

Background: Cardiac myosin-binding protein-C is a sarcomeric assembly protein necessary for the regulation of sarcomere structure and function.
Results: The cMyBP-C motif is composed of two subdomains, a largely disordered N-terminal portion and a more ordered C-terminal subdomain.
Conclusion: The C-terminal subdomain is capable of forming a three-helix bundle.
Significance: The three-helix bundle may provide a platform for actin binding.
Keywords: Actin, Cardiomyopathy, Heart Failure, Nuclear Magnetic Resonance, Structural Biology, CS-Rosetta, Actin Binding, Cardiac Myosin-binding Protein-C, Cardiac Myosin-binding Domain, Protein Structure
Abstract
The structural role of the unique myosin-binding motif (m-domain) of cardiac myosin-binding protein-C remains unclear. Functionally, the m-domain is thought to directly interact with myosin, whereas phosphorylation of the m-domain has been shown to modulate interactions between myosin and actin. Here we utilized NMR to analyze the structure and dynamics of the m-domain in solution. Our studies reveal that the m-domain is composed of two subdomains, a largely disordered N-terminal portion containing three known phosphorylation sites and a more ordered and folded C-terminal portion. Chemical shift analyses, dNN(i, i + 1) NOEs, and 15N{1H} heteronuclear NOE values show that the C-terminal subdomain (residues 315–351) is structured with three well defined helices spanning residues 317–322, 327–335, and 341–348. The tertiary structure was calculated with CS-Rosetta using complete 13Cα, 13Cβ, 13C′, 15N, 1Hα, and 1HN chemical shifts. An ensemble of 20 acceptable structures was selected to represent the C-terminal subdomain that exhibits a novel three-helix bundle fold. The solvent-exposed face of the third helix was found to contain the basic actin-binding motif LK(R/K)XK. In contrast, 15N{1H} heteronuclear NOE values for the N-terminal subdomain are consistent with a more conformationally flexible region. Secondary structure propensity scores indicate two transient helices spanning residues 265–268 and 293–295. The presence of both transient helices is supported by weak sequential dNN(i, i + 1) NOEs. Thus, the m-domain consists of an N-terminal subdomain that is flexible and largely disordered and a C-terminal subdomain having a three-helix bundle fold, potentially providing an actin-binding platform.
Introduction
Cardiac myosin-binding protein-C (cMyBP-C)2 is a sarcomeric assembly protein, which is necessary for the regulation of sarcomere structure and function. It constitutes 2% of the total myofibrillar protein (1). Exclusively found in the vertebrate heart, cMyBP-C is encoded by a distinct gene (MYBPC3) (2, 3) and is localized in the inner two-thirds of the A band of the so-called C-zone where it interacts with myosin, actin, and titin (4). Mutations in the cMyBP-C gene (MYBPC3) are associated with ∼34% of all cardiomyopathy cases (5, 6) and are inherited in 1% of the world's population (7), underscoring the necessity of determining the role(s) of these mutations in the pathogenesis of heart failure. Finally, cMyBP-C deficiency may underlie development of abnormal sarcomere shortening velocity, left ventricular systolic elastance, and cardiac dysfunction, suggesting the important role of cMyBP-C in myocardial systolic mechanics (8).
Structurally, cMyBP-C consists of 11 modules labeled C0 to C10 from the N to the C terminus and belongs to the intracellular immunoglobulin superfamily. Three isoforms of cMyBP-C have been reported: fast skeletal, slow skeletal, and cardiac, each encoded by a separate gene. The domain organization is conserved in all the isoforms, comprising seven immunoglobulin-like (IgG) and three fibronectin type III motifs. cMyBP-C weakly interacts with the S2 fragment of myosin (9, 10), and it is strongly anchored to light meromyosin via the C10 domain (11) and titin via C8–C10 domains (12). Importantly, the cardiac isoform differs from the other two isoforms in that it contains an extra N-terminal domain (C0), a Pro-Ala-rich linker connecting the C0 and C1 domains, a unique structural motif termed “m-domain” for myosin-binding motif located between the C1 and C2 domains, and an insertion of ∼30 amino acids in the C5 domain. The Pro-Ala-rich linker sequence has been shown to contain a potential actin binding sequence, and species-specific differences in the Pro-Ala linker have been postulated to play a role in modulating heart rate in different species (13). Recent data provide evidence for in vitro interaction between cMyBP-C and F-actin via the C0 domain (14), C1-m-C2 domains (15–19), and the C5 domain (20). In addition, the m-domain is unique in that it contains three serine residues (Ser-273, Ser-282, and Ser-302) that are differentially phosphorylated by the enzymes PKA, PKC, PKD, ribosomal S6 kinase, and Ca2+/calmodulin-dependent protein kinase II (21). Phosphorylation of the m-domain may modulate interactions of the C1-m-C2 region with the myosin S2 region and actin. cMyBP-C is extensively phosphorylated under basal conditions (22). However, the level of cMyBP-C phosphorylation decreases in animal models during injury-reperfusion injury (22), pathologic hypertrophy (22), and myocardial stunning (23, 24), atrial fibrillation (25), and heart failure (26). In vitro studies demonstrated that phosphorylated cMyBP-C displays a weakened interaction with the S2 region of myosin and that this state promotes myosin-actin interaction, activating cross-bridge cycling rates (9). Taken together, these studies support the idea that cMyBP-C modulates sarcomere assembly and stabilizes thick and thin filaments via its N-terminal region. However, little is known about the structural mechanisms by which the N-terminal region of cMyBP-C modulates cardiac contractility in different vertebrate species.
A complete understanding of cMyBP-C arrangement in the sarcomere is essential in determining the roles that post-translational modification plays in modulating cMyBP-C interaction with myosin and/or actin. Two structural models are currently being discussed in the literature: the trimeric collar model and the rod model. The collar-like model proposes that three cMyBP-C molecules interact with each other at the C5–C10 domains and form a collar-like structure around the core of the thick filament (27). The rod model proposes that the C-terminal region of cMyBP-C interacts along the axis of the thick filament with the N-terminal region, extending perpendicularly toward the thin filament (28). Although there are conflicting data for each of the models, all invoke interaction with the thick and thin filament systems, suggesting the necessity of further studies on cMyBP-C to determine the structure and arrangement of the N-terminal region, its interaction partners, and the role of phosphorylation.
Small angle x-ray scattering of the N-terminal region in combination with the NMR structures of the individual C0, C1, and C2 domains suggests an elongated rodlike particle of sufficient length to span the interfilament cross-bridge distance (29). Bioinformatics analyses and scattering data for C1-m-C2 suggest that the structure of the m-domain is compact and consistent with an immunoglobulin-like fold (29). These studies further show that interdomain interfaces between C0, C1, m-domain, and C2 have a degree of rigidity, whereas the Pro/Ala linker connecting the C0 and C1 domains provides structural flexibility at the extreme N terminus (30). In addition, atomic force microscopic analysis predicts that the m-domain is likely to be disordered, increasing the flexibility between the C1 and C2 domains (31). Small angle neutron scattering with contrast variation in the presence of actin demonstrated a specific interaction between the N-terminal region of cMyBP-C and actin (32). The best fit model predicts that C0-C1 lie at the interface between two adjacent actin molecules with C0 lying close to the DNase I binding loop and C1 lying close to the actin filament near subdomain 1 (32). In this configuration, the m-domain and C2 project away from the actin filament partially as a consequence of steric hindrance. Taken together, the available structural data suggest that the N-terminal region of cMyBP-C interacts with both myosin and actin, providing a means for cMyBP-C to modulate thick and thin filament interactions. However, the molecular details of these interactions and the structural consequences of m-domain phosphorylation on the interactions of cMyBP-C with the thick and thin filaments remain unknown. In the present study, we utilized NMR to analyze the structure and dynamics of the m-domain in solution. Surprisingly, we found the N-terminal subdomain to be largely disordered and the C-terminal subdomain containing three helices to be capable of forming a three-helix bundle, potentially providing a platform for interactions with actin.
EXPERIMENTAL PROCEDURES
m-domain Plasmid Construction
DNA coding for amino acids 255–357 of mouse cMyBP-C (UniProt accession number 070468), representing the m-domain, was amplified by PCR using C0C2-coding DNA as a template. The forward and reverse cMyBP-C m-domain primers were 5′-ATGCCGCATATGCATGAGGCCATTGGTTCTGG-3′ and 5′-ATGCATGCGGCCGCTCACTTCTTTTCATCCTG-3′. The PCR product was then cloned into pET 28a(+) using the NdeI and NotI restriction enzymes and subsequently transformed in DH5α cells immediately following successful ligation. Plasmid minipreps were done, and the authenticity of the m-domain insert was confirmed by DNA sequencing.
Protein Expression and Purification
Uniform 13C/15N isotope-labeled protein representing the cMyBP-C m-domain was expressed following transformation into BL21(DE3) host cells (Novagen) using M9 medium supplemented with 1.0 g/liter 15NH4Cl and 2 g/liter [13C]glucose. Cells were grown to an A600 of 1.0 and induced with 0.1 mm final concentration of isopropyl 1-thio-β-d-galactopyranoside for recombinant protein expression. Cells were harvested by centrifugation after 4 h of induction and stored at −80 °C until further use. Frozen cells were thawed, resuspended in lysis buffer (50 mm NaH2PO4, pH 8.0, 200 mm NaCl, 5 mm imidazole, 1 mm PMSF, 1 mm β-mercaptoethanol) containing 0.1 mg/ml lysozyme, and subsequently lysed by sonication. Following centrifugation, the clarified supernatant was loaded on a nickel-nitrilotriacetic acid column equilibrated with lysis buffer. The column was washed, and the m-domain was eluted with a 200-ml linear gradient of 5–300 mm imidazole. Fractions containing m-domain were pooled, concentrated, and loaded onto a Superdex 75 gel filtration column. The m-domain eluted as a single peak and was determined to be homogenous by SDS gel electrophoresis followed by Coomassie Brilliant Blue staining. Protein concentration was estimated by Bradford analysis. The purified m-domain was concentrated by ultrafiltration and exchanged into NMR buffer (20 mm Tris-d11, pH 6.8, 50 mm NaCl, 2 mm tris(2-carboxyethyl)phosphine, 1 mm EDTA, 10% 2H2O) to a final concentration of ∼1 mm.
NMR Experiments and Structure Calculation
Backbone and side-chain resonances for [13C,15N]cMyBP-C(255–357) were assigned using the following suite of NMR experiments: HNCαCβ, HNCO, HN(CA)CO, CβCα(CO)NH,HβHα(CO)NH, and C(CO)NH (Table 1). 15N-TOCSY (50-ms mixing time), 15N-edited NOESY-HSQC (100-ms mixing times), and 15N-edited HSQC-NOESY-HSQC (100-, 150-, and 200-ms mixing times) were also used (Table 1). 15N{1H}NOE values were collected at 800 MHz as described previously (33). Chemical shift assignments for the m-domain were deposited in the BioMagResBank the under the accession number 17867. The secondary structure of the m-domain was predicted using chemical shift index (CSI) analysis, which was generated using 1Hα, 13Cα,13Cβ, and 13C′ chemical shifts. Dihedral angle restraints were calculated from 1Hα, 13Cα, 13Cβ, 13C′, and 15N chemical shifts with the program TALOS+ (34). The tertiary structure for the C-terminal portion of the m-domain was calculated with CS-Rosetta using the complete 13Cα, 13Cβ, 13C′, 15N, 1Hα, and 1HN chemical shifts of those residues determined by TALOS+ to have an S2 value >0.7. A set of 200 fragment candidates matching these chemical shifts was used to calculate 5000 structures in Rosetta. The energy of these Rosetta structures was then rescored against the observed chemical shifts, and the 20 lowest rescored energy structures were selected for the ensemble. This produced an ensemble of 20 structures with a Cα root mean square deviation of 1.3 ± 0.4 against the lowest energy structure. The structure ensemble representing the C-terminal portion of the m-domain was deposited in the Protein Data Bank the under the accession number 2LHU.
TABLE 1.
NMR parameters for resonance assignment and secondary structure determination of m-domain from cMyBP-C
nt, number of transients; tocsy, total correlation spectroscopy.
| Experiment | nt | sw1 | sw2 | sw3 | np1 | np2 | np3 | Field |
|---|---|---|---|---|---|---|---|---|
| HNCO | 8 | 12,019 | 3,770 | 2,430 | 1,024 | 80 | 32 | 800 |
| HN(CA)CO | 32 | 12,019 | 3,770 | 2,430 | 1,024 | 64 | 32 | 800 |
| HNCACB | 16 | 12,019 | 16,079 | 2,430 | 1,024 | 80 | 32 | 800 |
| CBCA(CO)NH | 16 | 12,019 | 16,079 | 2,430 | 1,024 | 80 | 32 | 800 |
| HBHA(CO)NH | 32 | 12,019 | 6,010 | 2,430 | 1,024 | 60 | 32 | 800 |
| C(CO)NNH | 16 | 12,019 | 16,079 | 2,430 | 1,024 | 80 | 32 | 800 |
| noesyNhsqc | 32 | 8,381 | 8,385 | 1,830 | 1,024 | 80 | 28 | 600 |
| tocsyNhsqc | 32 | 8,385 | 8,385 | 1,830 | 1,024 | 80 | 28 | 600 |
| NhsqcNOESYNhsqc | 56/96 | 8,385 | 1,830 | 1,830 | 1,024 | 48/34 | 28 | 600 |
| gNhsqc | 32 | 12,019 | 2,430 | 1,024 | 128 | 800 | ||
| gNOE | 160 | 12,019 | 2,430 | 1,024 | 128 | 800 |
RESULTS AND DISCUSSION
Characterization of m-domain Secondary Structure
We have cloned, expressed, and purified the m-domain, corresponding to residues 255–357 (human 257–361), of mouse cMyBP-C in an effort to probe the structure, dynamics, and consequences of phosphorylation on the N terminus of this multidomain protein. A 20% SDS-polyacrylamide gel of the m-domain was used to visualize the purity of the protein before initiating structural studies (Fig. 1). The CD spectrum of the m-domain was consistent with a protein having a high content of undefined structural elements. Fitting the experimental data using the CDSSTR method gave 33% helix, 16% strand, 20% turns, and 31% unordered structure (35, 36). Consistent with the high percentage of unordered structure, the melting curve for the m-domain is gradual and noncooperative when ellipticity at 220 nm was monitored as a function of temperature. This suggested that a portion of the m-domain is disordered or flexible in solution without a compact hydrophobic core.
FIGURE 1.
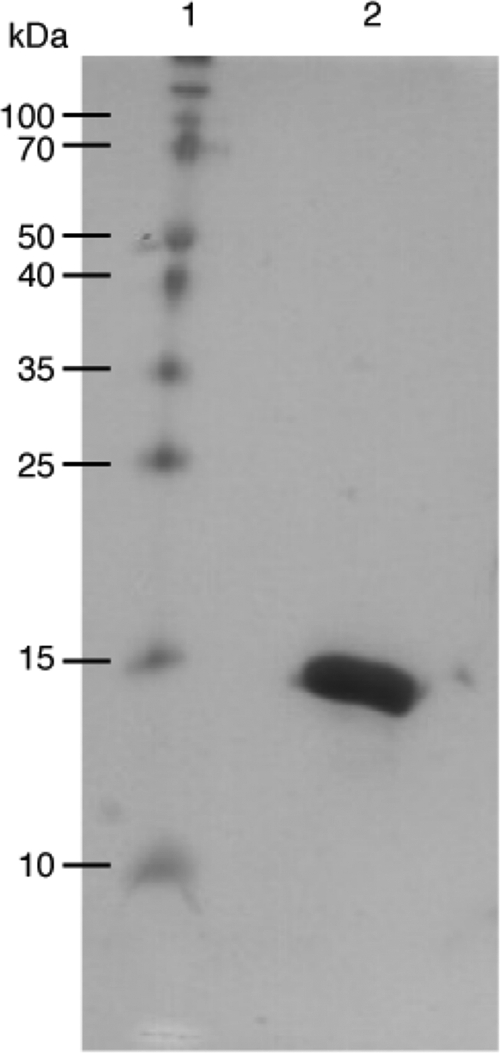
20% SDS-polyacrylamide gel of m-domain was used to evaluate purity of protein before initiating structural studies. The m-domain was visualized using Coomassie Brilliant Blue. Lane 1, molecular mass standards; Lane 2, 15 μg of [13C,15N]m-domain protein.
To further explore the structure and stability of the m-domain, isotope-labeled [13C, 15N]m-domain was prepared for analysis by heteronuclear multidimensional NMR. Backbone and side-chain NMR resonances were assigned for all 104 residues using standard triple resonance assignment strategies. Consistent with CD spectra, dispersion in the 1H-15N HSQC spectrum was significantly lower than that observed for a well structured protein (Fig. 2). To evaluate the dynamic properties of the m-domain, 15N{1H}NOE values at 800 MHz were collected. The 15N{1H}NOE data suggest that the protein can be divided into two regions, a well folded C-terminal portion having an average 15N{1H}NOE value of 0.78 and an N-terminal portion having 15N{1H}NOE values less then 0.6, suggesting a much more flexible structure (Fig. 3). Residues 309–314 appear to form a short linker between the less ordered N-terminal portion and the structured C-terminal portion (Fig. 3).
FIGURE 2.

Two-dimensional 1H-15N HSQC spectrum of [13C,15N]m-domain obtained at 800 MHz. Several of the more resolved correlations are labeled with residue assignments. Residues labeled with HT belong to the His-tag.
FIGURE 3.

1H-15N heteronuclear NOE values (15N{1H}NOE) for [13C,15N]m-domain are plotted versus residue number of m-domain in cMyBP-C. Data were collected as described under “Experimental Procedures.” The error bars represent the standard deviation of the heteronuclear NOE ratio.
Backbone and 13Cβ chemical shifts were used to evaluate m-domain secondary structure. Secondary chemical shifts, deviations in the observed chemical shifts from their random coil values, can be used to calculate the CSI, an accurate predictor of fully formed α- and β-structure (37, 38). Backbone chemical shifts for the Cβ, Cα, C′, and Hα resonances were used to determine CSI values for each m-domain residue (Table 2). The consensus CSI predicted five potential helical regions in the m-domain. Residues showing helical propensities were 265–268, 293–295, 314–322, 329–336, and 340–348 (Table 2). No regions of β-structure were detected. Backbone chemical shifts were also analyzed using a residue-specific secondary structure propensity (SSP) score (39). This approach has been highly successful in comparing secondary structure propensities in both intrinsically disordered and folded proteins. Residue-specific SSP scores of 1 or −1 reflect fully formed α- and β-structure, respectively. Scores between 0 and 1 are representative of the fraction of α-structure, whereas scores between 0 and −1 are representative of the fraction of β-structure. Thus, SSP scores qualitatively correspond to the secondary structural propensity at that residue. The SSP scores calculated for m-domain residues are shown in Fig. 4. Five regions with varying secondary structural propensities were identified. Residues 263–267 for helix 1 and residues 292–298 for helix 2 were found to have average structural propensities of 27 and 36%, respectively (Table 2). Lower structural propensities for these helices correspond to reduced 15N{1H}NOE values observed for the N-terminal region of the m-domain (Fig. 3). Residues 317–322, 324–339, and 341–351, enclosing helices 3, 4, and 5, exhibited 15N{1H}NOE values above 0.7, reflecting more stably formed secondary structures in the C-terminal region of the m-domain (Fig. 3). Table 2 summarizes secondary structure elements for the m-domain based on our experimental backbone and 13Cβ chemical shift assignments.
TABLE 2.
Secondary structure propensities for the m-domain of cMyBP-C determined using backbone chemical shifts obtained from multidimensional heteronuclear NMR experiments
Serine residues known to be phosphorylated are shown in red. seq, sequence.

1 Chemical shift index using secondary 1Hα chemical shifts to experimentally determine regions of secondary structure.
2 Consensus chemical shift index using secondary 1Hα, 13Cα, 13Cβ, and 13C′ chemical shifts to experimentally determine regions of secondary structure.
3 SSP scores were calculated as described under “Experimental Procedures.” SSP scores >0.20 and <0.5 are indicated as h for α-helix, and SSP scores >0.5 are indicated as H for α-helix. SSP scores <0.2 are indicated as C for coil.
4 Regions of secondary structure calculated using TALOS+ (43).
FIGURE 4.

SSP scores are plotted versus residue number of m-domain in cMyBP-C. Positive values represent α-structure propensity, and negative values represent β-structure propensity.
Computational Analysis of m-domain
NMR analysis of the m-domain demonstrated the presence of three regions of helical structure in the C-terminal portion, whereas the N-terminal portion exhibited significantly increased mobility and only two short regions characterized with a preference for occupying helical regions of φ/ψ space. Previously, Phyre homology modeling of C1-m-C2 modules suggested that the m-domain could be structurally related to an Ig fold (29). The authors noted that Phyre modeling of the m-domain alone failed to produce a useful model (29). We performed Phyre2 analysis on the m-domain to compare the secondary structural results obtained by NMR with the Phyre2 prediction (40). Although Phyre2 also failed to produce a reliable homology model, five helical regions largely consistent with those determined by NMR analysis were predicted. Secondary structure predictions were also performed using the SABLE, Psipred, and GOR V servers. All servers yielded five regions having a propensity for helical structure consistent with the NMR analysis (Table 3). SABLE, Psipred, and Phyre servers predicted helices 3–5 with high confidence and helices 1 and 2 at lower confidence levels (Table 3). The lower confidence in prediction of helices 1 and 2 may indicate regions of less stable structure capable of undergoing conformational transitions. This region contains three known phosphorylation sites at Ser-273/282/302. Serine residue 273 is a target for phosphorylation by both PKA and PKC (26). Interestingly, SABLE predicts that introducing negative charge at Ser-273 by mutation to Asp, mimicking phosphorylation, stabilizes and extends nascent helical structure in helix 1 between residues 265 and 270. Preliminary NMR data using a phosphorylation mimetic of the m-domain having Ser-273 mutated to Asp supports this prediction.3 In qualitative agreement with these results, SABLE predicted that introduction of negative charge at Ser-23/24 in the cardiac N-extension of cardiac troponin I, mimicking PKA phosphorylation, stabilized and extended the N-terminal cardiac troponin I helix (41). Amide NOEs confirmed this prediction (41). Bioinformatics analysis also suggested that introduction of negative charge at Ser-282 and Ser-302 by mutation to Asp has little effect on the stability of helices 1 and 2. Taken together, these studies suggest that the N-subdomain is a region of unstable structure that may switch conformations and possibly interacting partners upon phosphorylation. Similar phosphorylation-dependent molecular switches have previously been implicated in modulating cardiac contraction (41).
TABLE 3.
Bioinformatics analyses of secondary structural elements in m-domain of cMyBP-C
Serine residues known to be phosphorylated are shown in red. seq, sequence.

1 The predicted secondary structure and the secondary structure prediction confidence score obtained using the SABLE server.
2 The predicted secondary structure and the secondary structure prediction confidence score obtained using the Psipred server (55, 56).
3 The Phyre consensus predicted secondary structure and Phyre consensus probability obtained using the Phyre version 2 server (40).
4 The predicted secondary structure using the GOR V prediction algorithm (57).
5 Disorder prediction obtained using the Phyre version 2 server (40).
Structure Calculation of C-terminal Region of m-domain
Collection and analysis of NOE data for structure determination were hindered due to relatively poor chemical shift dispersion, a consequence of the natively disordered N-terminal region. Therefore, we choose to use CS-Rosetta (42), a program designed for structure calculation of proteins using backbone and 13Cβ chemical shift assignments, for structure determination of the stably folded C-terminal portion of the m-domain. Based on 15N{1H}NOE values, the C-terminal 35 residues corresponding to residues 315–349 were chosen for structure analysis. Complete 13Cα, 13Cβ, 13C′, 15N, 1Hα, and 1HN chemical shifts of those residues determined by TALOS+ (43) to have an S2 value >0.7 were used as input in CS-Rosetta structure calculations. A set of 200 fragment candidates matching these chemical shifts was used to calculate 5000 structures in Rosetta. The energy of these Rosetta structures was then rescored against the observed chemical shifts, and the 20 lowest rescored energy structures were selected for the ensemble, representing the three-dimensional structure of the C-terminal subdomain. The superposition of the 20 lowest energy structures having a Cα root mean square deviation of 1.3 ± 0.4 against the lowest energy structure is shown in Fig. 5A. The quality of the structures was analyzed by the MolProbity (44, 45) software package. Table 4 lists the statistics of the structure calculations. The low energy structure in the ensemble is shown in Fig. 5B. Overall, the structure reveals a novel three-helix bundle fold. The structure shows a tightly packed hydrophobic interior with all three helices participating in hydrophobic interactions along their length (Fig. 5B). Two antiparallel helices, spanning residues 317–322 (α1) and 327–335 (α2), lie nearly in the same plane, forming a helical hairpin (Fig. 5B). A third helix, corresponding to residues 341–348 (α3), is amphipathic and located across the top of the helical hairpin where it appears to stabilize the structure (Fig. 5B). All three helices are conserved across species. The two antiparallel helices, α1 and α2, are held in nearly the same plane by the short connecting loop containing two conserved Pro residues (Fig. 5B). The electrostatic surface of the C-terminal portion of the m-domain shows a largely hydrophobic belt around the middle of the structure, resulting from packing of the helix bundle (Fig. 5C). The solvent-exposed face of the α3 helix contains a basic cluster composed of Arg-342, Lys-346, Arg-347, and Lys-349 (Fig. 5C). This result is intriguing because it suggests the possibility of both hydrophobic and electrostatic interactions with either neighboring modules in cMyBP-C or additional sarcomeric proteins.
FIGURE 5.

Overall fold of C-terminal portion of m-domain generated using CS-Rosetta. A, backbone superposition of the 20 lowest energy NMR structures showing the three-helix bundle fold. B, ribbon representation of the low energy structure. The basic cluster composed of Arg-342, Lys-346, Arg-347, and Lys-349 forming the potential actin-binding motif is labeled. C, electrostatic surface representation of the low energy structure calculated using APBS (54). Positive and negative electrostatic potentials are shown in blue and red, respectively. The basic cluster composed of Arg-342, Lys-346, Arg-347, and Lys-349 forming the potential actin-binding motif is labeled. The hypertropic cardiomyopathy missense mutations, R236Q and E334K, are labeled in green.
TABLE 4.
Structural statistics for C-terminal portion of m-domain in cMyBP-C calculated using MolProbity
MolProbity server (45) was used.
| Clashscore, all atoms (percentile of structures) | 95.5 ± 3.9 |
| Poor rotamers (%) | 0.65 ± 1.03 |
| Ramachandran outliers (%) | 0 ± 0 |
| Ramachandran favored (%) | 100 ± 0 |
| Cβ deviations >0.25 Å | 0 ± 0 |
| MolProbity score (percentile of structures) | 98.1 ± 1.8 |
| Residues with bad bonds (%) | 0 ± 0 |
| Residues with bad angles (%) | 0 ± 0 |
There are four known human hypertropic cardiomyopathy missense mutations located in the C-terminal portion of the m-domain (46, 47). Two of these mutations, V338D and L348P (mouse sequence), are predicted to alter packing of the hydrophobic core in the three-helix bundle. The V338D mutation is located in the α1-α2 loop where the Val side chain normally contributes to the packing of the hydrophobic core. The L348P mutation is located in the α3 helix where the Leu side chain forms part of the hydrophobic core (Fig. 5B). Insertion of a Pro at this position is expected to disrupt packing of the hydrophobic core and destabilize the three-helix bundle. The impact of the L348P mutation on the stability of the three-helix bundle can be evaluated from a stability analysis performed with FoldX 3.0b5 (48). After repairing the low energy CS-Rosetta structure in FoldX and applying the BuildModel method to mutate Leu-348 to Pro, a stability analysis was performed. This revealed a ∼9.5 kcal/mol decrease in the favorable free energy of folding for the L348P mutation. In contrast, FoldX predicted a ∼1.6 kcal/mol decrease in the favorable free energy of folding for the V338D mutation located in the more mobile α1-α2 loop. The remaining two hypertropic cardiomyopathy missense mutations, R236Q and E334K (mouse sequence), are more surface-exposed and outside of the hydrophobic core (Fig. 5C).
Interestingly, the three-helix bundle fold is often found in actin-binding proteins. The headpiece domain from villin, a 35-residue actin-binding protein, is among the most studied three-helix bundle proteins. Sequence comparison of the villin headpiece with the C-terminal portion of the m-domain identifies a short sequence motif LK(R/K)XK in the third helix of both proteins. This motif is conserved between species in the m-domain of cMyBP-C proteins. Studies have shown this motif to be present in a number of actin-capping and -binding proteins and to bind actin (49–52). Taken together, our structural studies suggest that the C-terminal region of the m-domain may function as an actin-binding protein. Supporting this hypothesis, functional studies showed that the m-domain increased the Ca2+ sensitivity of tension and increased rates of tension redevelopment (53) and that C1-m binds F-actin in a saturable manner (15). Phosphorylation of the m-domain was shown to decrease the actin binding affinity (15). Both neutron scattering (32) and electron microscopy (16) are consistent with the binding of the N-terminal region of cMyBP-C (C0-C1-m-C2-C3) to actin.
Chemical shift mapping provides a powerful technique to monitor changes in protein structure and to map protein-protein interfaces. Amide proton and nitrogen chemical shift mapping was used to qualitatively monitor conformational changes in the m-domain induced by the presence of surrounding Ig domains, C1 and C2. Although complete amide resonance assignments are available for the isolated m-domain, only partial amide resonance assignments are available for C1-m and C0-C1-m-C2-C3. Amide resonance assignments for C-subdomain residues in the m-domain were mapped onto TROSY spectra of both C1-m and C0-C1-m-C2-C3. No significant chemical shift changes were detected for resolved m-domain resonances mapped onto the TROSY spectrum of C1-m. However, modest chemical shift differences were observed when resolved m-domain resonances were mapped onto the TROSY spectrum of C0-C1-m-C2-C3. The absence of chemical shift dispersion for resonances in the N-subdomain hindered chemical shift mapping of these resonances in C1-m and C0-C1-m-C2-C3. Although preliminary, chemical shift mapping suggests that addition of the C1 domain at the N terminus and the C2 domain at the C terminus of the m-domain does not greatly influence the conformational flexibility of the N-subdomain or the stability of the three-helix bundle in the C-subdomain. Modest chemical shift differences observed in the m-domain comparison with C0-C1-m-C2-C3 suggest protein-protein interactions between the C-subdomain and neighboring Ig modules.
In summary, we have shown that the N-terminal portion of the m-domain, containing phosphorylation sites Ser-273, Ser-282, and Ser-302, is conformationally flexible, containing two transiently formed helices that span residues 263–267 for helix 1 and residues 292–298 for helix 2. Bioinformatics analyses predict that introduction of negative charge at Ser-273, mimicking phosphorylation, will extend and stabilize helix 1. In contrast, 15N{1H}NOE and chemical shift analyses show that the C-terminal portion of the m-domain folds into a stable three-helix bundle. A known actin-binding motif, LK(R/K)XK, is positioned in the third helix (α3), similar to that found in villin and related proteins. These results suggest that the m-domain in cMyBP-C may alter actomyosin interactions in the heart through interactions with actin. Current studies are aimed at the role of the N-terminal portion of the m-domain in modulating actomyosin interactions via protein phosphorylation.
Acknowledgment
We thank Dr. Jarek Meller for valuable discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant R01HL105826 (to S. S.). This work was also supported by American Heart Association Scientist Development Grant 0830311N (to S. S.).
The atomic coordinates and structure factors (code 2LHU) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
J. W. Howarth, unpublished data.
- cMyBP-C
- cardiac myosin-binding protein-C
- m-domain
- myosin-binding motif (representing amino acids 255–357 of mouse cardiac myosin-binding protein-C)
- HSQC
- heteronuclear single quantum correlation
- SSP
- secondary structure propensity
- TROSY
- transverse relaxation-optimized spectroscopy
- CSI
- chemical shift index.
REFERENCES
- 1. Offer G., Moos C., Starr R. (1973) A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization. J. Mol. Biol. 74, 653–676 [DOI] [PubMed] [Google Scholar]
- 2. Yamamoto K., Moos C. (1983) The C-proteins of rabbit red, white, and cardiac muscles. J. Biol. Chem. 258, 8395–8401 [PubMed] [Google Scholar]
- 3. Fougerousse F., Delezoide A. L., Fiszman M. Y., Schwartz K., Beckmann J. S., Carrier L. (1998) Cardiac myosin binding protein C gene is specifically expressed in heart during murine and human development. Circ. Res. 82, 130–133 [DOI] [PubMed] [Google Scholar]
- 4. Oakley C. E., Hambly B. D., Curmi P. M., Brown L. J. (2004) Myosin binding protein C: structural abnormalities in familial hypertrophic cardiomyopathy. Cell Res. 14, 95–110 [DOI] [PubMed] [Google Scholar]
- 5. Spirito P., Seidman C. E., McKenna W. J., Maron B. J. (1997) The management of hypertrophic cardiomyopathy. N. Engl. J. Med. 336, 775–785 [DOI] [PubMed] [Google Scholar]
- 6. Barefield D., Sadayappan S. (2010) Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J. Mol. Cell. Cardiol 48, 866–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dhandapany P. S., Sadayappan S., Xue Y., Powell G. T., Rani D. S., Nallari P., Rai T. S., Khullar M., Soares P., Bahl A., Tharkan J. M., Vaideeswar P., Rathinavel A., Narasimhan C., Ayapati D. R., Ayub Q., Mehdi S. Q., Oppenheimer S., Richards M. B., Price A. L., Patterson N., Reich D., Singh L., Tyler-Smith C., Thangaraj K. (2009) A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat. Genet. 41, 187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Palmer B. M., Georgakopoulos D., Janssen P. M., Wang Y., Alpert N. R., Belardi D. F., Harris S. P., Moss R. L., Burgon P. G., Seidman C. E., Seidman J. G., Maughan D. W., Kass D. A. (2004) Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circ. Res. 94, 1249–1255 [DOI] [PubMed] [Google Scholar]
- 9. Gruen M., Prinz H., Gautel M. (1999) cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion. FEBS Lett. 453, 254–259 [DOI] [PubMed] [Google Scholar]
- 10. Sadayappan S., Osinska H., Klevitsky R., Lorenz J. N., Sargent M., Molkentin J. D., Seidman C. E., Seidman J. G., Robbins J. (2006) Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc. Natl. Acad. Sci. U.S.A. 103, 16918–16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gilbert R., Cohen J. A., Pardo S., Basu A., Fischman D. A. (1999) Identification of the A-band localization domain of myosin binding proteins C and H (MyBP-C, MyBP-H) in skeletal muscle. J. Cell Sci. 112, 69–79 [DOI] [PubMed] [Google Scholar]
- 12. Freiburg A., Gautel M. (1996) A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. Eur. J. Biochem. 235, 317–323 [DOI] [PubMed] [Google Scholar]
- 13. Shaffer J. F., Harris S. P. (2009) Species-specific differences in the Pro-Ala rich region of cardiac myosin binding protein-C. J. Muscle Res. Cell Motil. 30, 303–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kulikovskaya I., McClellan G., Flavigny J., Carrier L., Winegrad S. (2003) Effect of MyBP-C binding to actin on contractility in heart muscle. J. Gen. Physiol. 122, 761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shaffer J. F., Kensler R. W., Harris S. P. (2009) The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem. 284, 12318–12327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kensler R. W., Shaffer J. F., Harris S. P. (2011) Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin. J. Struct. Biol. 174, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mun J. Y., Gulick J., Robbins J., Woodhead J., Lehman W., Craig R. (2011) Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C). J. Mol. Biol. 410, 214–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Orlova A., Galkin V. E., Jeffries C. M., Egelman E. H., Trewhella J. (2011) The N-terminal domains of myosin binding protein C can bind polymorphically to F-actin. J. Mol. Biol. 412, 379–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu Y., Kwan A. H., Trewhella J., Jeffries C. M. (2011) The C0C1 fragment of human cardiac myosin binding protein C has common binding determinants for both actin and myosin. J. Mol. Biol. 413, 908–913 [DOI] [PubMed] [Google Scholar]
- 20. Rybakova I. N., Greaser M. L., Moss R. L. (2011) Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J. Biol. Chem. 286, 2008–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sadayappan S., Gulick J., Osinska H., Barefield D., Cuello F., Avkiran M., Lasko V. M., Lorenz J. N., Maillet M., Martin J. L., Brown J. H., Bers D. M., Molkentin J. D., James J., Robbins J. (2011) A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ. Res. 109, 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sadayappan S., Gulick J., Osinska H., Martin L. A., Hahn H. S., Dorn G. W., 2nd, Klevitsky R., Seidman C. E., Seidman J. G., Robbins J. (2005) Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 97, 1156–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Decker R. S., Decker M. L., Kulikovskaya I., Nakamura S., Lee D. C., Harris K., Klocke F. J., Winegrad S. (2005) Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation 111, 906–912 [DOI] [PubMed] [Google Scholar]
- 24. Yuan C., Guo Y., Ravi R., Przyklenk K., Shilkofski N., Diez R., Cole R. N., Murphy A. M. (2006) Myosin binding protein C is differentially phosphorylated upon myocardial stunning in canine and rat hearts—evidence for novel phosphorylation sites. Proteomics 6, 4176–4186 [DOI] [PubMed] [Google Scholar]
- 25. El-Armouche A., Pohlmann L., Schlossarek S., Starbatty J., Yeh Y. H., Nattel S., Dobrev D., Eschenhagen T., Carrier L. (2007) Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. J. Mol. Cell. Cardiol. 43, 223–229 [DOI] [PubMed] [Google Scholar]
- 26. Copeland O., Sadayappan S., Messer A. E., Steinen G. J., van der Velden J., Marston S. B. (2010) Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. J. Mol. Cell. Cardiol. 49, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 27. Flashman E., Korkie L., Watkins H., Redwood C., Moolman-Smook J. C. (2008) Support for a trimeric collar of myosin binding protein C in cardiac and fast skeletal muscle, but not in slow skeletal muscle. FEBS Lett. 582, 434–438 [DOI] [PubMed] [Google Scholar]
- 28. Squire J. M., Luther P. K., Knupp C. (2003) Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. J. Mol. Biol. 331, 713–724 [DOI] [PubMed] [Google Scholar]
- 29. Jeffries C. M., Whitten A. E., Harris S. P., Trewhella J. (2008) Small-angle x-ray scattering reveals the N-terminal domain organization of cardiac myosin binding protein C. J. Mol. Biol. 377, 1186–1199 [DOI] [PubMed] [Google Scholar]
- 30. Jeffries C. M., Lu Y., Hynson R. M., Taylor J. E., Ballesteros M., Kwan A. H., Trewhella J. (2011) Human cardiac myosin binding protein C: structural flexibility within an extended modular architecture. J. Mol. Biol. 414, 735–748 [DOI] [PubMed] [Google Scholar]
- 31. Karsai A., Kellermayer M. S., Harris S. P. (2011) Mechanical unfolding of cardiac myosin binding protein-C by atomic force microscopy. Biophys. J. 101, 1968–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Whitten A. E., Jeffries C. M., Harris S. P., Trewhella J. (2008) Cardiac myosin-binding protein C decorates F-actin: implications for cardiac function. Proc. Natl. Acad. Sci. U.S.A. 105, 18360–18365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gaponenko V., Abusamhadneh E., Abbott M. B., Finley N., Gasmi-Seabrook G., Solaro R. J., Rance M., Rosevear P. R. (1999) Effects of troponin I phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. J. Biol. Chem. 274, 16681–16684 [DOI] [PubMed] [Google Scholar]
- 34. Cornilescu G., Delaglio F., Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 [DOI] [PubMed] [Google Scholar]
- 35. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sreerama N., Woody R. W. (2000) Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 287, 252–260 [DOI] [PubMed] [Google Scholar]
- 37. Wishart D. S., Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 38. Wishart D. S., Sykes B. D. (1994) Chemical shifts as a tool for structure determination. Methods Enzymol. 239, 363–392 [DOI] [PubMed] [Google Scholar]
- 39. Marsh J. A., Singh V. K., Jia Z., Forman-Kay J. D. (2006) Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 41. Howarth J. W., Meller J., Solaro R. J., Trewhella J., Rosevear P. R. (2007) Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J. Mol. Biol. 373, 706–722 [DOI] [PubMed] [Google Scholar]
- 42. Shen Y., Lange O., Delaglio F., Rossi P., Aramini J. M., Liu G., Eletsky A., Wu Y., Singarapu K. K., Lemak A., Ignatchenko A., Arrowsmith C. H., Szyperski T., Montelione G. T., Baker D., Bax A. (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. U.S.A. 105, 4685–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C.(2003) Structure validation by Cα geometry: φ,ψ and Cβ deviation. Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 45. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harris S. P., Lyons R. G., Bezold K. L. (2011) In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 108, 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Richard P., Charron P., Carrier L., Ledeuil C., Cheav T., Pichereau C., Benaiche A., Isnard R., Dubourg O., Burban M., Gueffet J. P., Millaire A., Desnos M., Schwartz K., Hainque B., Komajda M. (2003) Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232 [DOI] [PubMed] [Google Scholar]
- 48. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L. (2005) The FoldX web server: an online force field. Nucleic Acids Res. 33, W382–W388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamamoto M., Nagata-Ohashi K., Ohta Y., Ohashi K., Mizuno K. (2006) Identification of multiple actin-binding sites in cofilin-phosphatase Slingshot-1L. FEBS Lett. 580, 1789–1794 [DOI] [PubMed] [Google Scholar]
- 50. Friederich E., Vancompernolle K., Huet C., Goethals M., Finidori J., Vandekerckhove J., Louvard D. (1992) An actin-binding site containing a conserved motif of charged amino acid residues is essential for the morphogenic effect of villin. Cell 70, 81–92 [DOI] [PubMed] [Google Scholar]
- 51. Hertzog M., Milanesi F., Hazelwood L., Disanza A., Liu H., Perlade E., Malabarba M. G., Pasqualato S., Maiolica A., Confalonieri S., Le Clainche C., Offenhauser N., Block J., Rottner K., Di Fiore P. P., Carlier M. F., Volkmann N., Hanein D., Scita G. (2010) Molecular basis for the dual function of Eps8 on actin dynamics: bundling and capping. PLoS Biol. 8, e1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vardar D., Buckley D. A., Frank B. S., McKnight C. J. (1999) NMR structure of an F-actin-binding “headpiece” motif from villin. J. Mol. Biol. 294, 1299–1310 [DOI] [PubMed] [Google Scholar]
- 53. Razumova M. V., Bezold K. L., Tu A. Y., Regnier M., Harris S. P. (2008) Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. J. Gen. Physiol. 132, 575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jones D. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 56. Bryson K., McGuffin L. J., Marsden R. L., Ward J. J., Sodhi J. S., Jones D. T. (2005) Protein structure prediction servers at University College London. Nucleic Acids Res. 33, W36–W38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sen T. Z., Jernigan R. L., Garnier J., Kloczkowski A. (2005) GOR V server for protein secondary structure prediction. Bioinformatics 21, 2787–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]