

Background: Retinoblastoma-binding protein 1 (RBBP1), a tumor suppressor, is involved in epigenetic regulation in cancer.
Results: The chromobarrel domain of RBBP1 binds methylated histone tails, whereas Tudor and PWWP domains do not.
Conclusion: The chromobarrel domain of RBBP1 is responsible for epigenetic regulation.
Significance: Our research provides a structural basis to understand the mechanism of RBBP1-mediated epigenetic regulation.
Keywords: Chromatin Histone Modification, Chromatin Regulation, Nuclear Magnetic Resonance, Protein Structure, Tumor Suppressor Gene, Retinoblastoma-binding Protein 1
Abstract
Retinoblastoma-binding protein 1 (RBBP1), also named AT-rich interaction domain containing 4A (ARID4A), is a tumor and leukemia suppressor involved in epigenetic regulation in leukemia and Prader-Willi/Angelman syndromes. Although the involvement in epigenetic regulation is proposed to involve its chromobarrel and/or Tudor domains because of their potential binding to methylated histone tails, the structures of these domains and their interactions with methylated histone tails are still uncharacterized. In this work, we first found that RBBP1 contains five domains by bioinformatics analysis. Three of the five domains, i.e. chromobarrel, Tudor, and PWWP domains, are Royal Family domains, which potentially bind to methylated histone tails. We further purified these domains and characterized their interaction with methylated histone tails by NMR titration experiments. Among the three Royal Family domains, only the chromobarrel domain could recognize trimethylated H4K20 (with an affinity of ∼3 mm), as well as recognizing trimethylated H3K9, H3K27, and H3K36 (with lower affinities). The affinity could be further enhanced up to 15-fold by the presence of DNA. The structure of the chromobarrel domain of RBBP1 determined by NMR spectroscopy has an aromatic cage. Mutagenesis analysis identified four aromatic residues of the cage as the key residues for methylated lysine recognition. Our studies indicate that the chromobarrel domain of RBBP1 is responsible for recognizing methylated histone tails in chromatin remodeling and epigenetic regulation, which presents a significant advance in our understanding of the mechanism and relationship between RBBP1-related gene suppression and epigenetic regulation.
Introduction
RBBP1 (retinoblastoma-binding protein 1) and its homolog RBBP1L1 (RBBP1-like protein 1) are leukemia and tumor suppressors (1–3). RBBP1 and RBBP1L1 are also known as ARID4A and ARID4B, respectively, because they each contain an AT-rich interaction domain (ARID)3 (4). RBBP1 specifically interacts with retinoblastoma protein (pRb), exhibiting both histone deacetylase (HDAC)-dependent and -independent repression activities (5–8). Two regions, named R1 and R2, of RBBP1 are responsible for the repression activities (8). The R1 region is located in the ARID domain, which can directly bind DNA to suppress certain genes, representing the HDAC-independent repression activity. The R2 region (or R2 domain) located in the RBBP1 C-terminal region is responsible for the HDAC-dependent repression activity of RBBP1 due to its interaction with SAP30, a component of the mSin3A co-repressor complex, which also contains HDAC and suppresses the transcription of certain genes (7). Further studies have indicated that both RBBP1 and RBBP1L1 are components of the mSin3A co-repressor complex (7, 9, 10) and can interact with each other in vivo (11).
RBBP1 and RBBP1L1 regulate epigenetic marks, such as methylation of lysines in histones H3 and H4, which are observed in leukemia and Prader-Willi/Angelman syndromes (3, 11). The epigenetic regulation functions of RBBP1 have been proposed to occur through its chromobarrel domain (a variant of a chromodomain) and/or its Tudor domain (3, 11) because these domains generally bind histone codes, an important epigenetic regulation pathway (12). The Royal Family domains, such as chromodomain, Tudor domain, malignant brain tumor domain and PWWP domain, are a structurally related group of protein folds believed to have descended from a common ancestor with a conserved methylated substrate binding ability (13, 14). A large number of Royal Family domains have been found in various epigenetic-related proteins and they recognize methylated histone tails with different affinities and specificities (13). However, to date, there is no direct evidence that the Royal Family domains of RBBP1 are able to interact with methylated histone tails, and the structural basis of this potential interaction is still unknown.
In this work, we analyzed the domain organization of RBBP1 by bioinformatics and found that RBBP1 contains three Royal Family domains, i.e. a Tudor domain (TD), a PWWP domain (PD), and a chromobarrel domain (CD). We found that these domains could fold independently and that there is no direct interaction between them. To explore the structural basis of the epigenetic regulation function of RBBP1, we detected the interaction of the three Royal Family domains of RBBP1 with methylated histone tails using NMR titration techniques. We found that only the chromobarrel domain of RBBP1 recognizes methylated lysines and histone tails. We then solved the solution structure of the RBBP1 CD, finding that it contains conserved aromatic residues that form an aromatic cage. Mutagenesis analysis confirmed the role of these conserved aromatic residues in methylated histone recognition. We found that the positively charged surface around the aromatic cage of the RBBP1 CD can interact with DNA and that the presence of DNA enhances significantly the recognition of methylated histones.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
DNA encoding the CD (residues 568–635) of human RBBP1 (UniProt ID P29374) was amplified by PCR and cloned into the pET30a (Novagen) expression vector between the NdeI and XhoI sites. The expressed protein contains a C-terminal His tag (LEHHHHHH). The resulting plasmid was transformed into Escherichia coli Rosetta (DE3) cells for protein expression. When the optical density at 600 nm (A600) of the cells grown in LB medium at 37 °C reached 0.7–0.8, the protein production was induced by addition of 0.6 mm isopropyl-β-d-thiogalactopyranoside, and the cells were grown for a further 6 h. The cells were harvested by centrifugation at 4800 × g at 4 °C for 30 min. The cell pellets were resuspended in 30 ml of buffer A (50 mm phosphate sodium buffer, pH 8.0, 200 mm NaCl) and then stored at −20 °C overnight. The resuspended cell pellets were thawed and then lysed by sonication. After centrifugation at 30,700 × g for 30 min, the supernatants were applied onto a Chelating Sepharose Fast Flow (GE Healthcare) column. The proteins were eluted with buffer A containing 300 mm imidazole. The eluted fraction was concentrated to 2 ml using Amicon Ultra-15 centrifugal filter units (3-kDa cut-off, Millipore) and further purified by gel filtration chromatography using a Superdex 75 column (GE Healthcare) pre-equilibrated in 50 mm phosphate sodium buffer (pH 7.0) containing 50 mm NaCl and 1 mm DTT.
DNA encoding RBBP1 TD (residues 4–121) was amplified by PCR and cloned into a modified pGBO expression vector (15) between the BamH I and XhoI sites. An additional cleavage site for PreScission protease (GE Healthcare) was introduced between the N-terminal tag and the target gene of the original pGBO vector. The expressed protein contained an N-terminal His6-tagged GB1 domain. The resulting plasmid was transformed into E. coli Rosetta (DE3) cells for protein expression. When the A600 of the cells grown in LB medium at 37 °C reached 0.6, the protein production was induced by addition of 0.6 mm isopropyl-β-d-thiogalactopyranoside, and the cells were grown for a further 48 h at 16 °C. After cell lysis, the protein was purified first by Ni2+ affinity chromatography. The eluted fraction was dialyzed against 50 mm Tris-HCl buffer containing 150 mm NaCl (pH 8.0), then 1 mg of PreScission protease was added to cleave the tag from TD, and the protein was incubated for 24 h at 4 °C. A second Ni2+ affinity chromatography step was used to remove the fusion tag. The flow-through was concentrated and further purified by gel filtration in 50 mm Tris-HCl buffer (pH 7.8) containing 50 mm NaCl. The cloning, expression, and purification of RBBP1 PD (residues 170–273) followed the same protocol as that for RBBP1 TD, except that the cells were grown for 16 h at 25 °C after induction of protein production.
15N- and 15N-13C-labeled RBBP1 CD, TD, and PD were prepared using the same procedures except cells were grown in M9 minimal medium containing 15NH4Cl and [13C]glucose as the sole nitrogen and carbon sources. The protein concentrations were determined by UV absorption at 280 nm using theoretical molar extinction coefficients 24,870 m−1 cm−1, 9,770 m−1 cm−1, and 14,180 m−1 cm−1 for RBBP1 CD, TD, and PD, respectively.
NMR Spectroscopy
All NMR experiments were performed at 298 K on a Bruker DMX 600 MHz spectrometer equipped with a z-gradient triple-resonance cryoprobe. NMR samples of RBBP1 CD and PD contained 0.2–0.5 mm protein in 50 mm phosphate sodium buffer, pH 7.0, 50 mm NaCl, 5 mm DTT, 0.02% (w/v) sodium 2,2-dimethylsilapentane-5-sulfonate, and 10% (v/v) D2O. NMR samples of RBBP1 TD contained 0.2–0.6 mm protein in 50 mm Tris-HCl, pH 7.8, 50 mm NaCl, 5 mm DTT, 0.02% (w/v) sodium 2,2-dimethylsilapentane-5-sulfonate, and 10% (v/v) D2O. Two-dimensional 1H-15N and 1H-13C HSQC, three-dimensional CBCA(CO)NH, HNCACB, HNCO, HN(CA)CO, HBHA(CO)NH, HBHANH, HCCH-TOCSY, CCH-COSY, and CCH-TOCSY experiments (16) were performed for backbone and side chain assignments of RBBP1 CD. Three-dimensional 1H-15N and 1H-13C NOESY-HSQC spectra with mixing times of 120 ms were collected to generate distance restraints. All data were processed with NMRPipe (17) and analyzed with NMRView (18). Proton chemical shifts were referenced to the internal sodium 2,2-dimethylsilapentane-5-sulfonate, and 15N and 13C chemical shifts were referenced indirectly (19).
Peptide and DNA Titration
The interacting ligands used in the titrations included methylated amino acids (mono-, di-, trimethylated lysines, and dimethylated arginine) (Sigma-Aldrich), chemically synthesized trimethylated and unmethylated histone N-terminal peptides (Sangon Biotech, Shanghai) (Table 1), as well as synthesized dsDNA (Sangon Biotech, Shanghai). The dsDNA contained two complementary DNA sequences (20): 5′-CTC AGG TCA AAG GTC ACG-3′ and 3′-AG TCC AGT TTC CAG TGC T-5′. The single-stranded DNA oligomers were dissolved in 50 mm phosphate sodium buffer, pH 7.0, 50 mm NaCl and mixed in a 1:1 molar ratio, heated to 94 °C for 3 min, and then allowed to anneal at room temperature, to obtain the dsDNA. The dsDNA was further purified by gel filtration and then lyophilized. The stock solution of dsDNA contained 5 mm dsDNA in the same buffer as that used for the RBBP1 CD sample.
TABLE 1.
Sequences of histone peptides used in this work
| Peptide | Sequence |
|---|---|
| H3K9 | 1ARTKQTARKSTGGKA15 |
| H3K9me3 | 1ARTKQTARK(Me3)STGGKA15 |
| H3K27 | 21ATKAARKSAPAT32 |
| H3K27me3 | 21ATKAARK(Me3)SAPAT32 |
| H3K36 | 31ATGGVKKPHRYR42 |
| H3K36me3 | 31ATGGVK(Me3)KPHRYR42 |
| H4K20 | 15AKRHRKVLRDN25 |
| H4K20me3 | 15AKRHRK(Me3)VLRDN25 |
The interactions between RBBP1 domains and ligands were detected by monitoring the two-dimensional 1H-15N HSQC spectra of proteins during the titration. The observed chemical shift perturbations (CSP) were calculated using the following formula,
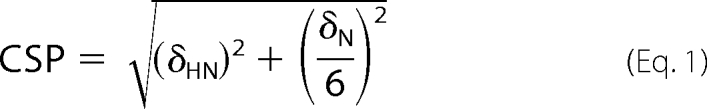 |
where δHN and δN are the changes of 1HN and 15N chemical shifts, respectively. The equilibrium dissociation constants (KD) were estimated by fitting the CSPs to the equation,
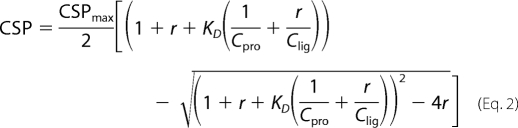 |
where CSPmax is CSP at the theoretical saturated condition, which was also obtained from the fit; r is the molar ratio of ligand to protein; Cpro is the concentration of initial protein solution; and Clig is the stock concentration of ligand.
Structure Calculations
The structures of RBBP1 CD were initially calculated with the program CYANA (21) and then refined using CNS (22) with manual assignments as well as semiautomated NOE assignments performed using SANE (23). Backbone dihedral angle restraints obtained using CSI (24) and TALOS (25), as well as hydrogen bond restraints according to the regular secondary structure patterns, were also incorporated into the structural calculation. From 100 initial structures, 50 lowest energy conformers of CD were selected to do water-refinement using CNS and RECOORDScript (26), and the 20 lowest energy conformers were selected to represent the final ensemble of structures for CD. The quality of the determined structures was analyzed using MOLMOL (27) and PROCHECK-NMR (Table 2) (28).
TABLE 2.
The experimental restraints and structural statistics for the 20 lowest energy structures of RBBP1 CD
| CD | |
|---|---|
| Distance restraints | |
| Intra-residue | 638 |
| Sequential | 353 |
| Medium | 111 |
| Long range | 341 |
| Ambiguous | 520 |
| Total | 1982 |
| Hydrogen bond restraints | 40 |
| Dihedral angle restraints | φ = 50, ψ = 50, total = 100 |
| Violations | |
| NOE violations (>0.3 Å) | 0 |
| Torsion angle violation (>3°) | 1 |
| Torsion angle violation (>5°) | 0 |
| PROCHECK statistics (%)a | |
| Most favored regions | 80.4 |
| Additional allowed regions | 16.6 |
| Generously allowed regions | 1.7 |
| Disallowed regions | 1.2 |
| R.m.s.d. from mean structure (Å) | |
| Backbone heavy atoms | |
| All residueb | 1.63 ± 0.33 |
| Regular secondary structurec | 0.39 ± 0.08 |
| All heavy atoms | |
| All residue | 2.10 ± 0.34 |
| Regular secondary structure | 0.84 ± 0.09 |
a Residues used to calculate PROCHECK statistics include 572–635 in CD.
b Residues used to calculate r.m.s.d. values of all residues include 572–635 in CD.
c Regular secondary structure regions are 578–583, 590–602, 605–612, and 621–629 for CD.
Data Deposition
The chemical shifts of RBBP1 CD have been deposited in the BioMagResBank database (University of Wisconsin) with accession no. 17606. Coordinates and NMR restraints for the structures of RBBP1 CD have been deposited in the Protein Data Bank under code 2LCC.
Isothermal Titration Calorimetry
ITC measurements were performed on an iTC-200 calorimeter (MicroCal, Inc.). All experiments were carried out at 25 °C in 50 mm sodium phosphate buffer (pH 7.0) containing 50 mm NaCl. The reactant (0.2 mm CD or the mixture of 0.2 mm CD and 0.3 mm dsDNA) was placed in the 200-μl sample chamber, and H4K20me3 peptide (6.7 mm) in the syringe was added in 20 successive additions of 2 μl each taking 4 s (with an initial injection of 0.5 μl). The interval between each injection lasted 150 s. Control experiments were performed under identical conditions to determine the heat signals that arise from addition of the peptide into the buffer. Data were fitted using the single-site binding model within the Origin software package (MicroCal, Inc.).
RESULTS
Full-length RBBP1 Contains Three Domains with Potential to Recognize Histone Codes
The domain organizations of RBBP1 and RBBP1L1 were analyzed by bioinformatic methods. Combining the results of the secondary structure prediction (supplemental Fig. S1), the disorder prediction (supplemental Fig. S2), and the sequence alignment of RBBP1 and RBBP1L1 (supplemental Fig. S3), we found that RBBP1 and RBBP1L1 each contain five structural domains: a Tudor domain (TD, residues 1–121), a PWWP domain (PD, previously named RBB1NT domain, residues 170–273), an ARID domain (AD, residues 307–411), a chromobarrel domain (CD, residues 568–635) and a C-terminal R2 domain (residues 1150–1257) (Fig. 1). The sequences of these domains are highly conserved between RBBP1 and RBBP1L1 with ∼40–80% identity, which is much higher than the other regions of the protein (supplemental Fig. S3), implying important functional roles for these domains.
FIGURE 1.

Domain organization of RBBP1 and RBBP1L1.
RBBP1 AD and R2 domain are responsible for the repression activities; their structures are still unknown. Blast searches in the PDB found that the RBBP1 AD shows ∼20–40% identity to a number of ARID domains in other proteins, whereas the RBBP1 R2 domain has no sequence homology to any protein with known structure. The RBBP1 TD possesses very low sequence identity to other TD domains. The RBBP1 PD, the least conserved of the five domains, shows little sequence homology with any known protein. The solution structure of the RBBP1 PD has been solved (PDB code 2YRV) by RIKEN Structural Genomics/Proteomics Initiative using NMR. Although the domain was named the RBP1NT domain by the depositor of the structure, we found that the structure shows significant structural similarity to several PWWP domains (PDB codes 1N27 and 2X4X with Z-score of 6.0) using Dali search (supplemental Fig. S4, A and B) (29). A Blast search in the PDB found that the RBBP1 CD shows ∼20–40% identity to a number of chromobarrel domains in other proteins, and contains the conserved residues forming the aromatic cage (Fig. 2).
FIGURE 2.

Sequence alignments of the RBBP1 CD with homologous proteins. A, sequence alignment of the CDs of RBBP1 and RBBP1L1 from various species. The species are abbreviated as follows: Hs, Homo sapiens; Gg, Gallus gallus; Bt, Bos taurus; Dr, Danio rerio; Tg, Taeniopygia guttata; Xt, Xenopus (Silurana) tropicalis; Mm, Mus musculus; Ci, Ciona intestinalis; Dm, Drosophila melanogaster; Bm, Brugia malayi. B, structure-based sequence alignment of the RBBP1 CD with homologous chromo/chromobarrel domains of other proteins. The structure-based alignments were made using secondary structure matching (37). The PDB codes used in the structural alignments are shown in parentheses. Residues forming an aromatic cage and structurally corresponding residues are indicated by asterisks.
The PD, CD, and TD domains of RBBP1 are all Royal Family domains and thus potentially recognize methylated lysines of histones (13, 30). Although Royal Family domains share a similar protein fold, they may bind to methylated histone tails with different affinities and specificities. No experimental data have been reported previously exploring the structures, binding affinities or binding specificities of RBBP1 Royal Family domains. Therefore, we performed further structural and interaction studies to confirm and gain insight into the specificity of binding.
CD, TD, and PD Can Each Fold Independently and There Is No Direct Interaction between These Domains
We cloned and expressed the CD, TD and PD of RBBP1 in E. coli. We found that each of these domains could be expressed in a soluble form when the expression was induced at low temperature (16 or 25 °C), whereas inclusion bodies were formed when the expression was induced at 37 °C. However, each of these domains could be purified either directly from the soluble form or from the inclusion bodies by refolding. The purified proteins from the soluble form and the inclusion bodies showed identical NMR spectra (data not shown), indicating these domains can fold independently. The two-dimensional 1H-15N HSQC spectra of CD, TD, and PD are well dispersed (supplemental Fig. S5), which demonstrates that each of these domains are well folded.
We further used NMR titrations to detect whether these domains can interact with each other. When adding unlabeled PD to 15N-labeled CD, or unlabeled CD and PD to 15N-labeled TD, no change was observed in the two-dimensional 1H-15N HSQC spectra (supplemental Fig. S5). This proves that these three domains do not interact directly with each other. Therefore, the potential interactions of these domains with the methylated histones were studied using each individual domain.
Only CD of RBBP1, Not TD or PD, Binds Methylated Histone Tails
To investigate the molecular basis by which RBBP1 participates in epigenetic regulation, we investigated their interactions with methylated compounds and histone tails by NMR chemical shift perturbation experiment. We first monitored the chemical shift perturbation of 1H-15N HSQC spectra of 15N-labeled TD, PD, or CD by gradually adding methylated compounds, including mono-, di-, trimethylated lysines and dimethylated arginine. The results showed that TD and PD did not bind any of these compounds, and CD did not bind dimethylated arginine. However, CD specifically bound trimethylated lysine, with an affinity of ∼10 mm (Fig. 3 and supplemental Fig. S6). The affinity of the CD for dimethylated lysine is ∼50 mm and that for mono-methylated lysine is weaker.
FIGURE 3.

KD values for binding of RBBP1 CD to methylated histone tails, methylated lysines M3L and M2L, and the unmethylated histone tail H4K20me0.
To investigate the specificity of the recognition of methylated histone tails by the CD, we performed a series of NMR titration experiments using trimethylated and unmethylated peptides derived from the N termini of histones H3 and H4 (Table 1). Similar to the titration of trimethylated lysine, no chemical shift perturbations were observed in NMR titration of RBBP1 TD and PD using histone tails (supplemental Fig. S7, A and B), whereas chemical shift perturbations were observed to a greater or lesser extent in the titration of RBBP1 CD with all methylated peptides tested (Fig. 3 and supplemental Fig. S6). The peptides H3K36me3 and H4K20me3 caused larger chemical shift perturbations than H3K9me3 and H3K27me3. The equilibrium dissociation constant KD for binding of H4K20me3 to the RBBP1 CD derived by fitting the chemical shift changes to the equation deduced from a 1:1 binding model was ∼3 mm (Fig. 3). H4K20me3 shows the highest affinity for the RBBP1 CD for all these peptides, and it binds slightly more strongly than H3K36me3 (KD ∼ 4 mm). Small chemical shift perturbations were also observed in the titration with the peptide derived from histone H4 with unmethylated Lys20 (H4K20me0) (supplemental Fig. S6), whereas no perturbation was observed in the titration with other unmethylated histone peptides. Therefore, the RBBP1 CD recognizes methylated histone peptides with the highest affinity and specificity for H4K20me3.
Solution Structure of RBBP1 CD Reveals Aromatic Cage for Binding to Methylated Histone Tails
To understand the structural basis of CD recognition of histone tails, we solved the solution structure of the RBBP1 CD using multidimensional NMR. The structure contains five β-strands (β0, 578–583; β1, 590–602; β2, 605–612; β3, 621–624; and β4, 628–629), forming a β-barrel (Fig. 4, A and B). β3 and β4 are intercalated with a 310-helix H1 (625–627). The overall structure of the RBBP1 CD is a typical chromobarrel domain, similar to the CDs of human MRG15 (r.m.s.d. of 1.03 Å), yeast Eaf3 (r.m.s.d. of 1.23 Å), and Drosophila MSL3 (r.m.s.d. of 0.85 Å) (Fig. 4C) but lacks the C-terminal helix of other CDs. The N and C termini and loops L23 and L01 are not well converged in the structure ensemble because of the flexible nature of these segments. The NH signals in loops L23 and L01 are quite weak, and the signals of several residues (Gly614, Asn616–Arg618) in this loop are absent in the 1H-15N HSQC spectrum, suggesting that these loops are flexible.
FIGURE 4.

Structure of the RBBP1 CD. A, stereo view of the backbone ensemble of 20 structures of RBBP1 CD. B, ribbon representation of the RBBP1 CD structure. C, superimposition of the CDs of human RBBP1 (red) and human MRG15 (blue). The residues involved in forming aromatic cages and structurally corresponding residues are shown as sticks.
Mapping the chemical shift perturbations to the structure shows that the residues with large perturbations are located in the C termini of the β0 strand, loop L01, the N termini of the β1 strand, the C termini of the β2 strand, and loop L23 (Fig. 5). These regions contain five aromatic residues, Tyr583, Tyr592, Tyr612, Trp615, and Tyr619, which potentially form an aromatic cage. Aromatic cages are generally utilized to recognize the methylated lysines of histones by Royal Family domains (13, 31). The five aromatic residues have the largest chemical shift perturbations among the perturbed residues (Fig. 5), indicating that the RBBP1 CD recognizes the methylated lysines of histone peptides through its aromatic cage.
FIGURE 5.

Recognition of the RBBP1 CD by methylated histone tails. A, 1H-15N HSQC spectra of RBBP1 CD titrated with H4K20me3 peptide. B, bar diagram of chemical shift perturbations versus residue number at molar ratio 1:12 of the RBBP1 CD to various peptides. C, mapping of chemical shift perturbation to the RBBP1 CD structure.
Mutagenesis Analysis of Aromatic Cage of RBBP1 CD
To investigate the role of each aromatic residue in methylated lysine recognition, we did further mutagenesis analysis and titration experiments. Five mutants, each of which contained one aromatic residue mutated to Ala (Y583A, Y592A, Y612A, W615A, Y619A), were obtained and titrated with trimethylated lysine (Fig. 6). All of the mutants have well dispersed spectra with similar peak distributions to the wild-type CD, indicating that none of the mutations disrupt the structure of the CD. In the titrations, Y583A showed an affinity for M3L (KD ∼ 10 mm) almost identical to that of wild-type RBBP1 CD. The mutants of the other four aromatic residues showed significant decreases in affinity for M3L. Y612A, W615A, and Y619A did not bind M3L in the titrations, and Y592A showed only very weak affinity (KD ∼ 93 mm). These results demonstrate that four of the five aromatic residues, Tyr592, Tyr612, Trp615, and Tyr619, are critical for the binding with methylated lysine, whereas Tyr583 is not.
FIGURE 6.

Mutagenesis analysis of the RBBP1 CD and trimethylated lysine interaction. 1H-15N HSQC spectra of RBBP1 CD mutants titrated with M3L are exhibited for Y583A (A) (the M3L:protein molar ratio is from 0:1 to 15:1), Y592A (the M3L:protein molar ratio is from 0:1 to 15:1) (B), Y612A (the M3L:protein molar ratio is from 0 to 15:1) (C), W615A (the M3L:protein molar ratio is from 0 to 12:1) (D), and Y619A (the M3L:protein molar ratio is from 0 to 18:1) (E). F, KD values for binding of Y583A (upper panel) and Y592A (lower panel) to M3L.
Mutagenesis analysis demonstrates that four of the five aromatic residues, Tyr592, Tyr612, Trp615, and Tyr619, form the aromatic cage to bind methylated histone tails, although the side chain conformations of these residues are not converged in the structure ensembles. Our results are consistent with previous studies on the aromatic cages of other chromodomains. The four residues are conserved in chromobarrel domains that recognize histone tails (30), whereas Tyr583 is only conserved in some of these domains and the corresponding residue (His18) in Saccharomyces cerevisiae Eaf3 chromodomain was also excluded from involvement in formation of the aromatic cage (32).
DNA Binding Enhances Interaction of RBBP1 CD with H4K20me3
The RBBP1 CD is positively charged with a pI of 8.8 and has a significant amount of positively charged surface area (supplemental Fig. S8). This led us to suspect that the function of the CD of RBBP1 may involve interaction with nucleic acids. It has been reported that co-recognition of DNA and methylated histone tails occurs in the case of the MSL3 CD (20). Therefore, we checked the possibility of an interaction between the RBBP1 CD and dsDNA by NMR. Titration with dsDNA caused chemical shift perturbations and intensity decreases for a few peaks in the RBBP1 CD HSQC spectra (supplemental Fig. S8). The affinity of the CD binding to dsDNA could not be determined accurately from the NMR titration data because a small degree of precipitation occurred during the titration. However, a rough estimate from the NMR data places the dissociation constant in the range 10–100 μm. The DNA-binding regions identified by chemical shift perturbation mapping are located around a positively charged region (C terminus of β0, N-terminal half of β1, C terminus of β2, and N terminus of β3) near the aromatic cage, while other regions remained undisturbed (supplemental Fig. S8).
The finding that dsDNA binds to the regions near the aromatic cage suggests that the interaction of the RBBP1 CD with methylated histone peptide may be affected by the binding of dsDNA. We therefore performed NMR titration and ITC experiments to detect the effects of dsDNA on the interaction. The NMR titration showed that the RBBP1 CD recognizes H4K20me3 in the presence of dsDNA, and the binding affinity was enhanced from ∼3 mm in its absence (Fig. 3) to ∼0.5 mm in its presence (Fig. 7 and supplemental Fig. S9). The chemical shift perturbation mapping indicates that the regions of the RBBP1 CD involved in recognition of H4K20me3 are similar in the absence and presence of dsDNA. A subset of the interacting regions, in particular the C terminus of the β0 strand, the loop L01, and the N terminus of the β1 strand, shows more significant perturbation in the presence of dsDNA, which suggests that the interaction of H4K20me3 with this region is enhanced by concomitant interaction with dsDNA. The results of ITC experiments also confirmed an enhancement effect of dsDNA on the binding of RBBP1 CD with H4K20me3 (Fig. 7, C and D). The KD values obtained for binding of the CD to H4K20me3 in the absence and presence of DNA were 6.0 ± 0.4 mm and 0.4 ± 0.02 mm, respectively. Therefore, the ITC measurements indicate a 15-fold difference in affinity, consistent with the NMR titration results, which also indicated an order of magnitude increase in affinity. Thus, the results of both NMR and ITC indicate that the presence of dsDNA results in a significant enhancement in the affinity of the RBBP1 CD for methylated histone tails.
FIGURE 7.

The enhanced binding of the RBBP1 CD with H4K20me3 in the presence of dsDNA. A, 1H-15N HSQC spectra of the RBBP1 CD in the presence of dsDNA during titration with H4K20me3. The spectra with CD:H4K20me3 in molar ratios of 1:0, 1:0.5, 1:1.5, 1:3.5, and 1:5.0 are in black, cyan, blue, green, and red, respectively. B, KD values for binding of RBBP1 CD to H4K20me3 in the presence of dsDNA from NMR titration experiments. C and D, ITC results of H4K20me3 titration with RBBP1 CD in the absence (C) and presence (D) of dsDNA (CD:dsDNA = 1:1.5), respectively.
DISCUSSION
Of the three domains (CD, TD, PD) of RBBP1 that could possibly bind to methylated histone tails and participate in epigenetic regulation, our results demonstrate that only the CD of RBBP1 is responsible for recognizing methylated histone tails. The RBBP1 CD binds H4K20me3 with higher affinity than H3 peptides or other H4 peptides with a lower degree of methylation. Interestingly, H4K20 modification changes are observed in both leukemia and Prader-Willi/Angelman syndromes (3, 11), although the relationship between binding affinity and in vivo modification changes needs further investigation. The RBBP1 TD and PD lack the ability to bind methylated lysine, so the functions of these domains remain to be identified.
Chromobarrel domains generally have much weaker binding affinity (∼ 1 mm) than canonical chromodomains (50 μm) (33). The RBBP1 CD binds methylated histone tails with affinity of∼ 3 mm, which is similar to that of the CD of Eaf3 with H3K36me2 and of the Brpf1 PWWP domain with H3K36me3 (32–34). As in the CDs of MRG15, Eaf3, and MSL3, the RBBP1 CD contains an extra β-strand (β0), blocking the binding groove of the histone peptide seen in the HP1/Pc chromodomains complexed with histone peptides (32), which may lead to the weaker binding affinity of these chromobarrel domains. Weak affinities of single domains for histone tails has been found in many proteins and the physiological relevance can be explained by a combination effect of multiple domains/partners to obtain high affinity, a susceptibility to competition, as well as a greater potential specificity through the synchronous recognition of several marks (35). Besides the combination effect, another mechanism to enhance binding affinity and specificity is to co-recognize an epigenetic marker and dsDNA within the nucleosome, which has been observed in the case of dsDNA and H4K20me1 co-recognization by MSL3 CD (20). Both mechanisms may occur in the case of RBBP1. The RBBP1 CD can recognize both dsDNA and methylated histone tails, and dsDNA binding enhances the binding affinity of RBBP1 CD for H4K20me3 by ∼15-fold. RBBP1 contains an ARID domain which is a DNA-binding domain and may further enhance the affinity with chromatin. Since the dsDNA used in this study was arbitrarily selected, it is also possible that dsDNA with a certain specific sequence may increase the affinity even further. Besides the co-recognization mechanism, RBBP1 utilizes its R2 domain to recruit the mSin3A complex, which acts as a platform to bind many chromatin-modification related proteins (36); therefore, combination effects between the RBBP1 CD and mSin3A-binding proteins could further enhance the affinity and specificity. RBBP1 and RBBP1L1 can also interact with each other (11), and each contain a CD, potentially providing an additional combination effect in binding to histone tails.
CDs often show different specificity for histone peptides. The MSL3 CD specifically recognizes H4K20me1 in favor of previously bound DNA (20). The MRG15 CD recognizes H3K36me2/3 but not methylated H3K4, H3K9, or H3K27 (37), whereas the Eaf3 CD recognizes H3K36me3 and H3K4me3, and it also binds H4K20me3 with weaker affinity. Sun et al. (32) have proposed that the C-terminal helix helps binding of histone tails in the case of Eaf3 CD, increasing its specificity for H3K36me3, and this binding mode is similar for MRG15 CD. The RBBP1 CD lacks the C-terminal helix, and exhibits similar binding affinity to Eaf3 CD. In NMR titration experiments, peptide binding causes a small perturbation of loop L01 (Gly584–Gln589) connecting β0 and β1, and almost no perturbation of β4 and 310-helix H1 (Fig. 5, B and C), indicating that peptide binding mainly occurs near the aromatic cage. This is consistent with the fact that the RBBP1 CD recognizes each of the peptides: H3K9me3, H3K27me3, H3K36me3, and H4K20me3. Loop L23 (613–620) may favor binding of H4K20me3 and therefore lead to the strongest binding affinity with H4K20me3 among the methylated peptides. DNA binding may cause some conformational changes to the RBBP1 CD binding site, which may then lead to enhanced binding to H4K20me3.
The data presented here demonstrate that the CD of RBBP1 is responsible for recognizing methylated histone tails, particularly H4K20me3. The chromatin remodeling function of the mSin3A complex is mainly related to its enzymatic activity in epigenetic modification (such as HDAC and demethylase activities), while the role of RBBP1 as a component of the mSin3A complex is in recognition of histone codes. The structure and histone tail recognition of the RBBP1 chromobarrel domain presented here provides a basis for future studies to elucidate the roles of RBBP1 in gene suppression and epigenetic regulation.
Supplementary Material
This work was supported by National Natural Science Foundation of China Grants 30970571 (to Y. F.) and 30770434 (to J. W.) and the China Postdoctoral Science Foundation (to W. G.).

This article contains supplemental Figs. S1–S9 and additional references.
The atomic coordinates and structure factors (code 2LCC) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- ARID
- AT-rich interaction domain
- HDAC
- histone deacetylase
- TD
- Tudor domain
- PD
- PWWP domain
- CD
- chromobarrel domain
- CSP
- chemical shift perturbation
- ITC
- isothermal titration calorimetry
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Cao J., Gao T., Stanbridge E. J., Irie R. (2001) RBP1L1, a retinoblastoma-binding protein-related gene encoding an antigenic epitope abundantly expressed in human carcinomas and normal testis. J. Natl. Cancer Inst. 93, 1159–1165 [DOI] [PubMed] [Google Scholar]
- 2. Meehan W. J., Samant R. S., Hopper J. E., Carrozza M. J., Shevde L. A., Workman J. L., Eckert K. A., Verderame M. F., Welch D. R. (2004) Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J. Biol. Chem. 279, 1562–1569 [DOI] [PubMed] [Google Scholar]
- 3. Wu M. Y., Eldin K. W., Beaudet A. L. (2008) Identification of chromatin remodeling genes Arid4a and Arid4b as leukemia suppressor genes. J. Natl. Cancer Inst. 100, 1247–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patsialou A., Wilsker D., Moran E. (2005) DNA-binding properties of ARID family proteins. Nucleic Acids Res. 33, 66–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lai A., Marcellus R. C., Corbeil H. B., Branton P. E. (1999) RBP1 induces growth arrest by repression of E2F-dependent transcription. Oncogene 18, 2091–2100 [DOI] [PubMed] [Google Scholar]
- 6. Defeo-Jones D., Huang P. S., Jones R. E., Haskell K. M., Vuocolo G. A., Hanobik M. G., Huber H. E., Oliff A. (1991) Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 352, 251–254 [DOI] [PubMed] [Google Scholar]
- 7. Lai A., Kennedy B. K., Barbie D. A., Bertos N. R., Yang X. J., Theberge M. C., Tsai S. C., Seto E., Zhang Y., Kuzmichev A., Lane W. S., Reinberg D., Harlow E., Branton P. E. (2001) RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol. Cell Biol. 21, 2918–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lai A., Lee J. M., Yang W. M., DeCaprio J. A., Kaelin W. G., Jr., Seto E., Branton P. E. (1999) RBP1 recruits both histone deacetylase-dependent and -independent repression activities to retinoblastoma family proteins. Mol. Cell Biol. 19, 6632–6641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fleischer T. C., Yun U. J., Ayer D. E. (2003) Identification and characterization of three new components of the mSin3A corepressor complex. Mol. Cell Biol. 23, 3456–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malovannaya A., Li Y., Bulynko Y., Jung S. Y., Wang Y., Lanz R. B., O'Malley B. W., Qin J. (2010) Streamlined analysis schema for high throughput identification of endogenous protein complexes. Proc. Natl. Acad. Sci. U.S.A. 107, 2431–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu M. Y., Tsai T. F., Beaudet A. L. (2006) Deficiency of Rbbp1/Arid4a and Rbbp1l1/Arid4b alters epigenetic modifications and suppresses an imprinting defect in the Prader-Willi/Angelman syndrome domain. Genes Dev. 20, 2859–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jenuwein T., Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 13. Yap K. L., Zhou M. M. (2010) Keeping it in the family: Diverse histone recognition by conserved structural folds. Crit. Rev. Biochem. Mol. Biol. 45, 488–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maurer-Stroh S., Dickens N. J., Hughes-Davies L., Kouzarides T., Eisenhaber F., Ponting C. P. (2003) The Tudor domain “Royal Family:” Tudor, plant Agenet, Chromo, PWWP, and MBT domains. Trends Biochem. Sci. 28, 69–74 [DOI] [PubMed] [Google Scholar]
- 15. Cheng Y., Patel D. J. (2004) An efficient system for small protein expression and refolding. Biochem. Biophys. Res. Commun. 317, 401–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferentz A. E., Wagner G. (2000) NMR spectroscopy: A multifaceted approach to macromolecular structure. Q. Rev. Biophys. 33, 29–65 [DOI] [PubMed] [Google Scholar]
- 17. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 18. Johnson B. A., Blevins R. A. (1994) J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 19. Markley J. L., Bax A., Arata Y., Hilbers C. W., Kaptein R., Sykes B. D., Wright P. E., Wüthrich K. (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Mol. Biol. 280, 933–952 [DOI] [PubMed] [Google Scholar]
- 20. Kim D., Blus B. J., Chandra V., Huang P., Rastinejad F., Khorasanizadeh S. (2010) Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat. Struct. Mol. Biol. 17, 1027–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 22. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 23. Duggan B. M., Legge G. B., Dyson H. J., Wright P. E. (2001) SANE (Structure Assisted NOE Evaluation): An automated model-based approach for NOE assignment. J. Biomol. NMR 19, 321–329 [DOI] [PubMed] [Google Scholar]
- 24. Wishart D. S., Sykes B. D. (1994) The 13C chemical shift index: A simple method for the identification of protein secondary structure using 13C chemical shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 25. Cornilescu G., Delaglio F., Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 26. Nederveen A. J., Doreleijers J. F., Vranken W., Miller Z., Spronk C. A., Nabuurs S. B., Güntert P., Livny M., Markley J. L., Nilges M., Ulrich E. L., Kaptein R., Bonvin A. M. (2005) RECOORD: A recalculated coordinate database of 500+ proteins from the PDB using restraints from the BioMagResBank. Proteins 59, 662–672 [DOI] [PubMed] [Google Scholar]
- 27. Koradi R., Billeter M., Wuthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 28. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 29. Holm L., Rosenström P. (2010) Dali server: Conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yap K. L., Zhou M. M. (2011) Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry 50, 1966–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams-Cioaba M. A., Min J. (2009) Structure and function of histone methylation binding proteins. Biochem. Cell Biol. 87, 93–105 [DOI] [PubMed] [Google Scholar]
- 32. Sun B., Hong J., Zhang P., Dong X., Shen X., Lin D., Ding J. (2008) Molecular basis of the interaction of Saccharomyces cerevisiae Eaf3 chromodomain with methylated H3K36. J. Biol. Chem. 283, 36504–36512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu C., Cui G., Botuyan M. V., Mer G. (2008) Structural basis for the recognition of methylated histone H3K36 by the Eaf3 subunit of histone deacetylase complex Rpd3S. Structure 16, 1740–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vezzoli A., Bonadies N., Allen M. D., Freund S. M., Santiveri C. M., Kvinlaug B. T., Huntly B. J., Göttgens B., Bycroft M. (2010) Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 17, 617–619 [DOI] [PubMed] [Google Scholar]
- 35. Ruthenburg A. J., Li H., Patel D. J., Allis C. D. (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 8, 983–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Silverstein R. A., Ekwall K. (2005) Sin3: A flexible regulator of global gene expression and genome stability. Curr. Genet. 47, 1–17 [DOI] [PubMed] [Google Scholar]
- 37. Krissinel E., Henrick K. (2004) Secondary structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.