

Abstract
A new type of rhodium-catalyzed [5+2] cycloaddition was developed for the synthesis of seven-membered rings with diverse functionalities. The ring formation was accompanied by a 1,2-acyloxy migration event. The 5- and 2-carbon components of the cycloaddition are 3-acyloxy-1,4-enynes (ACEs) and alkynes respectively. Cationic rhodium (I) catalysts worked most efficiently for the intramolecular cycloaddition, while only neutral rhodium (I) complexes could facilitate the intermolecular reaction. In both cases, electron-poor phosphite or phosphine ligands often improved the efficiency of the cycloadditions. The scope of ACEs and alkynes was investigated in both intra- and intermolecular reactions. The resulting seven-membered ring products have three double bonds that could be selectively functionalized.
1. INTRODUCTION
The Diels-Alder reaction is one of the most powerful tools for the construction of substituted six-membered rings. In contrast, it is difficult to find a cycloaddition method for the synthesis of seven-membered rings that can fully match the scope and impact of the Diels-Alder reaction, in spite of the prevalence of cycloheptanes in natural products and pharmaceutical agents.1 Efficient synthesis of seven-membered rings with diverse functionalities continues to stimulate the development of novel cycloaddition reactions. Three types of two-component cycloadditions exist for the synthesis of seven-membered rings: [4+3],2 [5+2],3 and [6+1]4 cycloadditions. The first two methods are more general since diverse 2- and 4-carbon synthons are readily available. The discovery of new 3-and 5-carbon synthons, therefore, would be highly desirable for the development of novel cycloaddition reactions that can lead to functionalized seven-membered rings.
Transition metal catalysts often facilitate reactions that are difficult or not possible under thermal conditions and they have proven to be particularly valuable in cycloaddition reactions. 5 The [5+2] cycloaddition of vinylcyclopropanes (VCPs) and alkynes (eq 1), a remarkable homo-Diels-Alder reaction, could be realized by different transition metal catalysts developed in the research groups of Wender,6–8 Trost,9 Louie,10 Fürstner,11 Yu,12 and Murai.13,14 The significance of the intramolecular [5+2] cycloaddition of VCPs and alkynes was highlighted in the synthesis of several natural products.15 The mechanism of this novel cycloaddition was extensively investigated by Houk, Wender, and Yu.16 A few other five-carbon (5C) synthons for [5+2] cycloadditions were also discovered by Stryker17 and Tanino18 when employing stoichiometric amount of cobalt.
Compared to other transition metal-catalyzed [m+n] cycloadditions, the [5+2] cycloaddition is still underdeveloped, especially with respect to the intermolecular version. Although various catalysts were discovered for the intramolecular [5+2] cycloaddition of VCPs and alkynes,6,9–13 only few examples of intermolecular counterparts are known due to challenging chemo- and regioselectivity issues.7 The first Rh-catalyzed intermolecular [5+2] cycloaddition was developed by Wender’s group utilizing an activated vinylcyclopropane. 7a Subsequently, the scope of the 5C synthon was expanded to unactivated systems.7b,7c The significance of the intermolecular reaction is substantial in the preparation of libraries of cycloheptenes from diverse commercially available alkynes.8
 |
(1) |
 |
(2) |
 |
(3) |
We have reported a Rh(I)-catalyzed cycloisomerization of 3-acyloxy-4-ene-1,9-diyne 1 to form bicyclo-[5.3.0]decatriene 2 (eq 2).19 One can view this transformation as an intramolecular [5+2] cycloaddition of 3-acyloxy-1,4-enyne (ACE) and a tethered alkyne accompanied by a 1,2-acyloxy migration of propargyl ester. In all previous examples, R3 was limited to hydrogen for ene-diyne 1. In this article, we presented the details for the development of the intramolecular [5+2] cycloaddition, expanded its scope to include ACEs bearing an internal alkyne (R3 = ester, ketone, or Br, eq 2), and disclosed our study on the intermolecular reaction of ACE 3 with alkyne 4 for the first time (eq 3). These two new [5+2] cycloadditions provide easy access to seven-membered rings with substituents and functionalities that were complementary to existing methods.
As a 5-carbon synthon, ACE has been used for the synthesis of five- and six-membered rings in transition metal catalyzed reactions. Rautenstrauch first reported that ACE 6 could undergo cycloisomerization to form cyclopentadiene 7 in the presence of a palladium catalyst (Scheme 1).20 The cascade reaction was initiated by a Pd(II)-promoted 1,2-acyloxy migration of the propargyl ester in complex 8 to form intermediate 9. Three pathways were proposed by Rautenstrauch for the conversion of this intermediate to cyclopentadiene 7. In pathway a, insertion of an olefin to the carbon-metal bond gave bicyclic intermediate 10, which underwent elimination to afford diene 7. Alternatively, product 7 could be formed by reductive elimination of metallacyclohexadiene 11, derived from either direct cyclization of metal complex 9 (pathway b), or through carbene intermediate 12 via a 6-π electrocyclization (pathway c). The scope of this rearrangement was later expanded using gold21 and platinum22 catalysts for the synthesis of functionalized five-membered rings.23 ACE was recently also employed as a 5C synthon in a novel Rh(I)-catalyzed [5+1] cycloaddition with CO for the synthesis of highly substituted phenols.24
Scheme 1.

Mechanism proposed by Rautenstrauch for the rearrangement of ACE 6 to cyclopentadiene 7
2. RESULTS AND DISCUSSION
During the search for alternative ways to generate cyclopropyl metal carbenes for cycloadditions,25 we found that [Rh(CO)2Cl]2 was able to catalyze 1,3-acyloxy migration of propargyl esters to form allenes,26 which was previously realized by π-acidic metals such as gold, platinum, or silver.27 We then decided to examine the possibility of Rh(I)-catalyzed 1,2-acyloxy migration of a simple propargyl esters for the formation of vinyl Rh(I) carbene.
Treatment of ester 13 with [Rh(CO)2Cl]2 catalyst in the presence of styrene indeed provided us known cyclopropane 1428 diastereoselectively (eq 4), demonstrating that Rh(I) carbene 15 could be generated by a 1,2-acyloxy migration of propargyl ester. We did not further optimize this reaction since it was well-documented. This type of cyclopropanation was first reported in a PtCl2-catalyzed intramolecular enyne cycloisomerization. 29 The intermolecular version was then extensively investigated using [RuCl2(CO)3]2 and other metal catalysts. 28,30 The enantioselective inter- and intramolecular cyclopropanations mediated by chiral gold complexes were subsequently developed.31 The atom-economical32 formation of metal carbenes from propargyl esters via 1,2-acyloxy migration has been applied in many other transformations catalyzed by gold,33–36 platinum,34,35,37 copper,35 and rhodium.38
 |
(4) |
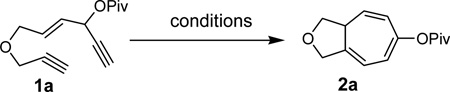
We envisioned that the combination of the novel reactivity of Rh(I) catalyst for facilitating 1,2-acyloxy migration and its well-known capability to catalyze cycloadditions might offer myriad opportunities for the design of new reactions. For example, if metal complex 11 could be formed in the presence of a Rh(I) catalyst and intercepted by an alkyne, a conceptually new [5+2] cycloaddition could then be capitalized. Inspired by Rautenstrauch’s pioneering work on the rearrangement of ACE to cyclopentadienes, we proposed that the ACE moiety in substrate 1a might be a suitable 5-carbon synthon for a Rhcatalyzed intramolecular [5+2] cycloaddition with concomitant 1,2-acyloxy migration to afford bicyclic product 2a (Table 1). There are a number challenges, however, for this transformation. The 1,6-enyne in substrate 1a might undergo cycloisomerization prior to 1,2-acyloxy migration. If a carbene intermediate similar to 12 was generated, it might undergo cyclopropanation or cyclopropenation with alkenes and alkynes presented in the system. Rautenstrauch rearrangement to form cyclopentadiene would be another potential competing pathway.
Table 1.
Screening of catalysts and conditions for the cycloisomerization of enyne 1a
 | ||
|---|---|---|
| entry | conditions | yield |
| 1 | [Rh(CO)2Cl]2 (5 mol %), toluene, 90 °C, 8h | 19%a |
| 2 | [Rh(CO)2Cl]2 (5 mol %), DCE, 90 °C, 8h | 48%a |
| 3 | [Rh(CO)2Cl]2 (5 mol %), TCE,b 90 °C, 1.5h | 43%a |
| 4 | [Rh(COD)Cl]2 (5 mol %), TCE, 90 °C, 8h | 21%a |
| 5 | Rh(PPh3)3Cl (5 mol %), TCE, 90 °C, 8h | 77%a |
| 6 | [Rh(COD)2]BF4 (5 mol %), DCE, rt, 8h | 70%a |
| 7 | [Rh(COD)2]BF4 (5 mol %), toluene, rt, 8h | NRc |
| 8 | [Rh(COD)2]BF4 (5 mol %), dioxane, rt, 8h | NR |
| 9 | [Rh(COD)2]BF4 (5 mol %), TCE, 50 °C, 20h | 81%a |
| 10 | [Rh(COD)2]BF4 (5 mol %), CH2Cl2, rt, 8h | 83%a |
| 11 | [Rh(COD)2]BF4 (3 mol %), CH2Cl2, rt, 16h | 85%d |
| 12 | AuCl(PPh3) (5 mol %), AgOTf (5 mol %), MeCN, rt, 20h | 0 |
| 13 | PtCl2, (10 mol %), DCE, 80 °C, 20h | 0 |
| 14 | HNTf2 (10 mol %), DCM, rt, 20h | 0 |
Yields were calculated based on 1H NMR using internal standard.
tetrachloroethane.
No reaction.
Isolated yield.
Substrate 1a was easily prepared from commercially available cis-2-butene-1,4-diol in just four operations.39 To our delight, when compound 1a was treated with catalytic amount of [Rh(CO)2Cl]2, product 2a was obtained in 19% and 48% yields in toluene and dichloroethane respectively (Table 1, entries 1 and 2). A number of other rhodium catalysts also worked for this cycloisomerization (entries 4–6). The cationic Rh(I) complex could catalyze the reaction even at room temperature (entry 6). The solvent-dependence was also investigated (entries 7–11). Bicyclic product 2a with a cycloheptatriene moiety was thus prepared efficiently from readily available linear substrate 1a under mild conditions (entry 11) using a Rh(I) catalyst. On the other hand, typical π-acidic metals such as Au(I) and Pt(II) and Brønsted acid did not promote the desired transformation (entries 12–14).
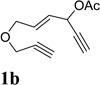
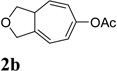
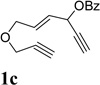
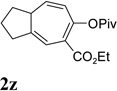
We then investigated the scope of this cycloisomerization (Table 2). The reaction worked well when the ester was changed from pivalate to acetate or benzoate (entries 1 and 2). Substrates with nitrogen or gem-diester linkers yielded bicyclic compounds 2d and 2e smoothly (entries 3 and 4). The structure of cycloisomerization product was unambiguously assigned by the X-ray analysis of product 2d (CCDC 823148).19 The reaction also tolerated mono- or gemsubstitution on the propargylic position of the tether (entries 5 and 6).
Table 2.
Scope of the cationic Rh(I)-catalyzed cycloisomerization of enyne 1a
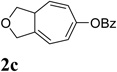
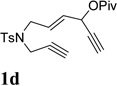
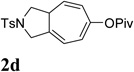
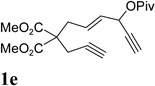
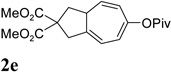
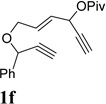
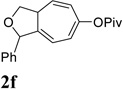
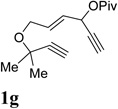
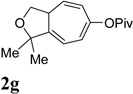
Condition: [Rh(COD)2]BF4 (3–5 mol %), CH2Cl2 (0.05 M), rt, 8–48h.
All yields were isolated yields.
50 °C.
The dr of the substrate was 1:1.
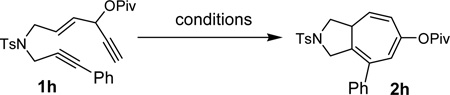
Only trace amount of product, however, was observed for substrate 1h, which has an internal alkyne for the 2-carbon component (Table 3, entry 1). This significantly limited the scope of the cycloisomerization. We therefore optimized the condition for this substrate by examining the effect of ligands using cationic [Rh(COD)2]BF4 complex. Mono- and bi-dentate phosphine ligands such as PPh3, (i-Bu)3P, or dppe had no effect (entries 2–4). The yield of product 2h was improved to 21% by the addition of phosphite ligand (entry 5). Similar improvement was also observed with electron-poor phosphine ligand (entries 6). Most of starting materials were recovered in these two cases. We then combined the above two structural features and tested electron-poor phosphite ligands. Gratifyingly, substrate 1h was completely consumed within 8h using (CF3CH2O)3P ligand and product 2h was isolated in 88% yield (entry 7). More sterically demanding electron-poor phosphite ligand [(CF3)2CHO]3P decreased the efficiency of this reaction (entry 8).
Table 3.
Screening of ligands for the cycloisomerization of substrate 1h bearing an internal alkynea
 | ||
|---|---|---|
| entry | ligand | yield |
| 1 | no ligand | trace |
| 2 | PPh3 | NRc |
| 3 | (i-Bu)3P | NR |
| 4 | dppe | NR |
| 5 | (EtO)3P | 21%b |
| 6 | (p-CF3C6H4)3P | 12% b |
| 7 | (CF3CH2O)3P | 88%d |
| 8 | [(CF3)2CHO]3P | 14%b |
[Rh(COD)2]BF4 (5 mol %), ligand (10 mol %), CH2Cl2, 50 °C, 8–16h.
Yields were calculated based on 1H NMR using internal standard.
No reaction.
Isolated yield.
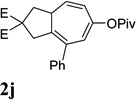
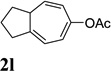
Dramatic improvements were also observed for other substrates with internal alkynes using the combination of [Rh(COD)2]BF4 and (CF3CH2O)3P (Table 4, entries 1–3). For substrates 1i–1k, no reaction or only trace amount of the desired products were observed using just the cationic Rh(I) catalyst. Moderate yields (40–50%) were obtained for substrate 1l with 3–10 mol % [Rh(COD)2]BF4 complex alone. The addition of (CF3CH2O)3P ligand again increased the yield of product 2l (entry 4).
Table 4.
Scope of [Rh(COD)2]BF4 / (CF3CH2O)3P-catalyzed cycloisomerization of enyne 1a
| entry | substrate | product | yieldb |
|---|---|---|---|
| 1 | 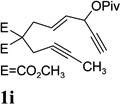 |
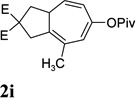 |
82% |
| 2 | 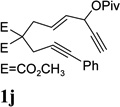 |
 |
70% |
| 3 | 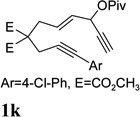 |
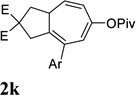 |
60% |
| 4 | 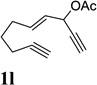 |
 |
76% |
| 5c | 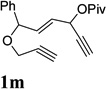 |
 |
76% |
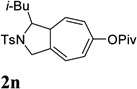
| 6c | 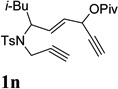 |
 |
80% |
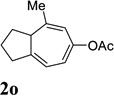
| 7 | 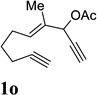 |
 |
80% |
| 8 | 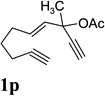 |
– | comlex mixture |
| 9 | 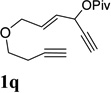 |
– | 0% |
| 10 | 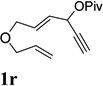 |
– | 0% |
Condition: [Rh(COD)2]BF4 (5–10 mol %), (CF3CH2O)3P (10–20 mol %), DCM (0.025–0.05 M), 50 °C, 8–4h.
All yields were isolated yields.
The dr of the substrate was 1:1.
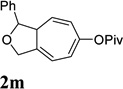
When we examined the substitution effect in the tether region of the 1,6-enyne, we found that substituents adjacent to alkyne had no apparent effect and the cycloisomerization worked well using the cationic Rh(I) catalyst alone (Table 2, entries 5 and 6). Substituents adjacent to alkene, however, slowed down the reaction significantly. The addition of (CF3CH2O)3P ligand was necessary to obtain good yields for substrates 1m and 1n (Table 3, entries 5 and 6). Substrate 1o with a trisubstituted olefin also worked well and afforded product 2o (entry 7). This new catalyst composed of cationic Rh(I) and electro-poor phosphite ligand as shown in Table 4 appeared to be more general than the previous catalytic system involving just the cationic [Rh(COD)2]BF4.
A complex mixture was observed for substrate 1p bearing a tertiary propargyl ester (entry 8). Under the standard condition in Table 4, no reaction was observed for substrates 1q and 1r, where the ACE was tethered with an alkyne by six atoms or tethered with an alkene (entries 9 and 10).
Propargyl esters with an internal alkyne tend to undergo 1,3-acyloxy migration in the presence of π-acidic transition metal catalysts.27k When we treated substrate 1s with cationic Rh(I) catalyst, only trace amount of product 2s was observed and most starting materials were recovered (Scheme 2). We previously found that [Rh(CO)2Cl]2 was an efficient catalyst for promoting 1,3-acyloxy migration of propargyl esters. Indeed, our preliminary study showed that product 2s could be isolated in 30–40% yields using [Rh(CO)2Cl]2 as the catalyst. Benzene derivative 2s was presumably derived from a cascade reaction involving 1,3-acyloxy migration of propargyl ester to form vinylallene 16, Diels-Alder cycloaddition to form isotoluene 17, and aromitization. This tandem process could be mediated efficiently by PtCl2 catalyst.40 Our results suggested that 1,2 and 1,3-acyloxy migration of propargyl esters were dependent on the substitution pattern of the substrates and the nature of the Rh(I) catalysts, which was consistent with observations using other transition metals.27k
Scheme 2.

Tandem 1,3-acyloxy migration, Diels-Alder reaction for ACEs with an internal alkyne
It has been reported that electron withdrawing ester may facilitate the 1,2-acyloxy migration of propargyl esters.38 Indeed, ACE 1t with an ester group at the terminal position of the alkyne underwent cycloisomerization with the tethered alkyne smoothly and led to the formation of bicyclic product 2t upon treatment of cationic Rh(I) catalyst (Table 5, entry 1). ACEs with different alkyl or aryl substituted ketones also participated in the cycloisomerization efficiently (entries 2–4). The sulfonamide tether could be replaced by ether for this type of substrates (entries 5 and 6). For substrate 1z, only 47% yield of desired cycloisomerization product was isolated using cationic catalyst alone. The addition of (CF3CH2O)3P ligand increased the yield to 64% (entry 7). This was consistent with results obtained for substrate 1l (Table 4, entry 4).
Table 5.
Scope of Rh-catalyzed cycloisomerization of enynes with an electron-withdrawing substituenta
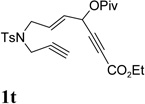
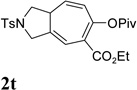
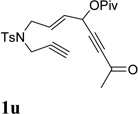
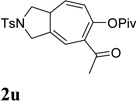
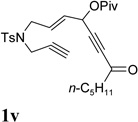
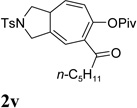
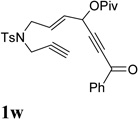
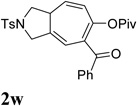
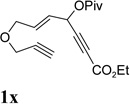
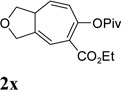
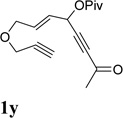
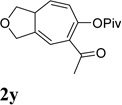
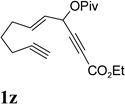
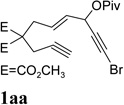
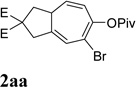
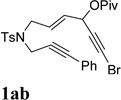
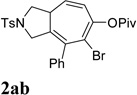
It was recently reported that propargyl esters with a haloalkyne underwent gold-catalyzed 1,2-acyloxy migration.36 We then prepared substrate 1aa containing a 1-bromo-3-acyloxy-1,4-enyne fragment (entry 8). A complex mixture, however, was obtained for the cycloisomerization of substrate 1aa using cationic Rh(I) alone. We were pleased to find that 69% yield of the cycloisomerization product 2aa could be isolated in the presence of [Rh(CO)2Cl]2 catalyst. The same catalyst also worked for substrate 1ab, which had internal alkynes for both 5 and 2-carbon components (entry 9).
In our previous communication for the Rh-catalyzed cycloisomerization of enediyne 1, ACEs were limited to terminal alkynes. Inspired by recent developments in the area of transition metal-catalyzed 1,2-acyloxy migration of propargyl esters, we have now expanded the scope of ACEs to internal alkynes with various electron-withdrawing groups including ester, ketone and bromine.
The enol olefin in triene 2 is a masked ketone. Direct hydrolysis of product 2b under basic conditions released the ketone functionality. Isomerization also occurred under this condition and it led to the formation of conjugated cycloheptadienone 18 (eq 5).
 |
(5) |
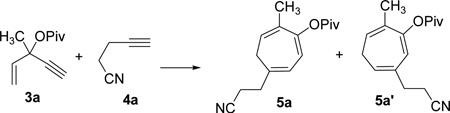
After successful development of the intramolecular [5+2] cycloaddition with concomitant 1,2-acyloxy migration between ACE and a tethered alkyne, we then turned our attention to the more challenging intermolecular reaction. Under optimized conditions described in Tables 2 and 4, we did not observe any cycloaddition product derived from ACE 3a and alkyne 4a (Table 6, entries 1 and 2). We were pleased to find that neutral rhodium(I) complexes catalyzed the intermolecular cycloaddition (Table 6, entries 3–5). Cycloheptatriene 5a could be isolated in 80% yield using Wilkinson’s catalyst (entry 6).
Table 6.
Screening of catalysts and conditions for the intermolecular reaction between ACE 1a and alkyne 4aa
 | ||
|---|---|---|
| entry | conditions | yield |
| 1 | [Rh(COD)]BF4 (5 mol %), DCM, 50 °C | 0 |
| 2 | [Rh(COD)]BF4 (5 mol %), (CF3CH2O)3P (10 mol %), DCM, 50 °C | 0 |
| 3 | [Rh(CO)2Cl]2 (5 mol %), DCE, 80 °C | 27% |
| 4 | [Rh(COD)Cl]2 (5 mol %), DCE, 80 °C | 38% |
| 5 | [Rh(PPh3)3Cl] (10 mol %), DCE, 80 °C | 79% |
| 6 | [Rh(PPh3)3Cl] (10 mol %), CHCl3, 65 °C | 89% (80%)b |
| 7 | [Rh(COD)Cl]2 (5 mol %), (CF3CH2O)3P (30 mol %), CHCl3, 65 °C | 0 |
| 8 | [Rh(COD)Cl]2 (5 mol %), (C6F5)3P (30 mol %), CHCl3, 65 °C | 33% |
| 9 | [Rh(COD)Cl]2 (5 mol %), (2-CH3C6H4)3P (30 mol %), CHCl3, 65 °C | 25% |
| 10 | [Rh(COD)Cl]2 (5 mol %), dppb (15 mol %), CHCl3, 65 °C | 67% |
| 11 | [Rh(COD)Cl]2 (5 mol %), (4-CF3C6H4)3P (30 mol %), CHCl3, 65 °C | 90% (84%)b |
| 12 | [Rh(PPh3)3Cl] (10 mol %), CHCl3, 65 °C, 1.2 equiv of 4a | 80% |
| 13 | [Rh(PPh3)3Cl] (10 mol %), CHCl3, 65 °C, 2.0 equiv of 4a | 90% (81%)b |
| 14 | PdCl2(CH3CN)2 (10 mol %), CH3CN, 80 °C | 0 |
| 15 | PtCl2 (10 mol %), DCE, 80 °C | 0 |
| 16 | Au(PPh3)Cl (5 mol %), DCM, 50 °C | 0 |
| 17 | Au(PPh3)Cl (5 mol %), AgSbF6 (5 mol %), DCM, rt | 0 |
Unless otherwise noted, 1.0 equiv of 3a and 3.0 equiv of 4a were employed and yields of 5a were determined based on 1H NMR using internal standard after 6h. For all entries, isomer 5a’ was not detected by 1H NMR of the crude product.
Isolated yields.
We also studied the effect of ligands using [Rh(COD)Cl]2 catalyst (entries 7–11). Surprisingly, no desired product was observed after the addition of electron-poor phosphite ligand (CF3CH2O)3P (entry 7). Phosphine ligands generally promoted the intermolecular cycloaddition (entries 8–11). We found that (4-CF3C6H4)3P ligand provided a comparable yield to Wilkinson’s catalyst (entry 11). The amount of alkyne 4a could be reduced to 2.0 equiv without significant change of the yield (entries 6, 12 and 13). Moreover, no cycloaddition product was observed in the presence of palladium, gold, or platinum catalysts (entries 14–17). ACE 3a was prepared in one step by esterification of the corresponding commercially available alcohol.39 Highly functionalized seven-membered ring 5a, therefore, could be accessed in just two steps regioselectively.
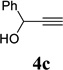
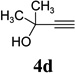
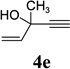
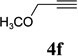
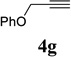
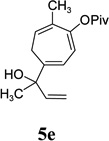
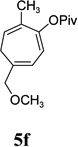
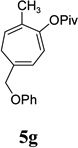
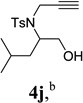
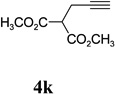
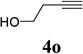
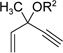
With the optimized condition in hand, we then studied the scope of terminal alkynes for the intermolecular [5+2] cycloaddition with concomitant 1,2-acyloxy migration (Table 7). The reaction between ACE 3a and free propargyl alcohol 4b proceeded smoothly to afford isomer 5b exclusively in the presence of Wilkinson’s catalyst. High regioselectivity was also achieved with secondary and tertiary propargyl alcohols (4c, 4d, and 4e). The olefin in 1,4-enyne 4e did not interfere with the reaction. When propargyl ether 4f was employed, regioisomer 5f’ became noticeable. Interestingly, higher regioisomeric ratios were observed for aryl propargyl ethers 4g and 4h. The formyl group in ether 4h was tolerated under the reaction condition. More than 10:1 regioisomeric ratios were generally obtained for propargyl amides and malonate derivatives 4i, 4j, and 4k. For more complex alkynes 4h and 4j, excess of ACE 1a was used (condition B).
Table 7.
Scope of intermolecular reaction between ACE 3a and different terminal alkynesa
| alkyne substrate | 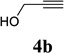 |
 |
 |
 |
 |
 |
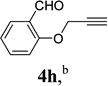 |
| cycloaddition product | 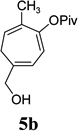 |
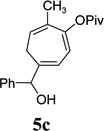 |
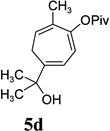 |
 |
 |
 |
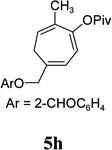 |
| yield, (5/5’) | 81%, (>20:1) | 83%, (>20:1) | 87%, (>20:1) | 93%, (>20:1) | 86%, (8:1) | 89%, (17:1) | 89%, (>20:1) |
| alkyne substrate | 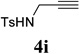 |
 |
 |
 |
 |
||
| cycloaddition product | 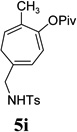 |
 |
 |
 |
 |
 |
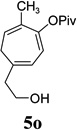 |
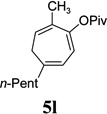
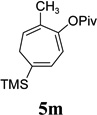
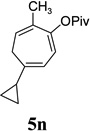
| yield, (5/5’) | 91%, (10:1) | 80%, (>20:1) | 85%, (14:1) | 81%, (10:1) | 53%, (10:1) | 5n, 64%, 5n’, 13%, (5:1) | 80%, (10:1) |
| alkyne substrate | 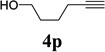 |
 |
 |
||||
| cycloaddition product | 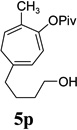 |
 |
 |
 |
 |
 |
 |
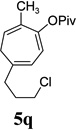
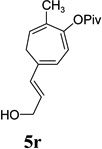
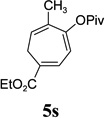
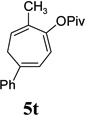
| yield, (5/5’) | 68%, (8:1) | 77%, (8:1) | 74%, (8:1) | 82%, (1.4:1) | 5t, 58%; 5t’, 17%, (4:1) | 83%, (6:1) | 76%, (3.6:1) |
| alkyne substrate | 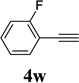 |
 |
 |
 |
 |
||
| cycloaddition product | 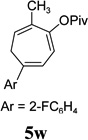 |
 |
 |
 |
 |
||
| yield, (5/5’) | 80%, (11:1) | 60%, (5:1) | 32% | 86%, (>20:1) | 92%, (>20:1) | ||
Unless otherwise noted, Condition A was employed: 1.0 equiv of 3a, 2.0 equiv of 4, Rh(PPh3)3Cl (10 mol %), CHCl3, 65 °C, 4–12h; Yields were isolated yields of combined 5 and 5’; The structure of isomer 5’ was analogous to 5a’ in Table 6; Regioisomeric ratios of 5/5’ were determined by 1H NMR of the crude product.
Condition B: The stoichiometry of two reactants in Conditions A was changed to 2.0 equiv of 3a and 1.0 equiv of 4.
Condition C: The catalyst in Condition A was changed to [Rh(COD)Cl]2 (5 mol %) and (4-CF3C6H4)3P (30 mol %).
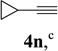
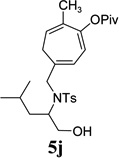
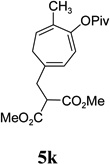
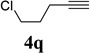
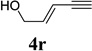
We also examined the regioselectivity for nonfunctionalized aliphatic 1-heptyne 4l. A regioisomeric ratio of 6:1 was observed using Wilkinson’s catalyst under condition A. The selectivity could be improved to 10:1 using the combination of [Rh(COD)Cl]2 and (4-CF3C6H4)3P under condition C. This condition also improved the regioisomeric ratio from 5:1 to 10:1 for TMS-acetylene 4m. A moderate change of ratio (4:1 under condition A and 5:1 under condition C) was observed for substrate 4n. However, similar yields and regioisomeric ratios were obtained for homopropargyl alcohol 4ounder conditions A or C. Alcohol 4p, alkyl chloride 4q, and conjugated enyne 4r all participated in the cycloaddition. Good regioselectivity was achieved for most terminal aliphatic alkynes with the exception of ynoate 4s. A similar electronic effect on regioselectivity was also observed in Rh-catalyzed intermolecular [5+2] cycloaddition of VCPs and alkynes.7c
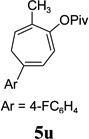
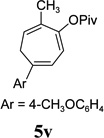
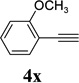
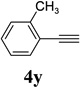
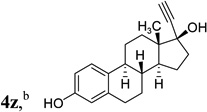
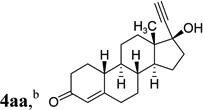
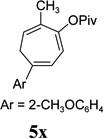
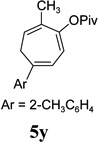
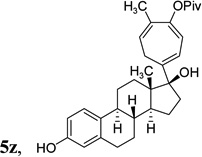
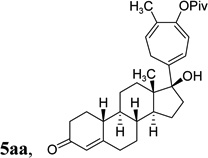
The two regioisomeric cycloheptatrienes 5t and 5t’ derived from phenyl acetylene could be isolated in 58% and 17% yields respectively. The ratio of these two isomers was 4:1 when the reaction was conducted under condition A. The regioselectivity, however, dropped to 2:1 under condition C. We then studied the electronic and steric effects of the substituents on the benzene ring under Condition A. An electron-withdrawing group improved the ratio of 5/5’ while an electron-donating group had opposite effect (4u vs 4v). The regioselectivity became higher when the heteroatoms were moved from para to ortho position in alkynes 4w and 4x. For substrate 4y with an ortho methyl group, the yield of product dropped to 32% and the 1H NMR of the crude products was too complex to identify the potential minor regioisomer. Highly functionalized terminal alkynes such as ethynylestradiol 4z and norethindrone 4aa also underwent cycloaddition with ACE 3a regioselectively. The free phenol and conjugated enone in these two substrates were compatible with the cycloaddition.
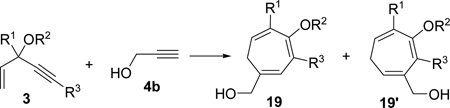
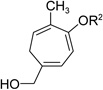
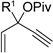
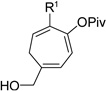
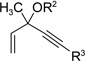
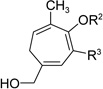
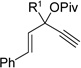
Having investigated the utility of ACE 3a for [5+2] cycloaddition with a wide variety of terminal alkynes, we then studied the scope of the 5C synthon (Table 8). The pivalate group in 3a could be replaced by acetate in 3b or benzoate in 3c without noticeable change of reaction rates (Table 8, entries and 2). ACEs with various alkyl or aryl groups on the 3-position could also participate in the intermolecular cycloaddition efficiently and regioselectively (entries 3–6).
Table 8.
Scope of intermolecular reaction between different ACEs and propargyl alcohol 4ba
 | |||
|---|---|---|---|
| entry | ACE substrate | product | yield of 19, (19/19’)b |
 |
 |
||
| 1 | 3b (R2=Ac) | 19b | 71%, (>20:1) |
| 2 | 3c (R2=Bz) | 19c | 58%, (>20:1) |
 |
 |
||
| 3 | 3d (R1=C2H5) | 19d | 87%, (>20:1) |
| 4 | 3e (R1=CH2OTBS) | 19e | 92%, (>20:1) |
| 5 | 3f (R1=Ph) | 19f | 81%, (20:1) |
| 6 | 3g (R1=4-BrC6H4) | 19g | 76%, (>20:1) |
| 7 | 3h (R1=H) | 19h | 16%,c (>20:1) |
 |
 |
||
| 8 | 3i, (R2=Ac, R3=CH2CH2Ph) | 19i | 0 |
| 9 | 3j (R2=Piv, R3=Br) | 19j | 34%, (>20:1)e |
| 10 | 3k (R2=Bz, R3=COPh) | 19k | 52%,d (5:1) |
 |
- | ||
| 11 | 3l (R1=H) | - | 0 |
| 12 | 3m (R1= CH3) | - | 0 |
| 13 | 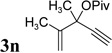 |
- | 0 |
| 14 | 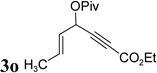 |
- | 0 |
See condition A in Table 7. All yields were isolated yield.
The regioisomeric ratio of 19/19’ was determined by 1H NMR of the crude product.
ACE 3h was recovered in 66% yield after 12h.
This was the isolated yield of isomer 19k.
rt, 14h.
The conversion for secondary ester 3h dropped significantly (entry 7), similar to gold-catalyzed reactions.27k Results for ACEs with internal alkynes were similar to the intramolecular reaction (entries 8 and 9). No desired product was observed for ACE 3i (entry 8). The halogen substituent in ACE 3j facilitated the cycloaddition and provided tetra-substituted cycloheptatriene 19j as a single regioisomer in moderate yield (entry 9). ACE 3k with a ketone group at the terminal position of the alkyne also underwent cycloaddition smoothly and led to the formation of 19k and its regioisomer 19k’ in a 5:1 ratio upon treatment of Rh(I) catalyst (entry 10). Isomer 19k was isolated in 52% yield. ACEs bearing substituents on the alkene (entries 11–14) did not participate in the cycloaddition reaction under the standard conditions A or C in Table 7.
The reaction between ACE 3a and internal alkyne 20a was very sluggish and only 21% yield of product 21a was isolated (Scheme 3). In contrast, the reaction between ACE 3a and 1,4-butynediol 20b proceeded smoothly to yield cycloheptatriene 21b in high yield under the identical condition. The hydroxyl group on the propargylic position improved the reactivity of 20b dramatically. For non-symmetric internal alkyne 20c, regioisomeric ratios of 3.3:1 and 5:1 were observed under condition A and C respectively. The reaction between ACE 3p and internal alkyne 20b afforded a moderate yield of bicyclic compound 23 after transesterification under condition C. Product 23 could be isolated in 71% yield by simply increasing the temperature to 80 °C and changing the solvent to DCE as shown in condition D (Scheme 3). ACEs bearing a secondary propargyl ester could therefore participate in the cycloaddition efficiently when the alkyne was substituted with an electron-withdrawing ester group.
Scheme 3.

Intermolecular reaction of ACE 3a and internal alkynes (see Table 7 for conditions A and C)
The intermolecular [5+2] cycloaddition with concomitant 1,2-acylox migration could be scaled up to prepare 0.76 g of product 5g with a more than 20:1 regioisomeric ratio (eq 6). The pivaloyl group in compound 5g could be removed by DIBALH as shown in eq 7. Further reduction41 of the resulting cycloheptadienone followed by directed epoxidation42 led to the isolation of highly functionalized cycloheptene 24. This demonstrated that the three alkenes in cycloheptatriene 5g could be further functionalized selectively. For the intermolecular cycloaddition, we could only place substituents to five out of seven possible positions on the cycloheptatriene skeleton as shown in compounds 5, 19, and 21–23. Through selective derivatization of the triene, however, more substituents and functionalities could be introduced to the seven-membered ring.
 |
(6) |
 |
(7) |
Based on the mechanism of Rautenstrauch rearrangement (Scheme 1), we proposed a mechanism for the Rh-catalyzed intramolecular [5+2] cycloaddition of ACEs and alkynes accompanied by a 1,2-acyloxy migration in Scheme 4. It first involved a Rh-promoted 1,2-acyloxy migration of the propargyl ester in metal complex 27 to form intermediate 28. Metallacyclohexadiene 29 could be derived from direct cyclization of metal complex 28, or through carbene 30 via a 6-π electrocyclization. Insertion of a tethered alkyne to metallacycle 29 followed by reductive elimination of metallacyclooctatriene 31 would then produce cycloheptatriene product 26. In one of the intramolecular cycloadditions, we isolated a small amount of cyclopropane byproduct 32 (Scheme 4),19 which was presumably derived from the reaction between Rh(I) carbene intermediate 30 and a cyclooctadiene present in the catalyst.
Scheme 4.

Proposed mechanism for the Rh-catalyzed intramolecular [5+2] cycloaddition of ACE and alkyne with a concomitant 1,2-acyloxy migration
The mechanism for the Rh-catalyzed intermolecular [5+2] cycloaddition of ACEs and alkynes is shown in Scheme 5. Following the same 1,2-acyloxy migration and cyclization sequence, metal complex 36 would be formed from ACE 33 and alkyne 34. The alkyne in intermediate 36 could insert into either the sp3-carbon-metal bond (pathway a) or sp2-carbon-metal bond (pathway b). For terminal alkyne 34, the R group could be either close (37a and 38b) or distal (37b and 38a) to the forming C-C bond. In all of our intermolecular [5+2] cycloadditions involving terminal alkynes, the major regioisomer we observed was product 35, which was presumably derived from intermediate 37a or 37b. In previous computational studies on Rh-catalyzed reactions involving unsymmetrical alkynes, the bulkier alkyne substituent prefers to be distal to the forming C-C bond.7c,43 The formation of product 35 via intermediate 37b might also be the favored pathway in our intermolecular [5+2] cycloaddition. We often obtained slightly higher regioselectivity for alkynes with heteroatoms on the propargylic or homopropargylic position. Coordination of the heteroatom to the rhodium in metal complex 37b might be responsible for the higher selectivity.
Scheme 5.

Proposed mechanism for the Rh-catalyzed intermolecular [5+2] cycloaddition of ACE and alkyne with a concomitant 1,2-acyloxy migration
To further understand the mechanism of the intermolecular cycloaddition, we also treated several ACEs in Table 8 with Wilkinson’s catalyst (10 mol %) in the absence of any external alkyne. A complex mixture together with a significant amount of starting material was observed in all cases. After careful analysis, we were able to isolate a small amount of Rautenstrauch rearrangement product 39 (~5% yield) from ACE 3c. Presumably, cyclopentadiene 39 was derived from reductive elimination of the corresponding metal-complex 36 prior to alkyne insertion. In the presence of external alkynes, we rarely observed the Rautenstrauch rearrangement product. The isolation of byproducts 32 and 39 was consistent with mechanisms proposed in Schemes 4 and 5 based on intercepting Rautenstrauch intermediates by alkynes.
3. CONCLUSION
In summary, we have demonstrated for the first time that 3-acyloxy-1,4-enynes (ACEs) can serve as 5-carbon synthons in Rh-catalyzed intra- and intermolecular [5+2] cycloadditions with alkynes. The ring formation was accompanied by a 1,2-acyloxy migration of propargyl ester. The 2-carbon component could be either terminal or internal alkynes. The ACE 5-carbon component had a terminal alkyne in most cases. ACEs bearing internal alkynes also participated in cycloadditions when the terminal substituent was an electron-withdrawing halogen, ketone or ester groups. Various substituted mono- and bicyclic compounds with a seven-membered ring were prepared from readily available starting materials through inter- and intramolecular [5+2] cycloadditions respectively. High regioselectivity was observed for most terminal alkynes in the intermolecular reaction. Applications of these new methods for the synthesis of natural products and pharmaceutical agents containing seven-membered rings are ongoing in this laboratory.
4. EXPERIMENTAL SECTION
4.1. General Information
Unless otherwise noted, all reactions in non-aqueous media were conducted under dry argon in glassware that had been oven-dried prior to use. Anhydrous solutions of reaction mixtures were transferred via an oven-dried syringe or cannula. All solvents were dried prior to use. Thin layer chromatography was performed using precoated silica gel plates (EMD Chemical Inc.60, F254). Flash column chromatography was performed with silica gel (Sillicycle, 40–63µm). Infrared spectra (IR) were obtained as neat oils on a Bruker Equinox 55 Spectrophotometer. 1H and 13C Nuclear magnetic resonance spectra (NMR) were obtained on a Varian Unity-Inova 400 MHz or 500 MHz recorded in ppm (δ) downfield of TMS (δ = 0) in CDCl3. Signal splitting patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quint), or multiplet (m), with coupling constants (J) in hertz. High resolution mass spectra (HRMS) were performed by the Analytical Instrument Center at the School of Pharmacy or the Department of Chemistry on an Electron Spray Injection (ESI) mass spectrometer.
4.1. General procedure for the Rh-catalyzed cycloisomerization of enynes in Table 2
To a solution of [Rh(COD)2]BF4 (2.5 mg, 3 mol %) in CH2Cl2 (0.05 M) was added propargylic ester (0.2 mmol). The solution was stirred at room temperature until the reaction was completed as determined by TLC analysis. The solvent was removed under reduced pressure. The residue was purified by chromatography on silica gel to afford the corresponding cycloheptatriene.
4.2. General procedure for the Rh-catalyzed cycloisomerization of enynes in Table 4
To a solution of [Rh(COD)2]BF4 (4.0 mg, 5 mol %) in CH2Cl2 (0.025–0.05 M) was added (CF3CH2O)3P (6.4 mg, 10 mol %). After stirring at room temperature for 5 min, propargylic ester (0.2 mmol) was added. The reaction mixture was allowed to stir at 50 °C until the reaction was completed as determined by TLC analysis. The solvent was removed under reduced pressure. The residue was purified by chromatography on silica gel to afford the corresponding cycloheptatriene.
4.3. General procedure for the Rh-catalyzed cycloisomerization of enynes 1aa and 1ab in Table 5
To a solution of [Rh(CO)2Cl]2 (3.9 mg, 5 mol %) in 1,2-dichloroethane (0.1 M) was added propargylic ester (0.2 mmol). The reaction mixture was allowed to stir at 80 °C until the reaction was completed as determined by TLC analysis. The solvent was removed under reduced pressure. The residue was purified by chromatography on silica gel to afford the corresponding cycloheptatriene.
4.4. General procedures for the intermolecular reaction between ACEs and alkynes in Tables 7 and 8
Condition A
To a solution of Rh(PPh3)3Cl catalyst (18.5 mg, 10 mol %) in CHCl3 (1 mL) was added 3-acyloxy-1,4-enyne (0.2 mmol) and alkyne (0.4 mmol). The reaction mixture was allowed to stir at 65 °C under argon until the reaction was completed as determined by TLC analysis. After removing the solvent under reduced pressure, the residue was purified by chromatography on silica gel to afford the corresponding product.
Condition B
The equivalents of substrates were changed as the following: 3-Acyloxy-1,4-enyne (0.4 mmol) and alkyne (0.2 mmol). Everything else was the same as condition A.
Condition C
To a solution of [Rh(COD)Cl]2 (5.2 mg, 5 mol %) in CHCl3 (1 mL) was added (4-CF3Ph)3P (28 mg, 30 mol %). After stirring at rt for 5 min, 3-acyloxy-1,4-enyne (0.2 mmol) and alkyne (0.4 mmol) were added. The reaction mixture was allowed to stir at 65 °C under argon until the reaction was completed as determined by TLC analysis. After removing the solvent under reduced pressure, the residue was purified by chromatography on silica gel to afford the corresponding product.
Supplementary Material
ACKNOWLEDGMENT
We thank the NIH (R01GM088285) and the University of Wisconsin for funding. W.T. is grateful for a Young Investigator Award from Amgen. S.H. is partially supported by a fellowship from Chinese Scholarship Council.
Footnotes
Supporting Information. Detailed experimental procedures and characterization of new compounds (IR, 1H NMR, 13C NMR, HRMS). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.For selected reviews on seven-membered ring synthesis see: Battiste MA, Pelphrey PM, Wright DL. Chem. Eur. J. 2006;12:3438. doi: 10.1002/chem.200501083. Butenschön H. Angew. Chem. Int. Ed. 2008;47:5287. doi: 10.1002/anie.200801738.
- 2.For recent reviews on [4+3] cycloadditions see: Harmata M. Chem. Commun. 2010;46:8886. doi: 10.1039/c0cc03620j. Harmata M. Chem. Commun. 2010;46:8904. doi: 10.1039/c0cc03621h. Lohse AG, Hsung RP. Chem. Eur. J. 2011;17:3812. doi: 10.1002/chem.201100260.
- 3.For a recent review on [5+2] cycloadditions see: Pellissier H. Adv. Synth. Catal. 2011;353:189.
- 4.Wender PA, Deschamps NM, Sun R. Angew. Chem. Int. Ed. 2006;45:3957. doi: 10.1002/anie.200600806. [DOI] [PubMed] [Google Scholar]
- 5.For selected reviews on transition metal catalyzed cycloadditions and cycloisomerizations see: Lautens M, Klute W, Tam W. Chem. Rev. 1996;96:49. doi: 10.1021/cr950016l. Ojima I, Tzamarioudaki M, Li ZY, Donovan RJ. Chem. Rev. 1996;96:635. doi: 10.1021/cr950065y. Fruhauf HW. Chem. Rev. 1997;97:523. doi: 10.1021/cr941164z. Trost BM, Krische MJ. Synlett. 1998:1. Yet L. Chem. Rev. 2000;100:2963. doi: 10.1021/cr990407q. Aubert C, Buisine O, Malacria M. Chem. Rev. 2002;102:813. doi: 10.1021/cr980054f. Evans PA. Modern Rhodium-Catalyzed Organic Reactions. Wiley-VCH: Weinheim; 2005. Michelet V, Toullec PY, Genet JP. Angew. Chem. Int. Ed. 2008;47:4268. doi: 10.1002/anie.200701589. Yu Z, Wang Y, Wang Y. Chem. Asian J. 2010;5:1072. doi: 10.1002/asia.200900712. Inglesby PA, Evans PA. Chem. Soc. Rev. 2010;39:2791. doi: 10.1039/b913110h. Aubert C, Fensterbank L, Garcia P, Malacria M, Simonneau A. Chem. Rev. 2011;111:1954. doi: 10.1021/cr100376w.
- 6.For representative Rh-catalyzed intramolecular [5+2] cycloadditions see: Wender PA, Takahashi H, Witulski B. J. Am. Chem. Soc. 1995;117:4720. Wender PA, Dyckman AJ, Husfeld CO. doi: 10.1021/ol0058691. Kadereit D, Love JA, Rieck H. J. Am. Chem. Soc. 1999;121:10442.
- 7.For representative Rh-catalyzed intermolecular [5+2] cycloadditions see: Wender PA, Rieck H, Fuji M. J. Am. Chem. Soc. 1998;120:10976. Wender PA, Barzilay CM, Dyckman AJ. J. Am. Chem. Soc. 2001;123:179. doi: 10.1021/ja0021159. Liu P, Sirois LE, Cheong PHY, Yu Z, Hartung IV, Rieck H, Wender PA, Houk KN. J. Am. Chem. Soc. 2010;132:10127. doi: 10.1021/ja103253d.
- 8.For selected applications of intermolecular [5+2] cycloadditions see: Wender PA, Dyckman AJ, Husfeld CO, Scanio MJC. Org. Lett. 2000;2:1609. doi: 10.1021/ol0058691. Wender PA, Gamber GG, Scanio MJC. Angew. Chem. Int. Ed. 2001;40:3895. doi: 10.1002/1521-3773(20011015)40:20<3895::AID-ANIE3895>3.0.CO;2-#. Wender PA, Sirois LE, Stemmler RT, Williams TJ. Org. Lett. 2010;12:1604. doi: 10.1021/ol100337m. Wender PA, Stemmler RT, Sirois LE. J. Am. Chem. Soc. 2010;132:2532. doi: 10.1021/ja910696x.
- 9.For representative Ru-catalyzed intramolecular [5+2] cycloadditions see: Trost BM, Toste FD, Shen H. J. Am. Chem. Soc. 2000;122:2379. Trost BM, Shen HC. Angew. Chem. Int. Ed. 2001;40:2313. doi: 10.1002/1521-3773(20010618)40:12<2313::AID-ANIE2313>3.0.CO;2-H. Trost BM, Shen HC, Horne DB, Toste FD, Steinmetz BG, Koradin C. Chem. Eur. J. 2005;11:2577. doi: 10.1002/chem.200401065.
- 10.For a Ni-catalyzed intramolecular [5+2] cycloaddition see: Zuo G, Louie J. J. Am. Chem. Soc. 2005;127:5798. doi: 10.1021/ja043253r.
- 11.For a Fe-catalyzed intramolecular [5+2] cycloaddition see: Fürstner A, Majima K, Martin R, Krause H, Kattnig E, Goddard R, Lehmann CW. J. Am. Chem. Soc. 2008;130:1992. doi: 10.1021/ja0777180.
- 12.Jiao L, Ye S, Yu Z. J. Am. Chem. Soc. 2008;130:7178. doi: 10.1021/ja8008715. [DOI] [PubMed] [Google Scholar]; b) Li Q, Jiang G, Jiao L, Yu Z. Org. Lett. 2010;12:1332. doi: 10.1021/ol100237h. [DOI] [PubMed] [Google Scholar]
- 13.Inagaki F, Sugikubo K, Miyashita Y, Mukai C. Angew. Chem. Int. Ed. 2010;49:2206. doi: 10.1002/anie.200906994. [DOI] [PubMed] [Google Scholar]
- 14.For representative Rh-catalyzed [5+2] cycloadditions of VCPs with alkenes and allenes see: Wender PA, Husfeld CO, Langkopf E, Love JA. J. Am. Chem. Soc. 1998;120:1940. Wender PA, Glorius F, Husfeld CO, Langkopf E, Love JA. J. Am. Chem. Soc. 1999;121:5348. Wegner HA, De Meijere A, Wender PA. J. Am. Chem. Soc. 2005;127:6530. doi: 10.1021/ja043671w. For related Rh-catalyzed hetero-[5+2] cycloadditions see: d) Wender PA, Pedersen TM, Scanio MJC. J. Am. Chem. Soc. 2002;124:15154. doi: 10.1021/ja0285013. Feng JJ, Zhang J. J. Am. Chem. Soc. 2011;133:7304. doi: 10.1021/ja2014604.
- 15.a) Wender PA, Fuji M, Husfeld CO, Love JA. Org. Lett. 1999;1:137. [Google Scholar]; b) Wender PA, Zhang L. Org. Lett. 2000;2:2323. doi: 10.1021/ol006085q. [DOI] [PubMed] [Google Scholar]; c) Wender PA, Bi FC, Brodney MA, Gosselin F. Org. Lett. 2001;3:2105. doi: 10.1021/ol0160699. [DOI] [PubMed] [Google Scholar]; d) Ashfeld BL, Martin SF. Org. Lett. 2005;7:4535. doi: 10.1021/ol051945u. [DOI] [PubMed] [Google Scholar]; e) Ashfeld BL, Martin SF. Tetrahedron. 2006;62:10497. [Google Scholar]; f) Trost BM, Hu Y, Horne DB. J. Am. Chem. Soc. 2007;129:11781. doi: 10.1021/ja073272b. [DOI] [PubMed] [Google Scholar]; g) Trost BM, Waser J, Meyer A. J. Am. Chem. Soc. 2007;129:14556. doi: 10.1021/ja076165q. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Trost BM, Waser J, Meyer A. J. Am. Chem. Soc. 2008;130:16424. doi: 10.1021/ja806724x. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Jiao L, Yuan C, Yu Z. J. Am. Chem. Soc. 2008;130:4421. doi: 10.1021/ja7100449. [DOI] [PubMed] [Google Scholar]; j) Trost BM, Nguyen HM, Koradin C. Tetrahedron Lett. 2010;51:6232. doi: 10.1016/j.tetlet.2010.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For computational studies on Rh(I)-catalyzed [5+2] cycloadditions see: Yu Z, Wender PA, Houk KN. J. Am. Chem. Soc. 2004;126:9154. doi: 10.1021/ja048739m. Wang Y, Wang J, Su JC, Huang F, Jiao L, Liang Y, Yang D, Zhang S, Wender PA, Yu Z. J. Am. Chem. Soc. 2007;129:10060. doi: 10.1021/ja072505w. Yu Z, Cheong PHY, Liu P, Legault CY, Wender PA, Houk KN. J. Am. Chem. Soc. 2008;130:2378. doi: 10.1021/ja076444d. Liu P, Cheong PHY, Yu Z, Wender PA, Houk KN. Angew. Chem. Int. Ed. 2008;47:3939. doi: 10.1002/anie.200800420. and 7c).
- 17.a) Dzwiniel TL, Etkin N, Stryker JM. J. Am. Chem. Soc. 1999;121:10640. [Google Scholar]; b) Dzwiniel TL, Stryker JM. J. Am. Chem. Soc. 2004;126:9184. doi: 10.1021/ja047852+. [DOI] [PubMed] [Google Scholar]; c) Witherell RD, Ylijoki KEO, Stryker JM. J. Am. Chem. Soc. 2008;130:2176. doi: 10.1021/ja710568d. [DOI] [PubMed] [Google Scholar]
- 18.Tanino K, Shimizu T, Miyama M, Kuwajima I. J. Am. Chem. Soc. 2000;122:6116. [Google Scholar]
- 19.Shu X-Z, Huang S, Shu D, Guzei IA, Tang W. Angew. Chem. Int. Ed. 2011;50:8153. doi: 10.1002/anie.201103136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Rautenstrauch V. J. Org. Chem. 1984;49:950. [Google Scholar]; b) Rautenstrauch V. Tetrahedron Lett. 1984;25:3845. [Google Scholar]
- 21.Shi X, Gorin DJ, Toste FD. J. Am. Chem. Soc. 2005;127:5802. doi: 10.1021/ja051689g. [DOI] [PubMed] [Google Scholar]
- 22.Prasad BAB, Yoshimoto FK, Sarpong R. J. Am. Chem. Soc. 2005;127:12468. doi: 10.1021/ja053192c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.For related studies see: Nakanishi Y, Miki K, Ohe K. Tetrahedron. 2007;63:12138. Dekorver KA, Hsung RP, Lohse AG, Zhang Y. Org. Lett. 2010;12:1840. doi: 10.1021/ol100446p. For computational study see: c) Faza ON, Lopez CS, Alvarez R, De Lera AR. J. Am. Chem. Soc. 2006;128:2434. doi: 10.1021/ja057127e.
- 24.Brancour C, Fukuyama T, Ohta Y, Ryu I, Dhimane AL, Fensterbank L, Malacria M. Chem. Commun. 2010;46:5470. doi: 10.1039/c0cc00747a. [DOI] [PubMed] [Google Scholar]
- 25.Xu H, Zhang W, Shu D, Werness JB, Tang W. Angew. Chem. Int. Ed. 2008;47:8933. doi: 10.1002/anie.200803910. [DOI] [PubMed] [Google Scholar]
- 26.Shu D, Li X, Zhang M, Robichaux PJ, Tang W. Angew. Chem. Int. Ed. 2011;50:1346. doi: 10.1002/anie.201006881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.For selected reviews on π-acidic metal catalyzed reactions see: Miki K, Uemura S, Ohe K. Chem. Lett. 2005;34:1068. Marion N, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2750. doi: 10.1002/anie.200604773. Fürstner A, Davies PW. Angew. Chem. Int. Ed. 2007;46:3410. doi: 10.1002/anie.200604335. Hashmi ASK. Chem. Rev. 2007;107:3180. doi: 10.1021/cr000436x. Hashmi ASK. Angew. Chem. Int. Ed. 2008;47:6754. doi: 10.1002/anie.200802517. Jimenez-Nunez E, Echavarren AM. Chem. Rev. 2008;108:3326. doi: 10.1021/cr0684319. Gorin DJ, Sherry BD, Toste FD. Chem. Rev. 2008;108:3351. doi: 10.1021/cr068430g. Abu Sohel SM, Liu R-S. Chem. Soc. Rev. 2009;38:2269. doi: 10.1039/b807499m. Fürstner A. Chem. Soc. Rev. 2009;38:3208. doi: 10.1039/b816696j. Shapiro ND, Toste FD. Synlett. 2010:675. doi: 10.1055/s-0029-1219369. Wang S, Zhang G, Zhang L. Synlett. 2010:692. Hashmi ASK. Angew. Chem. Int. Ed. 2010;49:5232. doi: 10.1002/anie.200907078. De Haro T, Nevado C. Synthesis. 2011:2530. Corma A, Leyva-Perez A, Sabater MJ. Chem. Rev. 2011;111:1657. doi: 10.1021/cr100414u. Krause N, Winter C. Chem. Rev. 2011;111:1994. doi: 10.1021/cr1004088.
- 28.a) Miki K, Ohe K, Uemura S. J. Org. Chem. 2003;68:8505. doi: 10.1021/jo034841a. [DOI] [PubMed] [Google Scholar]; b) Miki K, Ohe K, Uemura S. Tetrahedron Lett. 2003;44:2019. [Google Scholar]
- 29.Mainetti E, Mouries V, Fensterbank L, Malacria M, Marco-Contelles J. Angew. Chem. Int. Ed. 2002;41:2132. [PubMed] [Google Scholar]
- 30.Tenaglia A, Marc S. J. Org. Chem. 2006;71:3569. doi: 10.1021/jo060276a. [DOI] [PubMed] [Google Scholar]
- 31.Johansson MJ, Gorin DJ, Staben ST, Toste FD. J. Am. Chem. Soc. 2005;127:18002. doi: 10.1021/ja0552500. [DOI] [PubMed] [Google Scholar]; b) Watson IDG, Ritter S, Toste FD. J. Am. Chem. Soc. 2009;131:2056. doi: 10.1021/ja8085005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trost BM. Science. 1991;254:1471. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 33.a) Fürstner A, Hannen P. Chem. Commun. 2004:2546. doi: 10.1039/b412354a. [DOI] [PubMed] [Google Scholar]; b) Gorin DJ, Dube P, Toste FD. J. Am. Chem. Soc. 2006;128:14480. doi: 10.1021/ja066694e. [DOI] [PubMed] [Google Scholar]; c) Gorin DJ, Watson IDG, Toste FD. J. Am. Chem. Soc. 2008;130:3736. doi: 10.1021/ja710990d. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li G, Zhang G, Zhang L. J. Am. Chem. Soc. 2008;130:3740. doi: 10.1021/ja800001h. [DOI] [PubMed] [Google Scholar]; e) Moreau X, Goddard JP, Bernard M, Lemiere G, Lopez-Romero JM, Mainetti E, Marion N, Mouries V, Thorimbert S, Fensterbank L, Malacria M. Adv. Synth. Catal. 2008;350:43. [Google Scholar]; f) Zou Y, Garayalde D, Wang QR, Nevado C, Goeke A. Angew. Chem. Int. Ed. 2008;47:10110. doi: 10.1002/anie.200804202. [DOI] [PubMed] [Google Scholar]; g) Harrak Y, Makhlouf M, Azzaro S, Mainetti E, Romero JML, Cariou K, Gandon V, Goddard JP, Malacria M, Fensterbank L. J. Organomet. Chem. 2011;696:388. [Google Scholar]; h) Garayalde D, Kruger K, Nevado C. Angew. Chem. Int. Ed. 2011;50:911. doi: 10.1002/anie.201006105. [DOI] [PubMed] [Google Scholar]
- 34.Mamane V, Gress T, Krause H, Fürstner A. J. Am. Chem. Soc. 2004;126:8654. doi: 10.1021/ja048094q. [DOI] [PubMed] [Google Scholar]
- 35.a) Fehr C, Galindo J. Angew. Chem. Int. Ed. 2006;45:2901. doi: 10.1002/anie.200504543. [DOI] [PubMed] [Google Scholar]; b) Fehr C, Winter B, Magpantay I. Chem. Eur. J. 2009;15:9773. doi: 10.1002/chem.200901292. [DOI] [PubMed] [Google Scholar]
- 36.Wang YZ, Lu BA, Zhang LM. Chem. Commun. 2010;46:9179. doi: 10.1039/c0cc03669b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrak Y, Blaszykowski C, Bernard M, Cariou K, Mainetti E, Mouries V, Dhimane AL, Fensterbank L, Malacria M. J. Am. Chem. Soc. 2004;126:8656. doi: 10.1021/ja0474695. [DOI] [PubMed] [Google Scholar]; b) Pujanauski BG, Prasad BAB, Sarpong R. J. Am. Chem. Soc. 2006;128:6786. doi: 10.1021/ja061549m. [DOI] [PubMed] [Google Scholar]; c) Ji K, Shu X, Chen J, Zhao S, Zheng Z, Lu L, Liu X, Liang Y. Org. Lett. 2008;10:3919. doi: 10.1021/ol8015463. [DOI] [PubMed] [Google Scholar]
- 38.a) Shibata Y, Noguchi K, Tanaka K. J. Am. Chem. Soc. 2010;132:7896. doi: 10.1021/ja102418h. [DOI] [PubMed] [Google Scholar]; b) Shibata Y, Noguchi K, Tanaka K. Org. Lett. 2010;12:5596. doi: 10.1021/ol1025266. [DOI] [PubMed] [Google Scholar]
- 39.See Supporting Information for details.
- 40.Lu L, Liu X, Shu X, Yang K, Ji K, Liang Y. J. Org. Chem. 2009;74:474. doi: 10.1021/jo802043z. [DOI] [PubMed] [Google Scholar]
- 41.Gemal AL, Luche JL. J. Am. Chem. Soc. 1981;103:5454. [Google Scholar]
- 42.Itoh T, Jitsukawa K, Kaneda K, Teranishi S. J. Am. Chem. Soc. 1979;101:159. [Google Scholar]
- 43.For a review on theoretical study of regioselectivity of Rh-catalyzed C-C bond forming reactions with unsymmetrical alkynes see: Liu P, Houk KN. Inorg. Chim. Acta. 2011;369:2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.