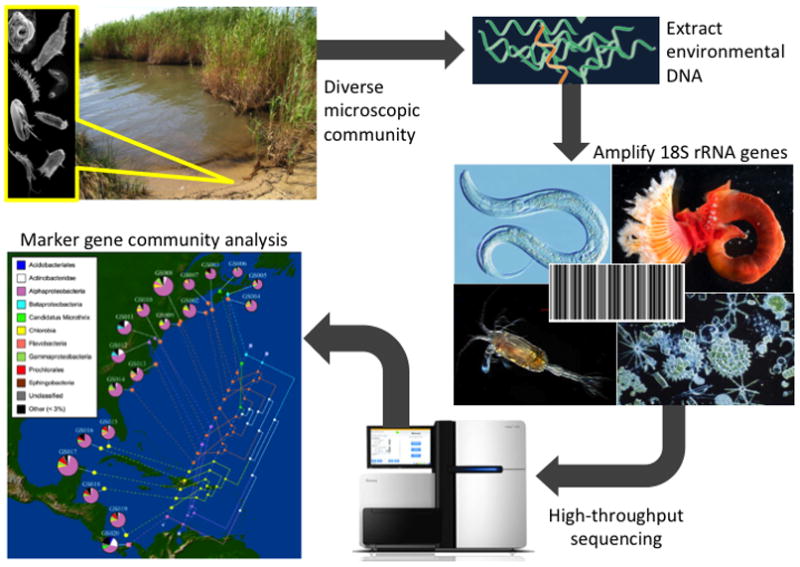
Typical standardized workflow (from environment to sequences) for high-throughput marker gene studies. Soils and sediments are typically frozen upon collection (−80°C to preserve RNA) and brought back to the lab for bulk extraction of environmental DNA. Marker genes (e.g. rRNA) are amplified from genomic extracts using barcoded, conserved primer pairs. Following high-throughput sequencing (typically conducted on 454 or Illumina platforms), datasets are processed and clustered into Operational Taxonomic Units (OTUs) under a range of pairwise identity cutoffs. OTUs are subsequently used to conduct alpha and beta diversity analyses, summarize community taxonomy, and interpret assemblages in a phylogenetic context. (Depiction of community analysis modified from Parks et al. [56])