

Abstract
Accumulation of misfolded proteins and alterations in calcium homeostasis induces endoplasmic reticulum (ER) stress, leading to apoptosis. Here we tested the hypothesis that β-AR stimulation induces ER stress, and induction of ER stress plays a pro-apoptotic role in cardiac myocytes. Using thapsigargin and brefeldin A, we demonstrate that ER stress induces apoptosis in adult rat ventricular myocytes (ARVMs). β-AR-stimulation (isoproterenol; 3h) significantly increased expression of ER stress proteins such as GRP-78, Gadd-153 and Gadd-34, while activating caspase-12 in ARVMs. In most parts, these effects were mimicked by thapsigargin. β-AR stimulation for 15 min increased PERK and eIF-2α phosphorylation. PERK phosphorylation remained higher, while eIF-2α phosphorylation declined thereafter, reaching to ~50% below basal levels 3 h after β-AR stimulation. This decline in eIF-2α phosphorylation was prevented by β1-AR, not by β2-AR, antagonist. Forskolin, adenylyl cyclase activator, simulated the effects of ISO on eIF-2α phosphorylation. Salubrinal, an ER stress inhibitor, maintained eIF-2α phosphorylation and inhibited β-AR-stimulated apoptosis. Furthermore, inhibition of caspase-12 using z-ATAD inhibited β-AR-stimulated and thapsigargin-induced apoptosis. In vivo, β-AR stimulation induced ER stress in the mouse heart as evidenced by increased expression of GRP-78 and Gadd-153, activation of caspase-12 and dephosphorylation of eIF-2α. Salubrinal maintained phosphorylation of eIF-2α, inhibited activation of caspase-12 and decreased β-AR-stimulated apoptosis in the heart. Thus β-AR stimulation induces ER stress in cardiac myocytes and in the heart, and induction of ER stress plays a pro-apoptotic role.
Keywords: ER stress, Apoptosis, Myocytes, Heart, eIF-2α
1. Introduction
Increased sympathetic activity to the heart is an early response to hemodynamic dysfunction [1;2] resulting in enhanced exposure of cardiac myocytes to the primary neurotransmitter, norepinephrine. Cardiac myocyte apoptosis has gained recognition as an important determinant of structure and function of the myocardium [3;4]. Stimulation of β-adrenergic receptors (β-AR) induces apoptosis in cardiac myocytes in vitro and in vivo [5–8]. β-AR-stimulated apoptosis in adult rat ventricular myocytes (ARVMs) is shown to occur via the JNK-dependent mitochondrial death pathway [9]. Our laboratory has provided evidence that β-AR stimulation activates glycogen synthase kinase-3β (GSK-3β), and activation of GSK-3β plays a pro-apoptotic role in β-AR-stimulated apoptosis [10].
The endoplasmic reticulum (ER or sarcoplasmic reticulum in cardiac myocytes) regulates protein synthesis, protein folding and trafficking, cellular responses to stress and intracellular Ca++ levels [11–13]. Specifically, ER is recognized as the site of synthesis and folding of secreted, membrane-bound, and some organelle-targeted proteins. Accumulation of misfolded proteins and alteration in Ca++ homeostasis initiate an adaptive response in the cell, termed the unfolded protein response (UPR, ER stress response). As a result, ER localized chaperones are induced, protein synthesis is slowed down and a protein degrading system is initiated [12]. Prolonged ER stress may take on the role of executioner by increasing expression of ER stress proteins such as Gadd-153 and Gadd-34, and activation of caspase-12. Prolonged ER stress triggers apoptosis in various cell types. A number of studies suggest that ER stress plays a critical role in the pathogenesis of heart failure. Expression of ER chaperones and accumulation of ubiquitylated proteins is demonstrated to be higher in failing human heart [14;15]. Pressure overload-induced cardiac myocyte apoptosis is shown to be associated with increased ER stress in the mouse myocardium [14]. Infusion of angiotensin II and induction of diabetes (using streptozotocin) are also shown to be associated with ER stress and apoptosis in the heart [16]. ER stressors (thapsigargin and tunicamycin) induced ER stress and apoptosis in neonatal cardiac myocytes [16]. Expression of a mutant KDEL (Lys-Asp-Glu-Leu) receptor, a retrieval receptor for ER chaperones such as GRP-78, in mice resulted in dilated cardiomyopathy with enhanced expression of Gadd-153 [17]. Anti-β1-AR antibodies are shown to induce ER stress and apoptosis in neonatal rat cardiac myocytes [18]
This study was undertaken to investigate if β-AR stimulation induces ER stress in ARVMs and in the heart, and if ER stress is involved in β-AR-stimulated apoptosis. It was hypothesized that induction of ER stress plays a pro-apoptotic role in β-AR-stimulated apoptosis. Our results show that β-AR stimulation induces ER stress in cardiac myocytes in vitro and in vivo. Using specific inhibitors of ER stress, we provide evidence that induction of ER stress plays a pro-apoptotic role in β-AR-stimulated myocyte apoptosis.
2. Methods and Material
2.1. Cell isolation and culture
Calcium-tolerant ARVMs were isolated from the hearts of adult male Sprague-Dawley rats (150–200 g) as described [19]. ARVMs were plated in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech) supplemented with HEPES (25 mM), BSA (0.2%), creatine (5 mM), L-carnitine (2 mM), taurine (5 mM) and 0.1% penicillin-streptomycin at a density of 30–50 cells/mm2 on 100-mm tissue culture dishes (Fisher Scientific) or coverslips precoated with laminin (1μg/cm2). The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The animal protocol was approved by the University Committee on Animal Care.
2.2. Cell treatment
ARVMs, cultured for 24 h, were treated with isoproterenol (ISO; 10 μM; Sigma), forskolin (10 μM; Sigma), thapsigargin (2 μM; Sigma) or brefeldin A (1 μM; Sigma) for 15 min, 3 h or 24 h. For treatment with ISO, dishes were supplemented with ascorbic acid (100 μM). CGP20712A (0.3 μM; Sigma), ICI 118551 (0.1 μM; Sigma), salubrinal (1 or 10 μM) or z-ATAD-FMK (10 μM) were added for 30 min prior to ISO treatment. Salubrinal was initially obtained from Dr Junying Yuan (Dept of Cell biology, Harvard Medical School) and then purchased from Tocris Bioscience. The concentrations of the inhibitors were chosen based on previously published reports [20–22]. The treatment times (3 and 24 h) were chosen based on the observation that an increase in the percentage of apoptotic ARVMs becomes apparent by 6 h which increases further 24 h after β-AR stimulation [7]. Increased levels of cytosolic cytochrome C are observed 6 h after β-AR stimulation [9], indicating induction mitochondrial death pathway at this time point. ER stress may act upstream in the activation of mitochondrial death pathway [23].
2.3. Isoproterenol infusion in mice
For in vivo studies, CD-1 mice (Harlan Lab.) were infused with ISO (400 μg/kg/day) for 7 days by subcutaneous implantation of mini-osmotic pumps (Alzet) as described [5]. The mice were 5–8 weeks old and weighed 25–32g. Saline (0.9% NaCl) infused mice served as sham. To investigate the role of ER stress, mice were infused with salubrinal (SAL; 1 mg/kg/day) in the presence (SAL+ISO) or absence of ISO (SAL alone). SAL was dissolved in DMSO followed by dilution in saline [24].
2.4. Terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay
TUNEL-staining was performed on ARVMs plated on thermanox coverslips using in situ death detection kit according to the manufacturer’s instructions (Roche Molecular Biochemicals). The percentage of TUNEL-positive cells (relative to total ARVMs) was determined by counting ~200 cells in 10 randomly chosen fields per coverslip for each experiment. To detect apoptosis in the heart, 5-μm-thick sections from the mid LV were stained using in situ death detection kit. To identify apoptosis associated with cardiac myocytes, the sections were immunostained using α-sarcomeric actin antibodies (1:50; 5C5 clone, Sigma). Hoechst 33258 (10 μM; Sigma) staining was used to count the total number of nuclei. TUNEL-positive nuclei clearly seen within cardiac myocytes were counted. The index of apoptosis was calculated as the percentage of apoptotic myocyte nuclei/total number of nuclei.
2.5. Western blot analyses
ARVMs were lysed in cell lysis buffer (10 mM Tris-HCl; pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 0.2 mM sodium orthovanadate, 0.5% Nonidet P-40, 1 % Triton X-100, and 1 mM phenylmethylsulfonyl fluoride). LV lysates were prepared in RIPA buffer (158 mM NaCl, 10 mM Tris-HCl; pH 7.2, 1 mM EGTA, 1mM sodium orthovanadate, 0.1% sodium dodecyl sulphate, 1.0% Triton X-100, 1% sodium deoxycholate and 1 mM phenyl-methylsulphonyl fluoride). Equal amounts of total proteins (50–100 μg) from cell or tissue lysates were analyzed by western blot as described [19]. The primary antibodies used were – GRP-78 (Stressgen), Gadd-34 (Santa Cruz), Gadd-153 (Santa Cruz), procaspase-12 (Sigma), PERK and p-PERK (Santa Cruz), eIF-2α (Santa Cruz) and p-eIF-2α (Cell Signaling). The membranes were stripped and probed for GAPDH (Santa Cruz; for cell lysates) or actin (Chemicon; for tissue lysates) as protein loading controls. Band intensities were quantified using Kodak photodocumentation system (Eastman Kodak Co.)
2.6. Statistical analyses
All data are expressed as mean ± SE. Statistical analysis was performed using the student’s t test or a one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test. Probability (p) values of <0.05 were considered to be significant. The number (n) indicates the number of biological replicates.
3. Results
3.1. ER stressors induce apoptosis in ARVMs
To demonstrate if induction of ER stress induces apoptosis in adult cardiac myocytes, ARVMs were treated with thapsigargin (THAP; 2 μM; sarcoplasmic reticulum calcium ATPase inhibitor) and brefeldin A (BFA; 1 μM; an inhibitor of protein transport between the ER and Golgi complex) as pharmacological ER stress inducers for 24 h. Isoproterenol (ISO; 10 μM) treatment served as a positive control. Analysis of apoptosis using TUNEL-staining assay demonstrated that both pharmacological ER stressors significantly increase the number of apoptotic ARVMs (Fig 1). The extent of apoptosis was found to be similar to that with ISO.
Fig 1.

Induction of ER stress induces apoptosis in ARVMs. ARVMs were treated with thapsigargin (THAP; 2 μM), brefeldin A (BFA; 1 μM) or isoproterenol (ISO; 10 μM) for 24 h. The number of apoptotic cells was measured using TUNEL staining assay. *p<0.05 vs. control (CTL); n=3.
3.2. β-AR stimulation induces ER stress in ARVMs
Increased Gadd-153 expression is considered as a hallmark of ER stress. Gadd-153 may promote ER stress-induced apoptosis by promoting expression of Gadd-34, a nonenzymatic cofactor of PP1 involved in dephosphorylation of eIF-2α [25]. To study expression of Gadd-153 and Gadd-34, ARVMs were treated with ISO (10 μM) or THAP (2 μM) for 3 or 24 h. Analysis of total cell lysates by western blot using anti- Gadd-153 and Gadd-34 antibodies indicated 1.55±0.18 and 1.62±0.15-fold increase in Gadd-153 and Gadd-34 protein levels, respectively, as compared to control 3 h after β-AR stimulation (*p<0.05 vs CTL; n=3–4; Fig 2A and B). THAP treatment for 3 h also increased expression of Gadd-153 and Gadd-34 (Fig 2A and B). The expression of these proteins declined thereafter, reaching basal levels 24 h after β-AR stimulation (data not shown). The expression of Gadd-153, not Gadd-34, remained higher 24 h after THAP treatment (data not shown).
Fig 2.

Expression of Gadd-153, Gadd-34 and GRP-78. ARVMs were treated with ISO or THAP for 3 h. Cell lysates were analyzed by western blot using anti-Gadd-153 (A), anti-Gadd-34 (B), anti-GRP-78 (C) antibodies. The lower panels exhibit the mean data normalized to GAPDH. *p<0.05 vs. CTL; n=3–8.
Glucose-regulated protein-78 (GRP-78; also called Bip) is a classical marker of UPR during ER stress. The induction of GRP-78 is required to alleviate ER stress and maintain ER function. During ER stress, misfolded proteins bind to GRP-78 and disrupt its interaction with the stress sensors, resulting in the activation of stress sensors (IRE1, PERK and ATF6) [11]. To study expression of GRP-78, ARVMs were treated with ISO (10 μM) or THAP (2 μM) for 3 or 24 h. Analysis of total cell lysates by western blot using anti-GRP-78 antibodies indicated 1.2±0.1-fold increase in GRP-78 protein levels as compared to control (*p<0.05 vs CTL; n=8; Fig 2C). No increase in GRP-78 protein levels was observed 24 h after β-AR stimulation or 3 and 24 h after THAP treatment (data not shown).
The main caspase associated with the ER stress-induced apoptosis is caspase-12. Procaspase-12 (~50 kDa) predominantly localizes on the surface of the ER or in the soluble compartment of the ER [26]. Activation of caspase-12 by ER stress and calcium release can promote apoptosis via the involvement of caspase-3. A decrease in the procaspase-12 signal on western is considered as activation of caspase-12 [27]. To study activation of caspase-12, ARVMs were treated with ISO (10 μM) or THAP (2 μM) for 3 or 24 h. Analysis of total cell lysates by western blot using anti-procaspase-12 antibodies indicated a significant decrease in the levels of procaspase-12 3 h after β-AR stimulation. THAP also decreased levels of procaspase-12 (Fig 3A). The levels of procaspase-12 remained lower 24 h after β-AR stimulation or THAP treatment (Fig 3B).
Fig 3.

Activation of caspase-12. ARVMs were treated with ISO or THAP for 3 (A) or 24 (B) h. Cell lysates were analyzed by western blot using anti-procaspase-12 antibodies. The lower panels exhibit the mean data normalized to GAPDH. *p<0.05 vs. CTL; n=3–6.
Promotion of eIF-2α (eukaryotic translation initiation factor 2 subunit alpha) phosphorylation reduces protein synthesis and protein folding load in the ER, permitting cells to recover from ER stress that can otherwise lead to apoptosis [11]. Therefore, we next investigated the phosphorylation status of eIF-2α. Analysis of total cell lysates using phospho-specific eIF-2α antibodies indicated a significant increase in the phosphorylation of eIF-2α within 15 min of ISO treatment (Fig 4A). However, a significant decline in eIF-2α phosphorylation was observed 3 h after ISO treatment. Phosphorylation of eIF-2α was ~50% lower than control (Fig 4B). Phosphorylation of eIF-2α remained lower 24 h after β-AR stimulation (Fig 4C). eIF-2α phosphorylation remained unchanged 3 h after THAP treatment, while it was significantly increased 24 h after THAP treatment (Fig 4B and C). Levels of total eIF-2α remained unchanged during these treatment times (data not shown).
Fig 4.

Phosphorylation of eIF-2α. ARVMs were treated with ISO for 15 min, and ISO or THAP for 3 or 24 h. Cell lysates were analyzed by western blot using anti-phospho-specific eIF-2α antibodies. A. β-AR stimulation for 15 min increased eIF-2α phosphorylation. *p<0.05 vs. CTL; n=4. B. β-AR stimulation for 3 h decreased eIF-2α phosphorylation. THAP treatment (3 h) had no eIF-2α phosphorylation. *p<0.05 vs. CTL; n=3–5. C. Phosphorylation of eIF-2α remained lower 24 h after β-AR stimulation. THAP treatment (24 h) increased eIF-2α phosphorylation. *p<0.05 vs. CTL; n=5–6. The lower panels exhibit the mean data normalized to GAPDH.
Activation of PERK (PKR-like ER kinase) and PERK-mediated phosphorylation of elF-2α on ser-51 leads to - 1) a decrease in protein folding load in the ER; 2) the expression of eIF-2α phosphorylation-dependent ER stress genes; and 3) promotion of cell survival [11]. On the other hand, Gadd-34 is a nonenzymatic cofactor of PP1 involved in dephosphorylation of eIF-2α [25]. Analysis of phosphorylation of PERK using phospho-specific PERK antibodies indicated a significant increase in PERK phosphorylation 15 min (Fig 5A) and 3 h after ISO treatment (Fig 5B). PERK phosphorylation declined thereafter reaching basal levels 24 h after ISO treatment (Fig 5C). THAP treatment for 3 or 24 h increased PERK phosphorylation (Fig 5B and C). Levels of total PERK remained unchanged during these treatment times (data not shown).
Fig 5.
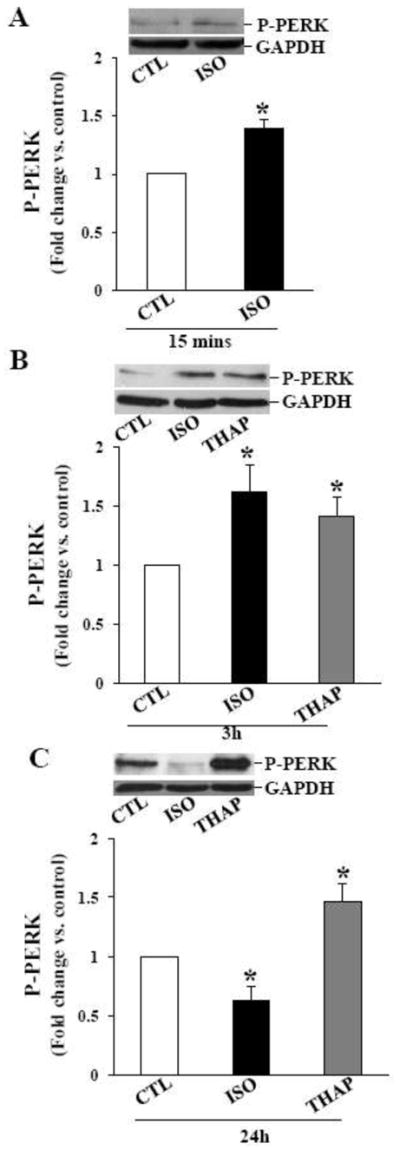
Phosphorylation of PERK. ARVMs were treated with ISO for 15 min, and ISO or THAP for 3 or 24 h. Cell lysates were analyzed by western blot using anti-phospho-specific PERK antibodies. A. β-AR stimulation for 15 min increased PERK phosphorylation. *p<0.05 vs. CTL; n=3. B. β-AR stimulation or THAP treatment for 3 h increased PERK phosphorylation; *p<0.05 vs. CTL; n=4. C. Phosphorylation of PERK decreased 24 h after β-AR stimulation, however PERK phosphorylation stayed higher 24 h after THAP treatment. *p<0.05 vs. CTL; n=5–6. The lower panels exhibit the mean data normalized to GAPDH.
3.3. Involvement of β-AR Subtypes and adenylyl cyclase in the phosphorylation of eIF-2α
Stimulation of β1-AR increases apoptosis, whereas stimulation of β2-AR inhibits apoptosis [20;28]. To study the involvement of β1- or β2-AR subtypes in the dephosphrylation of eIF-2α, ARVMs were pretreated with antagonist) or ICI 118,551 (ICI; CGP 20712A (CGP; 0.3 μM; β1-AR-selective antagonist) for 30 min followed by treatment with ISO for 3 h. Analysis of 0.1 μM; β2-AR-selective total cell lysates using phospho-specific antibodies showed that pretreatment with CGP, not ICI, prevents ISO-mediated decreases in the phosphorylation of eIF-2α (fold change vs CTL; ISO, 0.54±0.09* ICI+ISO, 0.78±0.10*; CGP+ISO, 1.56±0.29$,#; *p<0.05 vs CTL; $p<0.05 vs ISO; #p<0.05 vs ICI+ISO; n=6–8; Fig 6A). Similar to ISO, treatment with direct adenylyl cyclase activator forskolin (FSK; 10 μM) decreased phosphorylation of eIF-2α (Fig 6A).
Fig 6.

A. Involvement of adenylyl cyclase and β-AR subtypes in the phosphorylation of eIF-2α. ARVMs were treated with either forskolin or ISO for 3 h. ARVMs in CGP and ICI groups were pretreated for 30 min prior to ISO treatment for 3 h. Cell lysates were analyzed by western blot using anti-phospho-specific eIF-2α antibodies. *p<0.05 vs. CTL; $p<0.05 vs. ISO; n=6–8. The lower panels exhibit the mean data normalized to GAPDH. B. Salubrinal maintains eIF-2α phosphorylation. ARVMs were pretreated with SAL (10 μM) for 30 min followed by treatment with ISO for 3 h. Cell lysates were analyzed by western blot using anti-phospho-specific eIF-2α antibodies. *p<0.05 vs. CTL; $p<0.05 vs. ISO; n=3. The lower panels exhibit the mean data normalized to GAPDH. C. Salubrinal inhibits β-AR stimulated apoptosis. ARVMs were pretreated with SAL (1 μM) for 30 min followed by treatment with ISO for 24 h. The number of apoptotic cells was measured using TUNEL- staining assay. *p<0.05 vs. CTL; $p<0.05 vs. ISO; n=4. D. Inhibition of caspase-12 inhibits apoptosis. ARVMs were pretreated with z-ATAD for min followed by treatment with ISO or THAP for 24 h. The number of apoptotic cells was measured using TUNEL-staining assay. *p<0.05 vs. CTL; $p<0.05 vs. ISO or THAP; n=4–5.
3.4. Alleviation of ER stress maintains phosphorylation of eIF-2α and inhibits apoptosis
The drug salubrinal (SAL) is identified as a selective inhibitor of phosphatase (Gadd-34-PP1) that dephosphorylates eIF-2α [21], thereby maintaining eIF-2α phosphorylation and offering protection from the adverse effects of ER stress [29]. To investigate the role of SAL in maintenance of eIF-2α phosphorylation, ARVMs were pretreated with SAL (10 μM) for 30 min followed by treatment with ISO for 3 h. SAL pretreatment significantly inhibited β-AR-stimulated decreases in eIF-2α phosphorylation (Fig 6B). To study if SAL inhibits β-AR-stimulated apoptosis, ARVMs were pretreated with SAL for 30 min (1 μM) followed by treatment with ISO (10 μM) for 24 h. Analysis of apoptosis using TUNEL-staining assay indicated that pretreatment with SAL almost completely inhibits β-AR-stimulated apoptosis (CTL, 4.9±0.52; ISO, 8.5±0.94*; SAL+ISO, 4.80±0.36$; SAL, 4.11±0.33; *p<0.05 vs CTL; $p<0.05 vs ISO; Fig 6C; n=4).
3.5. Inhibition of caspase-12 inhibits apoptosis
ER stress-induced apoptosis involves activation of caspase-12 [12]. z-ATAD-FMK (z-ATAD) is considered as a specific inhibitor of caspase-12 [30]. To investigate the role of caspase-12, ARVMs were pretreated with z-ATAD for 30 min followed by treatment with ISO or THAP for 24 h. Analysis of apoptosis using TUNEL-staining assay indicated that z-ATAD almost completely inhibits β-AR-stimulated (CTL, 2.23±0.71; ISO, 6.90±1.47*; ISO+z-ATAD, 2.78±0.61$; z-ATAD, 2.48 ±0.46; *p<0.05 vs CTL; $p<0.05 vs ISO; n=4–5; Fig 6D) and THAP-induced (CTL, 2.23±0.71; THAP, 5.74±1.01*; THAP+z-ATAD, 2.92±0.65$; z-ATAD, 2.48 ±0.46; *p<0.05 vs CTL; $p<0.05 vs THAP; n=4–5; Fig 6D) apoptosis.
3.6. Chronic β-AR stimulation induces ER stress in the heart
To investigate, if β-AR stimulation induces ER stress in vivo, mice were infused with ISO for 7 days. Western blot analyses of LV lysates indicated that ISO-infusion significantly increased GRP-78 and Gadd-153 proteins levels (Fig 7A and B), while activating caspase-12 as analyzed by decreased procaspase-12 signal (Fig 7C). ISO-infusion decreased phosphorylation of eIF-2α (Fig 8A).
Fig 7. β-AR stimulation induces ER stress in vivo.

Mice were infused with vehicle (sham) or ISO for 7 days. LV lysates were analyzed by western blot using anti-GRP-78 (A), anti-Gadd-153 (B) or anti-caspase-12 (C) antibodies. In ISO+SAL samples, mice were infused with ISO in the presence of SAL for 7 days. The lower panel exhibits the mean data normalized to actin. *p<0.05 vs. sham; $p<0.05 vs. ISO; n=3–6.
Fig 8.

A. Salubrinal maintains eIF-2α phosphorylation in vivo. Mice were infused with ISO in the presence or absence of SAL for 7 days. LV lysates were analyzed by western blot using anti-phospho-specific eIF-2α antibodies. *p<0.05 vs. Sham; $p<0.05 vs. ISO; n=6–7. B. Salubrinal inhibits β-AR-stimulated apoptosis. Paraffin-embedded sections were used for TUNEL-staining assay. Upper panel demonstrates TUNEL-stained images obtained from ISO and ISO+SAL hearts. Yellow-green fluorescence represents apoptotic cells, while red fluorescence indicates α-sarcomeric actin staining (specific for myocytes). The lower panel demonstrates quantitative analysis of myocytes apoptosis with or without ISO-treatment; *p<0.05 vs sham; $p<0.05 vs. ISO; n=3–5.
3.7. Salubrinal inhibits activation of caspase-12, dephosphorylation of eIF-2α and myocyte apoptosis in the heart
To determine if SAL affects β-AR-stimulated activation of caspase-12, phosphorylation of eIF-2α, and myocyte apoptosis in the heart, mice were infused with ISO in the presence or absence of SAL. Western blot analysis of LV lysates demonstrated that SAL inhibits ISO-mediated increases in the activation of caspase-12 as analyzed by the increased intensity of procaspase-12 when compared to ISO group (Fig 7C). SAL treatment enhanced eIF-2α phosphorylation in the presence of ISO (Fig 8A).
ISO-infusion (7 days) increased the number of TUNEL-positive myocytes in the heart (Fig 8B). However, the percentage of apoptotic myocytes was significantly lower in the presence of SAL (percent apoptosis; sham, 0.05±0.02; ISO, 0.28±0.02*; ISO+SAL, 0.09±0.02$; SAL, 0.07±0.02; *p<0.05 vs CTL; $p<0.05 vs ISO; n=3–5).
4. Discussion
Stimulation of β-AR induces apoptosis in cardiac myocytes in vitro and in vivo [6;8;31–33]. However, molecular signals involved in β-AR-stimulated apoptosis are not yet completely understood. ER stress is suggested to play a critical role in the pathogenesis of heart failure [34]. The major findings of the present study are - 1) ER stress induces apoptosis in adult cardiac myocytes; 2) β-AR stimulation induces ER stress in adult cardiac myocytes in vitro and in vivo; 3) ER stress occurs via the involvement of β1-AR subtype as evidenced by phosphorylation of eIF-2α; 4) β-AR-stimulated induction of ER stress involves increases in cellular concentration of cAMP as evidenced by decreased phosphorylation of eIF-2α using forskolin; 5) inhibition of caspase-12 inhibits β-AR-stimulated apoptosis; 6) inhibition of ER stress inhibits β-AR-stimulated apoptosis in vitro and in vivo.
Stress conditions that interfere with the homeostasis of the ER initiate diverse signaling responses, resulting in a decreased rate of protein translation to prevent further accumulation of unfolded proteins. There is simultaneous activation of transcription factors to induce the expression of ER-resident chaperones to deal with accumulated protein aggregates. To halt the build-up of proteins, ER-specific protein-degrading apparatus also becomes activated [11–13]. These processes allow time to re-establish cellular homeostasis. However, if the stress cannot be resolved, the cell may die due to apoptosis. ER stress is shown to be involved in the apoptosis of neonatal rat cardiac myocytes [35]. In neonatal rat cardiac myocytes, antibodies against β1-AR induced ER stress and apoptosis [18]. Neonatal and adult cardiac myocytes are suggested to be physiologically different with respect to growth/hypertrophy and adrenergic physiology [36–38]. Here, we provide evidence that pharmacological inducers of ER stress (thapsigargin and brefeldin A) induce apoptosis in adult cardiac myocytes. Tunicamycin, which induces accumulation of misfolded proteins and ER stress, also increased the number of apoptotic ARVMs (not shown). Furthermore, thapsigargin-induced apoptosis was inhibited by the inhibition of caspase-12. Thus, induction of ER stress may represent a common signaling pathway leading to apoptosis in neonatal and adult cardiac myocytes.
Stress conditions initiate an adaptive response called unfolded protein response (UPR or ER stress response). The UPR is characterized by the coordinated activation of multiple proteins, including PERK and eIF-2α. The activation of the UPR may lead either to cell survival (by triggering the synthesis of ER chaperone proteins such as GRP-78 or to cell demise via the activation of apoptosis [11]. The molecular components involved in the UPR leading to cell survival or cell death signaling are classified as sensors, modulators and effectors. In this study, we analyzed expression of GRP-78, and phosphorylation of PERK and eIF-2α (as examples of ER stress sensors), expression of Gadd-153 and Gadd-34 (as examples of ER stress modulators) and activation of caspase-12 (as an example of ER stress effectors) to investigate if β-AR stimulation induces ER stress in ARVMs. The data presented here provide evidence for the induction of ER stress in vitro in adult cardiac myocytes and in vivo in the mouse heart following β-AR stimulation. It is interesting to note that both ISO and THAP induce ER stress in ARVMs. However, time points at which different ER stress molecules are activated or expressed appear different. Both ISO and THAP increased Gadd-153 and Gadd-34 protein levels 3 h after treatment. However, Gadd-153 expression remained higher 24 h after THAP treatment. β-AR stimulation, not THAP, for 3 h increased expression of GRP-78. Both ISO and THAP increased PERK phosphorylation 3 h after treatment. PERK phosphorylation was decreased 24 h after ISO treatment, while PERK phosphorylation remained higher 24 h after THAP treatment. eIF-2α phosphorylation remained lower than control 3 and 24 h after ISO treatment, while eIF-2α phosphorylation remained unchanged 3 h after THAP treatment and was higher than control 24 h after THAP treatment. Of note, activation of caspase-12 remained higher 3 and 24 h after ISO and THAP treatment. Inhibition of caspase-12 inhibited apoptosis induced by both these agents. These data highlight the previously observed phenomenon in tumor cells where kinetic and magnitude of the ER stress response varied between drug treatment and cell lines [39].
Phosphorylation of ser51 residue in eIF-2α is considered as an important signaling event of ER stress. This phosphorylation of eIF-2α prevents initiation of protein translation and transiently halts global protein synthesis, while upregulating the translation of selected stress-induced mRNAs [21;40]. The apoptotic stimuli are shown to induce transient phosphorylation of eIF-2α. In neuro-ectodermal tumor cells, ER stressors including THAP induced transient phosphorylation of eIF-2α [39]. Likewise, transient eIF-2α phosphorylation was observed in response to glutamate receptor agonist kainic acid [41]. The data presented here demonstrate that β-AR stimulation also induces transient phosphorylation of eIF-2α. It was interesting to note that phosphorylation of eIF-2α was greater 15 min after β-AR stimulation. However, it declined below control levels 3 h after β-AR stimulation. The initial increase in eIF-2α phosphorylation suggests cellular attempt to relieve ER stress, while decreased phosphorylation of eIF-2α 3 h β-AR stimulation suggests ER stress, leading to cell demise.
Activation of PERK phosphorylates eIF-2α [11], while Gadd-34 (a non-enzymatic cofactor of PP1) dephosphorylates eIF-2α [25]. β-AR stimulation for 15 min activated PERK (Fig 5) and increased phosphorylation of eIF-2α (Fig 4) with no effect on the expression of Gadd-34 (data not shown). On the other hand, PERK phosphorylation remained higher 3 h after β-AR stimulation, while eIF-2α phosphorylation declined with concomitant increase in Gadd-34 expression. These data suggest a role for PERK in the increased phosphorylation of eIF-2α 15 min after β-AR stimulation and a role for Gadd-34/PP1 complex in the dephosphorylation of eIF-2α 3 h after β-AR stimulation. SAL is identified as a selective inhibitor of Gadd-34/PP1 complex [21], thereby maintaining eIF-2α phosphorylation and offering protection from the adverse effects of ER stress [29]. SAL pretreatment improved phosphorylation of eIF-2α, and inhibited β-AR-stimulated increase in cardiac myocyte apoptosis in vitro and in vivo. Thus, maintenance of eIF-2α phosphorylation may represent an important signaling event offering protection against ER stress.
Stimulation of β1-AR-Gs pathway increases apoptosis in ARVMs, while stimulation of β2-AR-Gi pathway plays an anti-apoptotic role in β-AR-induced apoptosis [20;28]. CGP, not ICI, inhibited β-AR-stimulated decreases in eIF-2α phosphorylation, suggesting involvement of β1-AR subtype in the dephosphorylation of eIF-2α. Activation of β-AR in cardiac myocytes increases the cellular concentration of cAMP [42]. Previously, direct stimulation of adenylyl cyclase using forskolin is shown to induce apoptosis in ARVMs [7]. Similar to β-AR stimulation, forskolin treatment for 3h decreased phosphorylation of eIF-2α, suggesting involvement of cAMP in this process.
β-AR-stimulation increases intracellular calcium concentration [7]. This increase in intracellular may alter ER Ca++ homeostasis and activation of m-calpain, leading to activation of caspase-12 [11]. Activated caspase-12 translocates from ER to cytosol and contributes to the activation of caspase-9 and caspase-3 leading to apoptosis [12]. Caspase-12 is shown to be an important regulator of ER stress-induced apoptosis, since caspase-12 deficient cells and mice exhibit reduced sensitivity to apoptosis induced by ER stress agents such as brefeldin A and tunicamycin [43]. Using specific inhibitor of caspase-12, we provide evidence for the involvement of caspase-12 in β-AR-stimulated apoptosis. The involvement of caspase-12 is supported by the observation that THAP-induced apoptosis is inhibited by the inhibition of caspase-12 (Fig 6D).
5. Conclusion
The data presented here are of clinical significance since activation of the sympathetic nerves to the heart is a common feature of myocardial failure and stimulation of β-AR induces cardiac myocyte apoptosis in the heart. Inhibition of myocyte apoptosis using SAL suggests that a critical balance of phosphorylated eIF-2α may be necessary for myocyte survival during sympathetic overstimulation. A clear understanding of the signaling pathways involved in the regulation of expression and activity of components of ER stress and their role in cardiac myocyte apoptosis may uncover novel therapies for the treatment of heart failure.
Acknowledgments
Funding
This work is supported by National Institutes of Health (Grant numbers HL-091405 and HL-092459) and a Merit Review Grant (award number IO1BX000640) from the Biomedical Laboratory Research & Development Service of the VA office of Research and Development.
Technical help received from Barbara A. Connelly is appreciated.
Footnotes
Conflicts of interest
None.
References
- 1.Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73:615–621. doi: 10.1161/01.cir.73.4.615. [DOI] [PubMed] [Google Scholar]
- 2.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 3.Kajstura J, Bolli R, Sonnenblick EH, Anversa P, Leri A. Cause of death: suicide. J Mol Cell Cardiol. 2006;40:425–437. doi: 10.1016/j.yjmcc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 4.Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res. 2003;92:139–150. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- 5.Krishnamurthy P, Subramanian V, Singh M, Singh K. Beta1 integrins modulate Beta-adrenergic receptor-stimulated cardiac myocyte apoptosis and myocardial remodeling. Hypertension. 2007;49:865–872. doi: 10.1161/01.HYP.0000258703.36986.13. [DOI] [PubMed] [Google Scholar]
- 6.Shizukuda Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN, Sonnenblick EH. Beta-adrenergic stimulation causes cardiocyte apoptosis: influence of tachycardia and hypertrophy. Am J Physiol. 1998;275:H961–H968. doi: 10.1152/ajpheart.1998.275.3.H961. [DOI] [PubMed] [Google Scholar]
- 7.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation. 1998;98:1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 8.Iwai-Kanai E, Hasegawa K, Araki M, Kakita T, Morimoto T, Sasayama S. Alpha- and beta-adrenergic pathways differentially regulate cell type-specific apoptosis in rat cardiac myocytes. Circulation. 1999;100:305–311. doi: 10.1161/01.cir.100.3.305. [DOI] [PubMed] [Google Scholar]
- 9.Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, Singh K, Colucci WS. Beta-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circ Res. 2003;92:136–138. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 10.Menon B, Johnson JN, Ross RS, Singh M, Singh K. Glycogen synthase kinase-3beta plays a pro-apoptotic role in beta-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes: Role of beta1 integrins. J Mol Cell Cardiol. 2007;42:653–661. doi: 10.1016/j.yjmcc.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 12.Szegezdi E, FitzGerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 13.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 15.Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E, Hayakawa Y, Zimmermann R, Bauer E, Klovekorn WP, Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Wang G, Wang Y, Liu Q, Xu W, Tan Y, Cai L. Diabetes- and angiotensin II-induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med. 2009;13:1499–1512. doi: 10.1111/j.1582-4934.2009.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, Suzuki M, Nishino T, Nakaya H, Koseki H, Aoe T. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–8017. doi: 10.1128/MCB.24.18.8007-8017.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang CS, Mao W, Liu J. Pro-apoptotic effects of anti-beta1-adrenergic receptor antibodies in cultured rat cardiomyocytes: actions on endoplasmic reticulum and the prosurvival PI3K-Akt pathway. Autoimmunity. 2008;41:434–441. doi: 10.1080/08916930802031710. [DOI] [PubMed] [Google Scholar]
- 19.Menon B, Singh M, Ross RS, Johnson JN, Singh K. Beta-Adrenergic receptor-stimulated apoptosis in adult cardiac myocytes involves MMP-2-mediated disruption of beta1 integrin signaling and mitochondrial pathway. Am J Physiol Cell Physiol. 2006;290:C254–C261. doi: 10.1152/ajpcell.00235.2005. [DOI] [PubMed] [Google Scholar]
- 20.Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin-sensitive G protein. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 21.Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 22.Luthra S, Dong J, Gramajo AL, Chwa M, Kim DW, Neekhra A, Kuppermann BD, Kenney MC. 7-Ketocholesterol activates caspases-3/7, -8, and -12 in human microvascular endothelial cells in vitro. Microvasc Res. 2008;75:343–350. doi: 10.1016/j.mvr.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu Y, Fenik P, Zhan G, Sanfillipo-Cohn B, Naidoo N, Veasey SC. Eif-2a protects brainstem motoneurons in a murine model of sleep apnea. J Neurosci. 2008;28:2168–2178. doi: 10.1523/JNEUROSCI.5232-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiseman RL, Balch WE. A new pharmacology--drugging stressed folding pathways. Trends Mol Med. 2005;11:347–350. doi: 10.1016/j.molmed.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 26.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 27.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shizukuda Y, Buttrick PM. Subtype specific roles of beta-adrenergic receptors in apoptosis of adult rat ventricular myocytes. J Mol Cell Cardiol. 2002;34:823–831. doi: 10.1006/jmcc.2002.2020. [DOI] [PubMed] [Google Scholar]
- 29.Kessel D. Protection of Bcl-2 by salubrinal. Biochem Biophys Res Commun. 2006;346:1320–1323. doi: 10.1016/j.bbrc.2006.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu HY, Minamino T, Tsukamoto O, Sawada T, Asai M, Kato H, Asano Y, Fujita M, Takashima S, Hori M, Kitakaze M. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by proteasome inhibition. Cardiovasc Res. 2008;79:600–610. doi: 10.1093/cvr/cvn128. [DOI] [PubMed] [Google Scholar]
- 31.Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol. 2001;189:257–265. doi: 10.1002/jcp.10024. [DOI] [PubMed] [Google Scholar]
- 32.Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ, Siddiqui MA. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000;102:344–350. doi: 10.1161/01.cir.102.3.344. [DOI] [PubMed] [Google Scholar]
- 33.Colucci WS, Sawyer DB, Singh K, Communal C. Adrenergic overload and apoptosis in heart failure: implications for therapy. J Card Fail. 2000;6:1–7. [PubMed] [Google Scholar]
- 34.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 35.Nickson P, Toth A, Erhardt P. PUMA is critical for neonatal cardiomyocyte apoptosis induced by endoplasmic reticulum stress. Cardiovasc Res. 2007;73:48–56. doi: 10.1016/j.cardiores.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kajstura J, Mansukhani M, Cheng W, Reiss K, Krajewski S, Reed JC, Quaini F, Sonnenblick EH, Anversa P. Programmed cell death and expression of the protooncogene bcl-2 in myocytes during postnatal maturation of the heart. Exp Cell Res. 1995;219:110–121. doi: 10.1006/excr.1995.1211. [DOI] [PubMed] [Google Scholar]
- 37.Simpson P. Stimulation of hypertrophy of cultured neonatal rat heart cells through an alpha 1-adrenergic receptor and induction of beating through an alpha 1- and beta 1-adrenergic receptor interaction. Evidence for independent regulation of growth and beating. Circ Res. 1985;56:884–894. doi: 10.1161/01.res.56.6.884. [DOI] [PubMed] [Google Scholar]
- 38.Clark WA, Rudnick SJ, LaPres JJ, Andersen LC, LaPointe MC. Regulation of hypertrophy and atrophy in cultured adult heart cells. Circ Res. 1993;73:1163–1176. doi: 10.1161/01.res.73.6.1163. [DOI] [PubMed] [Google Scholar]
- 39.Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. J Biol Chem. 2010;285:6091–6100. doi: 10.1074/jbc.M109.014092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suragani RN, Ghosh S, Ehtesham NZ, Ramaiah KV. Expression and purification of the subunits of human translational initiation factor 2 (eIF2): phosphorylation of eIF2 alpha and beta. Protein Expr Purif. 2006;47:225–233. doi: 10.1016/j.pep.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sperelakis N, Xiong Z, Haddad G, Masuda H. Regulation of slow calcium channels of myocardial cells and vascular smooth muscle cells by cyclic nucleotides and phosphorylation. Mol Cell Biochem. 1994;140:103–117. doi: 10.1007/BF00926749. [DOI] [PubMed] [Google Scholar]
- 43.Hoppe V, Hoppe J. Mutations dislocate caspase-12 from the endoplasmatic reticulum to the cytosol. FEBS Lett. 2004;576:277–283. doi: 10.1016/j.febslet.2004.09.012. [DOI] [PubMed] [Google Scholar]