

Abstract
An infectious etiology has been proposed for many human cancers, but rarely have specific agents been identified. One difficulty has been the need to propagate cancer cells in vitro to produce the infectious agent in detectable quantity. We hypothesized that genome amplification from small numbers of cells could be adapted to circumvent this difficulty. A patient with concomitant chronic lymphocytic leukemia (CLL) and polycythemia vera (PV) requiring therapeutic phlebotomy donated a large amount of phlebotomized blood to test this possibility. Using genome amplification methods, we identified a new isolate (BIS8-17) of torque teno virus (TTV) 10. The presence of blood isolate sequence 8-17 (BIS8-17) in the original plasma was confirmed by polymerase chain reaction (PCR), validating the approach, since TTV is a known plasma virus. Subsequent PCR testing of plasmas from additional patients showed that BIS8-17 had a similar incidence (~20%) in CLL (n = 48) or PV (n = 10) compared with healthy controls (n = 52). CLL cells do not harbor BIS8-17; PCR did not detect it in CLL peripheral blood genomic deoxyribonucleic acid (DNA) (n = 20). CLL patient clinical outcome or prognostic markers (immunoglobulin heavy chain variable region [IGHV ] mutation, CD38 or zeta-chain associated protein kinase 70kDa [ZAP-70]) did not correlate with BIS8-17 infection. Although not causative to our knowledge, this is the first reported isolation and detection of TTV in either CLL or PV. TTV could serve as a covirus with another infectious agent or TTV variant with rearranged genetic components that contribute to disease pathogenesis. These results prove that this method identifies infectious agents and provides an experimental methodology to test correlation with disease.
INTRODUCTION
The direct role of infectious agents in the development of cancer was initially demonstrated in 1911 by the discovery of Rous sarcoma virus, which causes sarcomas in chickens (1). Such agents have also been identified in a few human cancers, such as human papilloma virus (HPV) in cervical carcinoma, human T-cell lymphotropic virus-1 (HTLV-1) in adult T-cell leukemia and human herpesvirus 8 (HHV-8) in Kaposi’s sarcoma (2). Additionally, infectious agents may have an indirect role in cancer development by promoting an inflammatory environment that favors tumor growth, as exemplified by the association of Helicobacter pylori infection with stomach cancer and mucosa-associated lymphoma (2). Despite these examples, the identification of infectious agents in human cancer has been difficult, in part because cell culture techniques are needed to produce large quantities of infectious agents in vitro. Use of modern genomic amplification techniques circumvents this difficulty and provides the ability to isolate and identify unique deoxyribonucleic acid (DNA) sequences of putative infectious agents obtained from small tissue samples containing few infectious particles (3).
To test such an approach, we used plasma from a patient (CLL831) with concomitant B-cell chronic lymphocytic leukemia (CLL) and polycythemia vera (PV), who donated blood made available from periodic phlebotomy for PV. Because CLL and PV result in the accumulation of cells in the blood, either leukemic B lymphocytes or erythrocytes, the plasma could be a rich and convenient source of potential infectious agents in these two diseases. The pathogenic initiation of either CLL or PV is currently not known.
CLL is a monoclonal expansion of a CD5+CD19+ B lymphocytes expressing a unique antibody or B-cell antigen receptor (BCR), for which sequence can be characterized by the degree of immunoglobulin heavy chain variable region (IGHV) somatic mutation (4). CLL patients with an unmutated (≤2% difference from the germline IGHV DNA sequence) BCR tend to have a worse clinical outcome (5,6), suggesting that antigen binding to the BCR is important for the initiation and/or progression of CLL. Furthermore, at least 27% of CLL patients share similar BCR amino acid sequences (7), implicating some common antigenic stimulant in CLL. This antigen could be an infectious agent (8), since stimulation through the BCR by an infectious agent (Helicobacter pylori, Borrelia burgdorferi, Chlamydophila psittaci and Campylobacter jejuni) has been implicated in the growth and development of marginal zone lymphoma of the stomach, skin, ocular adnexa and small intestine, respectively (9). Interestingly, decreased risk of CLL among subjects with chronic rheumatic heart disease suggested that long-term antibiotic prophylaxis may decrease occurrence of bacterial infections that may induce CLL (10), and increased risk of CLL was found in subjects with prior pneumonia (11). Thus, CLL could follow a similar pattern of infection stimulating growth and proliferation.
Alternatively, a pathogen could directly infect a B lymphocyte, leading to transformation into CLL. In humans, HTLV-1 causes T-cell lymphoma, and Epstein-Barr virus (EBV) may be involved in the development of Burkitt lymphoma (2). For CLL, many viral associations have been suggested, including HTLV-1 (12), EBV (13), HPV (14), Merkel cell polyomavirus (MCPyV) (15), hepatitis C virus and cytomegalovirus (16), but no definitive causation has been shown. Although there are no known human B-cell leukemia viruses, both bovine leukemia virus and feline leukemia virus cause a B-cell leukemia in appropriate animals with features similar to those of human CLL (17,18). Interestingly, one study detected transcripts of human endogenous retrovirus type K (HERV-K) in CLL cells (19); however, we were not able to detect evidence of HERV-K particles in CLL plasma (3).
Human PV is associated with an acquired somatic Janus kinase 2 (JAK2) point mutation that leads to erythropoietin receptor growth signals, resulting in erythrocytosis (20). Because of this breakthrough in the understanding of the molecular basis of human PV, studies designed to detect infectious agents in human PV appear to have been neglected. However, studies in mice indicate that Friend leukemia virus can cause PV (21); perhaps, a virus also drives PV in humans and leads to subsequent acquisition of JAK2 mutations.
Furthermore, a common infectious agent could be involved in the development of concomitant CLL and PV; such an agent might trigger the transformation of a pluripotent stem cell that could subsequently differentiate down both erythroid and lymphoid pathways (22,23). However, in one report, a patient’s PV cells had the characteristic PV-associated gain-of-function JAK2 point mutation, whereas the CLL cells did not, suggesting that CLL and PV do not necessarily share a common malignant precursor (23). We report a similar finding for patient CLL831.
Our initial results after using genome amplification techniques on CLL831 plasma yielded 49 particle nucleic acid sequences that were all nonhuman, documenting that our preparation method avoided significant contamination with human DNA (3). The predominant species, found in over 20% of the obtained sequences, represented a single virus. We have further characterized this virus and report herein that this species is a type of torque teno virus (TTV) or anellovirus, TTV10, which heretofore has not been identified in CLL and/or PV. Interestingly, because TTV may be involved in other hematologic neoplasias (24), we examined the relationship of TTV infection with CLL and/or PV. In summary, the identification of a new TTV10 species from patient plasma validates the use of genome amplification techniques to discover new infectious agents in disease.
MATERIALS AND METHODS
Human Samples
After informed consent was gained, as approved by the North Shore–Long Island Jewish Health System (NSLIJHS) Institutional Review Board and according to the Helsinki Declaration, peripheral blood samples and corresponding clinical data were collected in the NSLIJHS (healthy control, n = 9; CLL, n = 48; PV, n = 10) or by the New York Cancer Project (healthy control, n = 43) (25). Plasmas were prepared by centrifugation (800g) of blood collected in tubes containing 1.8 mg ethylenediaminetetraacetic acid (EDTA)/mL blood to prevent coagulation and stored at −80ºC. Plasma from patient CLL831 was collected at two time points: 26 June 2006 and 26 July 2006. Peripheral blood mononuclear cells (PBMCs) were purified from heparinized venous blood by Ficoll-Paque PLUS (GE Healthcare, Piscataway, NJ, USA) density gradient centrifugation and cryopreserved for future use. Characteristics of healthy control plasma, CLL plasma, CLL PBMCs (n = 20) and PV plasma donors are shown in Tables 1–4. CLL IGHV sequences were determined as previously described (26) and were assigned GenBank accession numbers (Table 1). CD38 and zeta-chain associated protein kinase 70kDa (ZAP-70) percentages of CD5+CD19+ CLL cells were determined by flow cytometry as previously described (27,28). JAK2 V617F mutation was tested in whole blood DNA by real-time PCR (29) or in purified CLL cell DNA by the PCR and restriction fragment–length polymorphism method.
Table 1.
Clinical and molecular characteristics of CLL plasma donors.
| CLL IDa | Sex | Birth year | % Mutationb | IGHV c GenBank | % CD38d | % ZAP-70d | TFTe | Overall survivalf | BIS8-17g | TTVh | Kgagi |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 277 | F | 1967 | 4.1 | HM152485 | 2.8 | 40.0 | >160 | >167 | + | + | + |
| 360 | M | 1942 | 0.0 | AY553647 | 31.4 | 68.1 | >138 | >141 | − | − | + |
| 403 | M | 1952 | 0.0 | HM152486 | 52.0 | ND | 31 | >129 | − | − | + |
| 462 | M | 1934 | 10.4 | HM152487 | ND | ND | >98 | >99 | − | + | + |
| 569 | M | 1952 | 0.7 | HM152488 | 86.5 | 45.8 | 65 | 67 | − | − | + |
| 581 | M | 1939 | 0.7 | HM152489 | 89.9 | 13.0 | 20 | 127 | − | + | + |
| 584 | M | 1953 | 6.6 | HM152490 | 1.4 | 1.0 | >82 | >87 | + | ND | + |
| 606 | M | 1956 | 6.3 | HM152491 | 6.0 | 1.7 | >103 | >103 | − | ND | + |
| 616 | M | 1956 | 5.9 | HM152492 | 1.8 | 1.0 | >83 | >83 | + | − | + |
| 625 | M | 1952 | 0.0 | HM152493 | 41.7 | 32.3 | 22 | >82 | − | ND | + |
| 647 | M | 1954 | 0.0 | HM152494 | 41.0 | 83.5 | 42 | >111 | − | − | + |
| 657 | M | 1942 | 0.3 | FJ039790 | 96.4 | 65.9 | 10 | 53 | − | − | + |
| 689 | F | 1937 | 0.7 | HM152495 | 93.6 | 50.1 | 85 | 169 | − | + | + |
| 701 | M | 1948 | 0.0 | HM152496 | 73.5 | ND | >102 | >105 | − | ND | + |
| 711 | M | 1953 | 6.9 | HM152497 | 9.8 | 13.5 | >153 | >153 | − | − | + |
| 720 | F | 1940 | 4.9 | HM152498 | 18.4 | 6.4 | 81 | >203 | − | + | + |
| 745 | M | 1953 | 0.3 | HM152499 | 35.1 | 22.0 | 70 | >74 | − | + | + |
| 746 | M | 1931 | 0.0 | HM152500 | 86.6 | 20.2 | 0 | 68 | + | + | + |
| 748 | M | 1933 | 8.3 | HM152501 | 6.7 | ND | >78 | >78 | − | + | + |
| 806 | M | 1950 | 0.0 | HM152502 | 3.0 | 35.9 | 30 | >95 | − | − | + |
| 831 | F | 1925 | 4.6 | HM152503 | 3.0 | ND | >28 | 60 | + | + | + |
| 934 | F | 1949 | 0.0 | HM152504 | 7.7 | 61.1 | 19 | >65 | + | + | + |
| 936 | F | 1941 | 5.9 | HM152505 | 52.8 | 8.8 | 39 | >59 | + | + | + |
| 937 | M | 1953 | 7.4 | HM152506 | 98.5 | 5.0 | >49 | >60 | − | − | + |
| 942 | M | 1951 | 4.6 | HM152507 | 10.3 | 23.3 | >85 | >142 | − | + | + |
| 943 | M | 1932 | 0.0 | HM152508 | ND | ND | >1 | >58 | + | − | + |
| 945 | F | 1959 | 7.0 | HM152509 | 59.5 | 3.9 | >59 | >62 | − | + | + |
| 946 | F | 1938 | 4.2 | HM152510 | 7.3 | 17.1 | 53 | >171 | − | − | + |
| 947 | M | 1953 | 6.0 | HM152511 | ND | ND | 36 | >79 | − | − | + |
| 948 | F | 1947 | 0.0 | HM152512 | ND | ND | >129 | 137 | − | − | + |
| 949 | M | 1940 | 3.4 | HM152513 | 4.6 | 1.4 | 0 | >462 | − | + | + |
| 950 | M | 1937 | 0.0 | HM152514 | ND | ND | 29 | >58 | − | + | + |
| 951 | F | 1959 | ND | — | ND | ND | >17 | >58 | − | + | + |
| 952 | M | 1932 | 6.3 | HM152515 | 0.8 | 1.1 | >55 | >58 | − | + | + |
| 953 | M | 1948 | 4.9 | HM152516 | ND | ND | >30 | >68 | − | − | + |
| 954 | M | 1934 | 0.0 | HM152517 | 37.8 | 68.1 | 27 | >61 | − | − | + |
| 956 | M | 1946 | 1.0 | HM152518 | 74.6 | 91.9 | 4 | 95 | − | + | + |
| 957 | F | 1946 | 0.0 | HM152519 | 86.0 | 3.4 | >57 | >62 | − | + | + |
| 958 | M | 1940 | ND | — | ND | ND | >41 | >60 | − | − | + |
| 962 | F | 1960 | 1.0 | HM152520 | 3.0 | 9.7 | >9 | >60 | + | − | + |
| 964 | M | 1962 | 0.0 | HM152521 | 99.7 | 58.7 | — | — | − | + | + |
| 966 | M | 1929 | 0.0 | HM152522 | 64.6 | 29.3 | >55 | >92 | − | + | + |
| 968 | M | 1938 | 2.4 | HM152523 | 12.9 | 28.1 | 103 | 128 | − | + | + |
| 969 | M | 1935 | 6.6 | HM152524 | 3.5 | 8.2 | >16 | >57 | + | + | + |
| 970 | M | 1952 | 10.8 | HM152525 | 7.2 | 32.9 | >53 | >56 | − | + | + |
| 971 | M | 1942 | 0.3 | HM152526 | 9.5 | 70.9 | 28 | >71 | − | + | + |
| 973 | M | 1962 | 0.0 | HM152527 | 58.2 | 65.6 | 29 | 53 | − | + | + |
| 977 | F | 1941 | 10.1 | HM152528 | ND | ND | >21 | >76 | + | + | + |
| Total + | 11 | 27 | 48 |
ND, not determined.
Arbitrarily assigned CLL identification number. All samples collected at NSLIJHS.
Percent mutation of IGHV as compared with germline according to ImMunoGeneTics (IMGT) (53).
GenBank accession numbers for IGHV nucleotide sequence. —, Not determined.
Percentage positive by flow cytometry.
TFT in months after diagnosis of CLL. >, Patient is still untreated as of last patient contact; —, not known.
Overall survival in months after diagnosis of CLL. >, Patient is still alive as of 23 May 2011; —, not known.
Visible PCR product for BIS8-17–specific primers CCC560/CCC561 or CCC562/CCC563, as in Figure 1C, indicated by +. Not detectable PCR product is indicated by −.
+, Visible PCR product for TTV primers T801/T935; −, PCR product is not detectable.
+, Visible PCR product for Kgag-specific primers, as in Figure 1C.
Table 4.
Characteristics of plasmas from PV patients.
| PV IDa | Sex | Birth year | BIS8-17b | TTVc | Kgagd |
|---|---|---|---|---|---|
| 001 | F | 1925 | + | + | + |
| 002 | M | 1953 | + | + | + |
| 005 | M | 1931 | − | − | + |
| 006 | F | 1950 | − | − | + |
| 007 | M | 1957 | − | − | + |
| 008 | M | 1942 | − | + | + |
| 009 | F | 1963 | − | − | + |
| 010 | F | 1936 | − | − | + |
| 012 | M | 1935 | − | − | + |
| 013 | M | 1942 | − | − | + |
| Total + | 2 | 3 | 10 |
Arbitrarily assigned PV identification number. PV001 = CLL831. All samples collected at NSLIJHS.
Visible PCR product for BIS8-17–specific primers CCC560/CCC561 or CCC562/CCC563, as in Figure 1C, indicated by +. Not detectable PCR product is indicated by −.
+, Visible PCR product for TTV primers T801/T935; −, PCR product is not detectable.
+, Visible PCR product for Kgag-specific primers, as in Figure 1C.
TTV10 Cloning and Sequencing
EcoRI or HindIII DNA fragments from whole genome amplified DNA prepared from particles isolated from the plasma of patient CLL831 were cloned into plasmids, and the ends of these DNA fragments were sequenced (3). Of the 49 plasmid clones, 11 contained sequences similar to TTV10. Five clones contained a 284-bp HindIII fragment that was completely sequenced, and these blood isolate sequences (BIS8-8, BIS8-9, BIS8-19, BIS8-20, BIS8-31) were deposited into GenBank (accession numbers GU797355–GU797359). Two clones contained a 3.5-kb HindIII fragment (BIS8-5, BIS8-36). One clone contained both the ~284-bp and 3.5-kb HindIII fragments, resulting in the expected size (3.8 kb) for the TTV10 genome (BIS8-17). Two clones contained HindIII fragments, adding up to sizes larger than expected for the TTV10 genome (BIS8-23, two 3.8-kb fragments; BIS8-28, 284-bp and 3.8-kb fragment). Initial DNA sequencing revealed that the fragments of these last two clones did not have the expected TTV10 genome orientation, whereas the BIS8-17 fragments did. Finally, one clone contained a 3.8-kb EcoRI fragment (BIS7-36). Because BIS8-17 had the expected TTV10 genome size and contained the frequently obtained small HindIII fragment in the correct orientation, we sequenced the entire DNA insert of this clone by a primer walking strategy (Figure 1A). Oligonucleotide primers (with DNA sequence in parentheses) used were as follows: CCC560 (5′ CCC AAG TAG CCT GAA CCA CAG C), CCC561 (5′ AAC CGT TTG TGG CCA TCC AG), CCC562 (5′ CAC CTG GTA GTC GCG TGT CC), CCC563 (5′ ACG ACG GGC TAG ACG AGC TG), CCC568 (5′ ACG GCC TGC CAG CTA CAA AG), CCC569 (5′ TTG GGA TTG CCC CCT ATT CC), CCC572 (5 ′ CAG GTT GTT AGA GAC CCC TGC AC), CCC573 (5′ GTG GGG ACC GAG GAG TTT CG), CCC574 (5′ GGC CGT GGG AGT TTC ACT TG), CCC575 (5′ GCG GCT CCG AAG AAG TTG AG), CCC576 (5′ CCC CCG TGC TAC GTC ACT AA), CCC577 (5′ CCC CTT GAC TGC GGT GTG TA), CCC578 (5′ GGT TGC CAT TGG ACA CTG GAG) and CCC579 (5′ GGA AGG AAG TCG GCC ATT TTG). The Molecular Biology Core at the Feinstein Institute for Medical Research (FIMR) synthesized these primers and then sequenced BIS8-17 using the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit with Ampli-Taq DNA Polymerase, FS and an ABI 373 DNA Sequencer (Applied Biosystems, Foster City, CA, USA). Overlapping sequences were assembled using AssemblyLIGN (MacVector, Cary, NC, USA) and SeqMan (DNASTAR, Madison, WI, USA). The GC-rich region of TTV10 in BIS8-17 contained a stretch of ~37 nucleotides that could not be determined. The small HindIII fragment of clone BIS8-17 contained an extra base pair that introduced a frame shift destroying open reading frame (ORF)1 and was not found in the 284-bp HindIII fragment from other clones. Therefore, this extra base pair was deleted from the derived BIS8-17 sequence. The resulting complete 3,767-bp sequence for BIS8-17 was deposited in GenBank (accession number GU797360).
Figure 1.

TTV confirmed in CLL831 plasma. (A) Schematic of BIS8-17 genomic DNA. The 3,767-bp BIS8-17 genomic DNA is shown as a circle with the origin (1/3,767) starting after the GC-rich region (box and thick line). The gray box indicates the area of GC-rich region that could not be sequenced. Location and direction of primers used for sequencing are shown as arrowheads, with labels indicating the CCC primer number positioned outside of the circle. Location and direction of PCR primers T801 and T935 are shown as arrowheads and labeled within the circle. ORFs 1–4 are shown as thick lines with arrows indicating direction of translation. The putative splice donor (SD) site just before the translation stop of ORF2 that potentially joins with the putative splice acceptor (SA) sites of ORF3 and ORF4 is shown. HindIII restriction enzyme sites are indicated. (B) CLL831 plasma contains putative TTV particles. An electron micrograph (original magnification 100,000×) of plasma particle preparation from CLL831 patient is shown, where the black bar indicates 100 nm. The average diameter of putative TTV particles (n = 13) was 36.6 ± 5.6 nm. (C) BIS8-17 nucleic acid is detectable in CLL831 plasma. Nucleic acid was prepared from two CLL831 plasma samples, one originally used to isolate BIS8-17 (6/26) and one obtained 1 month later (7/26) and from two healthy controls (AG52 and AG128). Nucleic acids were PCR amplified to specifically detect BIS8-17 using primer pairs CCC560/CCC561 or CCC562/CCC563 (251 and 303 bp fragments, respectively). In addition, PCR amplification with no added nucleic acid template (Blank) was performed to ensure that PCR products depended on template addition. All samples were also PCR amplified with primer pairs CCC549/CCC550 (Kgag), which produces a 214-bp product from human genomic DNA to ensure that the template was added.
Electron Microscopy
Plasma particles from patient CLL831 were isolated from 22-mL plasma as described (3). In brief, differential low-speed centrifugation, DNase (Promega, Madison, WI, USA) treatment and 0.45-μm filtration (Nalgene, Rochester, NY, USA) were used to collect plasma and remove cellular debris. Plasma particles were collected by ultracentrifugation (100,000g), washed one time in phosphate-buffered saline (PBS, Mediatech, Manassas, VA, USA) and resuspended in 500 μL PBS. A total of 5-μL particle preparation was applied to a formvar-coated, carbon-tabilized, copper grid (Electron Microscopy Sciences, Hatfield, PA, USA) for 1 min at room temperature, washed once with distilled water (dH2O), stained with fresh 1% uranyl acetate (Electron Microscopy Sciences) in dH2O for 2 min and then examined by transmission electron microscopy (JEM-100 CXII, JEOL, Peabody, MA, USA) at 80 kV.
Nucleic Acid Preparation
Nucleic acid was prepared using the High Pure Viral Nucleic Acid Kit (Roche, Mannheim, Germany) from 200 μL plasma, which was not purified for particles as above to retain cell debris containing genomic DNA as a positive control for subsequent PCR analysis. From the resulting 50-μL nucleic acid preparation, 1 μL was used for subsequent PCR analysis, allowing detection of 103 viral particles/mL plasma.
Genomic DNA was prepared from 3–5 × 106 cryopreserved PBMCs, using the Gentra Puregene Cell Kit (Qiagen, Valencia, CA, USA) according to the manufacturer, except for omitting RNAse A treatment. Genomic DNA was quantitated by absorption at 260 nm and resuspended at 200 ng/μL for PCR testing.
PCR
DNA (1 μL) was PCR amplified in a 25-μL reaction (2 mmol/L MgCl2, 50 mmol/L KCl, 10 mmol/L Tris [pH 8.3], 0.2 mmol/L dGTP, 0.2 mmol/L dATP, 0.2 mmol/L dTTP, 0.2 mmol/L dCTP, 1.0 U AmpliTaq Gold [Applied Biosystems] and 0.4 mmol/L of each primer pair) in a GeneAmp PCR System 9600 (Applied Biosystems) under the following conditions: 95ºC for 9 min, 40 × (95ºC for 5 s, 65ºC annealing for 10 s, 72ºC for 30 s), 72ºC for 30 s and soak at 10ºC. A portion of the gag gene from HERV-K (Kgag) was amplified using primer pairs CCC549 (5′ AGC AGG TCA GGT GCC TGT AAC ATT)/CCC550 (5′ TGG TGC CGT AGG ATT AAG TCT CCT) to produce a 214-bp fragment (3). Parts of BIS8-17 were specifically amplified using primer pairs CCC560/CCC561 and CCC562/CCC563 to produce 251-bp and 303-bp fragments, respectively. To detect nearly all TTV species, PCR amplification with primer pair T801/T935 was performed as described (30).
Gel Electrophoresis
PCR fragments and 1 Kb Plus DNA Ladder size standard (Invitrogen, Carlsbad, CA, USA) were separated by size on a 2% agarose gel (Thermo Scientific Owl Separation Systems, Rochester, NY, USA) using a PowerPac200 power supply (Bio-Rad Laboratories, Hercules, CA, USA), stained with ethidium bromide (Invitrogen), visualized and imaged with ultraviolet transillumination using a Gel Doc 2000 System and Quantity One software (Bio-Rad Laboratories).
Statistical Analyses
The significance of differences in virus incidence between disease and healthy populations was determined in a 2 × 2 contingency table with InStat (GraphPad Software, La Jolla, CA, USA) to calculate the two-sided P value using Fisher exact test and the odds ratio (OR) with 95% confidence interval (CI) using the approximation of Woolf. The statistical significance of differences in Kaplan-Meier survival curves was evaluated with Prism (GraphPad Software) using the log-rank test to calculate a two-tailed P value. Correlations between virus infection and CLL clinical parameters (IGHV mutation [>2%], CD38 positivity [>30%] and ZAP-70 positivity [>20%]) were tested in a 2 × 2 contingency table with InStat.
RESULTS
Patient CLL831 Case Report
A 79-year-old female (CLL831), with a history of stage I right breast cancer treated with lumpectomy and radiation therapy at age 58, presented with erythrocytosis and erythromelalgia. Physical examination revealed neither hepato-splenomegaly nor lymphadenopathy. Laboratory evaluation disclosed a white blood cell count of 17.5 × 106/μL with a normal differential, hemoglobin 16.6 g/dL, hematocrit 49% and platelets 430 × 106/μL; further studies showed an elevated red cell mass in a normal plasma volume with no apparent cause for a secondary erythrocytosis. Therefore, an initial primary diagnosis of PV was made. Therapy with 325 mg aspirin daily and periodic phlebotomy to maintain a hemoglobin level <14 g/dL was instituted.
Nine months after the diagnosis of PV, left axillary adenopathy was noted. Biopsy revealed B-cell small lymphocytic lymphoma. White blood cell count was 18.7 × 106/μL with a normal differential. Serum LDH was 560 U/mL and β-2 microglobulin was 2.2 mg/mL. Flow cytometry on the lymph node and peripheral blood lymphocytes revealed a monoclonal population expressing CD5, CD19, CD20, CD23 and dim surface Ig kappa light chain and lacking CD10 and CD38, features consistent with CLL (31). Fluorescence in situ hybridization (FISH) evaluation on peripheral blood lymphocytes for common mutations associated with CLL revealed deletion of 13q14.3. Subsequent analysis of CLL cells revealed a mutated IGHV3-53 (4.56%). One year after the PV diagnosis, peripheral blood cells were found to express both wild-type and mutated JAK2, whereas purified CLL cells exhibited only wild-type JAK2. At this time, platelets had risen to ~700 × 106/μL, which decreased to ~300 × 106/μL after administration of hydroxyurea.
Two years after initial diagnosis, intermittent episodes of waxing and waning choreiform movements began, for which no etiology was defined. Four years after diagnosis, despite well-controlled hemoglobin and platelet counts, left calf deep venous thrombosis and multiple pulmonary emboli occurred. A lupus-type anticoagulant was detected. Anticoagulation was changed from aspirin to warfarin. The patient subsequently developed pulmonary hypertension requiring supplemental oxygen and multiple ulcerations on her lips, tongue and perineum, despite continued warfarin administration. Because an underlying vasculitis was suspected, hydroxyurea was discontinued and steroids were begun. However, gangrene of her distal lower extremities developed and clopidogrel and rituximab were administered. Despite aggressive therapy, she progressively deteriorated. The patient expired 6 years after the initial PV diagnosis.
Identification of TTV10 in Plasma from Patient CLL831
To identify potential infectious agents in this patient, 49 DNA fragments from isolated plasma particles were sequenced and found to be nonhuman, with 11 representing a single virus family (3). Comparisons to the public sequence database found the best similarity with TTV in the anellovirus family. One DNA fragment, BIS8-17, which had an insert size (3.8 kb) expected for the entire TTV genome (32,33) was completely sequenced. When circularized, the derived BIS8-17 sequence had the expected TTV genome structure (Figure 1A).
The genome structure of BIS8-17 has a typical GC-rich region with stem-and-loop structures that are empirically difficult to sequence and are functionally important for viral replication (32,33). BIS8-17 has the customary four ORFs: a large ORF1, a short ORF2 and the spliced open reading frames, ORF3 and ORF4, which are spliced into ORF2. TTV sequences are highly variable and have been segregated into five major genetic groups with genotypes >50% different (32,33). The current taxonomic classification defines 29 human TTV species (34,35). BIS8-17 is most similar to the TTV10 species (with only 15% nucleotide sequence dissimilarity to TTV10 ORF1; GenBank accession number NC_014076).
To confirm the presence of TTV, we examined the plasma from CLL831 by electron microscopy and found particles close to the expected size for TTV (36.6 ± 5.6 nm, which is in the range previously observed for TTV particles [30–32 nm]) [36]; Figure 1B). To specifically confirm the presence of BIS8-17, CLL831 plasma was tested by PCR amplification with primers specific for BIS8-17 DNA (Figure 1C). Nucleic acid prepared from the original CLL831 plasma sample (6/26) used to isolate and identify BIS8-17 was confirmed positive using two different specific primer pairs (Figure 1C). Furthermore, BIS8-17 was detected in a subsequent CLL831 plasma sample obtained 1 month later (7/26). In comparison, the BIS8-17 sequence could not be amplified from nucleic acids prepared from the plasmas of two healthy control subjects, although the control Kgag sequence that is present in human genomic DNA was detected in all plasma samples.
TTV Exists in Plasmas from Other CLL Patients
Because BIS8-17 was isolated from a patient with CLL, we examined if this virus is present in other patients with CLL. Plasmas from 47 additional patients were tested by PCR amplification with specific primer pairs (Figure 1C), and the plasmas from 10 of these patients contained BIS8-17 (Table 1). All 47 samples were also positive for Kgag control. Overall, 23% (11/48) of CLL patients screened were positive for BIS8-17.
To determine if the incidence of BIS8-17 in CLL patients differed from that in a healthy population without CLL, plasmas from 50 additional healthy subjects were tested for BIS8-17, and 13 of these were found to be positive (Table 2). Again, Kgag was detected in all samples. Overall, 25% (13/52) of plasmas from healthy control subjects contained BIS8-17. Thus, there was no significant difference in BIS8-17 incidence between CLL and healthy populations (P = 0.917, OR 0.892, 95% CI 0.355–2.239).
Table 2.
Characteristics of plasmas from healthy controls.
| IDa | Sex | Birth year | Source | BIS8-17b | TTVc | Kgagd |
|---|---|---|---|---|---|---|
| 825 | M | 1931 | NYCP | + | + | + |
| 827 | M | 1932 | NYCP | + | + | + |
| 902 | M | 1931 | NYCP | - | + | + |
| 972 | F | 1968 | NYCP | − | − | + |
| 1006 | F | 1943 | NYCP | + | − | + |
| 1084 | M | 1937 | NYCP | + | ND | + |
| 4320 | M | 1944 | NYCP | − | + | + |
| 5843 | F | 1931 | NYCP | − | − | + |
| 5998 | F | 1960 | NYCP | − | + | + |
| 6257 | F | 1931 | NYCP | − | + | + |
| 6290 | M | 1931 | NYCP | − | + | + |
| 7195 | M | 1932 | NYCP | + | + | + |
| 9199 | F | 1966 | NYCP | − | + | + |
| 9391 | M | 1966 | NYCP | − | + | + |
| 9457 | M | 1955 | NYCP | − | + | + |
| 9507 | M | 1932 | NYCP | − | + | + |
| 9684 | F | 1932 | NYCP | + | + | + |
| 9740 | F | 1931 | NYCP | + | + | + |
| 10404 | F | 1932 | NYCP | + | + | + |
| 10551 | F | 1967 | NYCP | − | + | + |
| 10714 | F | 1970 | NYCP | − | ND | + |
| 10884 | M | 1932 | NYCP | + | + | + |
| 11604 | M | 1932 | NYCP | − | + | + |
| 11779 | M | 1932 | NYCP | − | + | + |
| 11858 | M | 1932 | NYCP | − | + | + |
| 11880 | F | 1931 | NYCP | − | + | + |
| 12042 | M | 1932 | NYCP | − | + | + |
| 12328 | F | 1947 | NYCP | − | − | + |
| 12478 | F | 1931 | NYCP | − | − | + |
| 12847 | F | 1955 | NYCP | − | + | + |
| 13204 | M | 1932 | NYCP | − | + | + |
| 13531 | F | 1951 | NYCP | − | ND | + |
| 13630 | M | 1932 | NYCP | + | + | + |
| 14973 | M | 1931 | NYCP | − | + | + |
| 14978 | F | 1931 | NYCP | − | + | + |
| 17317 | M | 1932 | NYCP | − | + | + |
| 17356 | M | 1932 | NYCP | − | + | + |
| 18470 | M | 1932 | NYCP | − | + | + |
| 19020 | M | 1931 | NYCP | − | + | + |
| 19227 | F | 1931 | NYCP | − | + | + |
| 20931 | F | 1931 | NYCP | − | + | + |
| 24733 | F | 1931 | NYCP | − | + | + |
| 24884 | F | 1931 | NYCP | − | − | + |
| AG51 | M | 1934 | NSLIJHS | − | + | + |
| AG52 | M | 1945 | NSLIJHS | − | + | + |
| AG53 | M | 1932 | NSLIJHS | − | + | + |
| AG54 | M | 1952 | NSLIJHS | + | − | + |
| AG82 | F | 1963 | NSLIJHS | + | ND | + |
| AG128 | F | 1951 | NSLIJHS | − | ND | + |
| AG189 | F | 1965 | NSLIJHS | − | − | + |
| AG190 | F | 1965 | NSLIJHS | − | − | + |
| AG193 | M | 1960 | NSLIJHS | + | − | + |
| Total + | 13 | 37 | 52 |
ND, not determined. NYCP, New York Cancer Project.
Arbitrarily assigned identification number.
Visible PCR product for BIS8-17–specific primers CCC560/CCC561 or CCC562/CCC563, as in Figure 1C, indicated by +. Not detectable PCR product is indicated by −.
+, Visible PCR product for TTV primers T801/T935; −, PCR product is not detectable.
+, Visible PCR product for Kgag-specific primers, as in Figure 1C.
To test if TTV species in general have a different incidence between CLL and healthy populations, most plasma samples were also tested with PCR primers (T801/T935) that amplify a region conserved in the majority of TTV isolates (30). As expected (37), a high incidence of TTV was observed in healthy individuals (37/47, 79%). CLL patients had a slightly lower overall incidence of TTV (27/44, 61%), but this difference was not statistically significant (P = 0.107, OR 0.429, 95% CI 0.170–1.083).
Although the mechanism of TTV replication is incompletely understood, TTV DNA has been detected in a wide variety of human tissues and cells, including bone marrow, lymph node, spleen and PBMCs (38). To test if CLL cells harbor BIS8-17, we prepared genomic DNA from 20 CLL patients’ PBMCs, which were predominantly composed of CLL cells. PCR amplification of genomic DNA with both sets of primer pairs specific for BIS8-17 was negative for all samples (Table 3), including two CLL patients (CLL584 and CLL746) who had BIS8-17 present in their plasma. Control amplification with Kgag PCR primers was positive for all samples (Table 3). Furthermore, PCR amplification with primer pair T801/T935 was negative for all 20 genomic DNA samples (Table 3). Thus, TTV neither exists nor replicates in a large fraction of CLL cells, suggesting that plasma virus particles are produced at another tissue site and that direct TTV infection of a CLL cell or its leukemic progenitor does not occur.
Table 3.
Characteristics of CLL genomic DNA samples.
| CLL IDa | Sex | Birth year | BIS8-17b | TTVc | Kgagd |
|---|---|---|---|---|---|
| 38 | F | 1923 | − | − | + |
| 66 | F | 1911 | − | − | + |
| 68 | M | 1923 | − | − | + |
| 353 | M | 1931 | − | − | + |
| 385 | M | 1947 | − | − | + |
| 584 | M | 1953 | − | − | + |
| 585 | M | 1953 | − | − | + |
| 657 | M | 1942 | − | − | + |
| 661 | M | 1928 | − | − | + |
| 680 | F | 1945 | − | − | + |
| 682 | F | 1944 | − | − | + |
| 699 | M | 1949 | − | − | + |
| 705 | F | 1949 | − | − | + |
| 706 | M | 1932 | − | − | + |
| 710 | M | 1938 | − | − | + |
| 721 | M | 1934 | − | − | + |
| 732 | M | 1946 | − | − | + |
| 742 | M | 1935 | − | − | + |
| 743 | M | 1950 | − | − | + |
| 746 | M | 1931 | − | − | + |
| Total + | 0 | 0 | 20 |
Arbitrarily assigned CLL identification number. All samples collected at NSLIJHS.
Visible PCR product for BIS8-17–specific primers CCC560/CCC561 or CCC562/CCC563, as in Figure 1C, indicated by +. Not detectable PCR product is indicated by −.
+, Visible PCR product for TTV primers T801/T935; −, PCR product is not detectable.
+, Visible PCR product for Kgag-specific primers, as in Figure 1C.
TTV Is Detectable in Plasmas of Other PV Patients
Because BIS8-17 was isolated from a patient who had PV, perhaps TTV could be detected in other PV patients. To test this, plasmas from nine additional PV patients were screened for BIS8-17. One additional PV plasma was positive for BIS8-17 (Table 4), whereas all samples were positive for Kgag. Thus, 20% (2/10) of PV patients were positive for BIS8-17. Furthermore, the incidence of TTV species in PV was tested with PCR primer pair T801/T935 (Table 4). TTV was detected in 30% (3/10) of PV plasmas. Thus, BIS8-17 and TTV can be detected in PV patients. However, because the incidence of BIS8-17 and TTV in PV is low, a larger PV population is required to test if the incidence is significantly different from healthy controls.
TTV Infection Does Not Correlate with CLL Clinical Parameters
Perhaps TTV incidence correlates with CLL disease severity, analogous to the situation observed for chicken anemia virus and Marek’s leukemia (39). To test this possibility, we compared time to first treatment (TFT) and overall survival of CLL patients (Table 1) with BIS8-17 or general TTV infection. No significant differences in Kaplan-Meier TFT curves between BIS8-17 infected and uninfected patients (Figure 2A) or between TTV infected and uninfected CLL patients (Figure 2B) were observed. Similarly, Kaplan-Meier overall survival curves between BIS8-17 or TTV infected and uninfected CLL patients were very similar (not shown). Thus, neither BIS8-17 nor TTV incidence correlated with CLL disease severity, as measured by TFT or overall survival.
Figure 2.
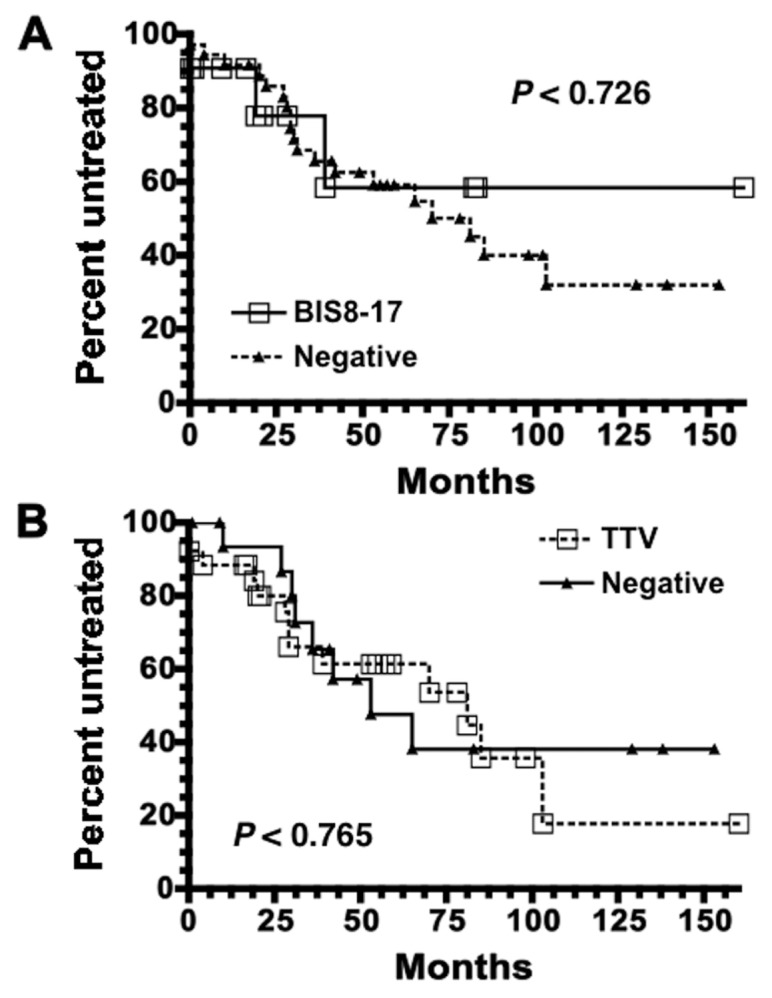
Presence of TTV has no effect on CLL patient TFT. TFT data were available for 47 of 48 CLL patients. (A) Kaplan-Meier curves comparing CLL patients whose plasma was positive for BIS8-17 by PCR (solid line with open squares) versus those that were negative (dotted line with black triangles). CLL patients positive for BIS8-17 (n = 11) had not yet reached the median TFT, whereas those patients negative for BIS8-17 had a median TFT time of 81 months (n = 36). (B) Same analysis as in (A), except CLL patients were divided into those who were positive for TTV by PCR using the T801/T935 primer pair (dotted line with open squares) versus those who were negative (solid line with black triangles). CLL patients positive for TTV (n = 26) had a median TFT of 81 months, whereas median TFT for those patients negative for TTV (n = 17) was 53 months. These curves are not statistically different (P values are shown).
We next examined if prognostic factors associated with poor CLL outcome (IGHV mutation status and CD38 or ZAP-70 levels) (5,28,40) correlated with specific BIS8-17 or general TTV infection. CLL patients with an unmutated IGHV (U-CLL) have shorter survival times than patients with a mutated IGHV (M-CLL). However, no correlation of IGHV mutation status between specific BIS8-17 infection (M-CLL = 7, U-CLL = 4) and uninfected (M-CLL = 15, U-CLL = 20) was observed. Similarly, no correlation between general TTV infection (M-CLL = 14, U-CLL = 12) and uninfected (M-CLL = 6, U-CLL = 10) existed. CLL patients with a high (≥30%) CD38 level (CD38+) generally have shorter survival times than patients with low (<30%) levels (CD38−) (5). As with IGHV mutation status, no correlation of CD38 level between specific BIS8-17 infection (CD38− = 7, CD38+ = 2) and uninfected (CD38− = 13, CD38+ = 17) or between general TTV infection (CD38− = 13, CD38+ = 10) and uninfected (CD38− = 5, CD38+ = 7) was observed. Finally, CLL patients with high (≥20%) ZAP-70 levels (ZAP-70+) have shorter survival times than patients with low (<20%) levels (ZAP-70−). Again, no correlation was seen between specific BIS8-17 infection (ZAP-70− = 5, ZAP-70+ = 3) and uninfected (ZAP-70− = 10, ZAP-70+ = 17) or between general TTV infection (ZAP-70− = 8, ZAP-70+ = 13) and uninfected (ZAP-70− = 5, ZAP-70+ = 6) and ZAP-70 levels. Thus, although the CLL patient populations studied are small, no significant correlations were observed between TTV infection and CLL prognostic markers.
DISCUSSION
Applying modern genome amplification techniques (3), we report the successful identification and characterization of a new TTV10 species, BIS8-17, from the plasma of a patient with concomitant CLL and PV (Figure 1). TTV10 is a type of anellovirus, a family of viruses with small circular single-stranded DNA genomes that are found in plasma (32–35). The discovery of BIS8-17 in this patient validates the use of ge-nome amplification techniques to identify new infectious agents from plasma. As further evidence that genome amplification techniques can identify infectious agents from human blood and tissue samples, we have repeated our method on plasma samples from three CLL patients (CLL220, CLL258 and CLL831) and a spleen sample from one CLL patient (CLL257) and identified additional TTV variants in all cases (data not shown). Thus, we have adapted modern genome amplification techniques to the identification of human infectious agents from patient samples and provide the first report of detecting TTV in CLL and PV. Furthermore, this method could be used on other types of tissues, providing broad utility for uncovering infectious agents in clinical disease.
The abundance of TTV in blood suggests a potential role in hematological malignancies. However, TTV infection did not associate with pediatric acute lymphoblastic leukemia (41) or B-cell non-Hodgkin lymphoma (42), although a significant prevalence of TTV has been observed in non-Hodgkin lymphomas (43), including follicular lymphoma and diffuse large B-cell lymphoma, as well as in Hodgkin lymphoma (44). Another report hypothesized that maternal TTV infection leads to increased risk for children developing leukemia or lymphoma (24). In support of this model, rearranged TTV genomes were detected in sera taken from pregnant mothers whose children later developed childhood leukemia (45). However, this was not significantly different from sera of mothers with unaffected children. Similarly, in CLL, no significant correlation with BIS8-17 or overall TTV infection (Table 1) was found, as compared with healthy control plasmas (Table 2). Additionally, TTV DNA was not detectable in PBMCs from CLL patients (Table 3). Furthermore, using quantitative real-time PCR with Set A primers analogous to T801/T935 (46), TTV was not detectable in PBMCs from CLL720, CLL831 and CLL934 (data not shown). Thus, not only does the presence of plasma TTV not correlate with CLL, but TTV infection and replication also do not appear to occur in the majority of PBMCs in CLL.
The isolation of TTV from plasma also suggests a potential role in benign hematological disorders, such as hepatitis-associated aplastic anemia (33). However, this association is weak because of the high incidence of TTV in unaffected individuals (Table 2) (37). We examined the association of TTV incidence with PV and found no increase in BIS8-17 or overall TTV infection in PV (Table 4), as compared with healthy control plasmas (Table 3). Larger PV populations will be required for statistical evaluation.
The lack of BIS8-17 and overall TTV correlation with CLL or PV does not necessarily indicate that TTV plays no role in CLL or PV. Larger patient and control populations may provide sufficient statistical power to discern finer differences in viral prevalence. Furthermore, because of the extreme variability of TTV sequences (32,33), screening with multiple TTV primer sets may provide a more precise measure of overall TTV infection (30,37). In line with this, we found evidence for TTV sequence variability in our CLL and healthy control PCR results (Tables 1 and 2), with several cases showing BIS8-17 positivity, yet displaying TTV negativity as assessed by T801/T935 primers. Thus, PCR amplification with other general TTV primers may provide more comprehensive detection (37). Additionally, with regard to a specific TTV species, perhaps one of the other 28 TTV species (34,35), other than TTV10 or BIS8-17, may associate with CLL or PV and correlate with disease. Moreover, a TTV containing a specific genomic rearrangement could associate with disease; such a possibility has been entertained for childhood leukemias (45).
Another possibility for TTV involvement in CLL or PV is as a covirus. For example, chicken anemia virus is a similar TTV-like circovirus that frequently infects commercial chicken flocks; although the infection is asymptomatic, it causes anemia in chickens coinfected with a second virus such as the herpes virus responsible for Marek’s leukemia (39). Additionally, Friend leukemia virus, which causes PV in mice, is actually a combination of two viruses: Friend murine leukemia virus and a spleen focus forming virus (21). Similarly, the lack of correlation between TTV incidence and CLL or PV (Tables 1, 2 and 4), CLL patient overall survival or TFT (Figure 2) or CLL prognostic markers (IGHV mutation, CD38%, ZAP-70%) may be due to the association of a second, as yet unidentified, virus. If so, further application of these genome amplification techniques may lead to its identification.
The occurrence of concomitant CLL and PV suggests a common malignant precursor. Patient CLL831 blood cells had the characteristic PV-associated JAK2 point mutation, but the CLL cells did not, suggesting that CLL and PV in this patient did not share a common transformed precursor. This result agrees with the lack of detection of this JAK2 mutation in CLL (47), as well as a case report demonstrating the absence of JAK2 mutation in CLL cells from a patient with concomitant CLL and PV (23). In contrast, the JAK2 mutation was detected in B lymphocytes of a subpopulation of PV patients (48–50), suggesting that JAK2 mutations might be found in CLL with concomitant PV in some cases, as has been reported (51,52). Thus, the rare patients with concomitant CLL and PV require further study to ascertain the potential existence of a common transformed precursor.
In conclusion, the identification of BIS8-17, a new TTV10 species, validates the use of genome amplification techniques for discovery of new infectious agents. This approach obviates the need to produce large quantities of infectious agent in vitro. Further study of BIS8-17 or other new infectious agents identified by this method may reveal correlations with CLL or PV disease. Interestingly, previously suggested CLL-associated viruses, such as HTLV-1, EBV, HPV, Merkel cell polyomavirus, hepatitis C virus and cytomegalovirus, were not identified in plasma by this method (data not shown). Application of this method to cells or other tissues may identify additional CLL- or PV-associated viruses. Finally, this method has clear utility for identifying infectious agents in a wide range of additional diseases.
ACKNOWLEDGMENTS
We thank Craig Gawel and Dorothy Guzowski (Molecular Biology Core, FIMR) for help with oligonucleotides and sequences; Erin Boyle (FIMR) for sample collection; Angela Tse Chuang Chu for support and encouragement; and Bettie Steinberg, Rosa Catera, Patricia Mongini and Sophia Yancopoulos for scientific discussions. This work was supported in part by a R01 grant from the National Institutes of Health (NIH) (CA81554), a M01 General Clinical Research Center grant from NIH (RR018535), the Ruth E. Raskin Fund of the Jewish Community Foundation, the Karches Foundation, the Prince Family Foundation, the Marks Foundation, the Jerome Levy Foundation, the Leon Levy Foundation and the Joseph Eletto Leukemia Research Fund.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Javier RT, Butel JS. The history of tumor virology. Cancer Res. 2008;68:7693–706. doi: 10.1158/0008-5472.CAN-08-3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziegler JL, Buonaguro FM. Infectious agents and human malignancies. Front Biosci. 2009;14:3455–64. doi: 10.2741/3464. [DOI] [PubMed] [Google Scholar]
- 3.Chu CC, et al. Plasma from a B-cell chronic lymphocytic patient with polycythemia vera contains viral and bacterial DNA. In: Kalil J, Cunha-Neto E, Rizzo LV, editors. 13th International Congress of Immunology. Medimond Srl, Monduzzi Editore; Bologna, Italy: 2007. pp. 589–95. [Google Scholar]
- 4.Fais F, et al. Chronic lymphocytic leukemia B cells express restricted sets of mutated and un-mutated antigen receptors. J Clin Invest. 1998;102:1515–25. doi: 10.1172/JCI3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Damle RN, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7. [PubMed] [Google Scholar]
- 6.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V (H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54. [PubMed] [Google Scholar]
- 7.Murray F, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111:1524–33. doi: 10.1182/blood-2007-07-099564. [DOI] [PubMed] [Google Scholar]
- 8.Hamblin T. Is chronic lymphocytic leukemia a response to infectious agents? Leuk Res. 2006;30:1063–4. doi: 10.1016/j.leukres.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 9.Zucca E, Bertoni F, Stathis A, Cavalli F. Marginal zone lymphomas. Hematol Oncol Clin North Am. 2008;22:883–901. viii. doi: 10.1016/j.hoc.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Landgren O, et al. Patterns of autoimmunity and subsequent chronic lymphocytic leukemia in Nordic countries. Blood. 2006;108:292–6. doi: 10.1182/blood-2005-11-4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landgren O, et al. Respiratory tract infections and subsequent risk of chronic lymphocytic leukemia. Blood. 2007;109:2198–201. doi: 10.1182/blood-2006-08-044008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann DL, et al. HTLV-I: associated B-cell CLL: indirect role for retrovirus in leukemogenesis. Science. 1987;236:1103–6. doi: 10.1126/science.2883731. [DOI] [PubMed] [Google Scholar]
- 13.Dolcetti R, Carbone A. Epstein-Barr virus infection and chronic lymphocytic leukemia: a possible progression factor? Infect Agent Cancer. 2011;5:22. doi: 10.1186/1750-9378-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flynn JM, Andritsos L, Lucas D, Byrd JC. Second malignancies in B-cell chronic lymphocytic leukaemia: possible association with human papilloma virus. Br J Haematol. 2010;149:388–90. doi: 10.1111/j.1365-2141.2010.08110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teman CJ, Tripp SR, Perkins SL, Duncavage EJ. Merkel cell polyomavirus (MCPyV) in chronic lymphocytic leukemia/small lymphocytic lymphoma. Leuk Res. 2011;35:689–92. doi: 10.1016/j.leukres.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 16.Chumak AA, et al. Persistent infections and their relationship with selected oncologic and non-tumor pathologies. J Immunotoxicol. 2010;7:279–88. doi: 10.3109/1547691X.2010.489528. [DOI] [PubMed] [Google Scholar]
- 17.Gillet N, et al. Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in human. Retrovirology. 2007;4:18. doi: 10.1186/1742-4690-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy J, et al. American Association of Feline Practitioners’ feline retrovirus management guidelines. J Feline Med Surg. 2008;2008;10:300–16. doi: 10.1016/j.jfms.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Depil S, Roche C, Dussart P, Prin L. Expression of a human endogenous retrovirus, HERV-K, in the blood cells of leukemia patients. Leukemia. 2002;16:254–9. doi: 10.1038/sj.leu.2402355. [DOI] [PubMed] [Google Scholar]
- 20.Tefferi A. JAK2 mutations in polycythemia vera: molecular mechanisms and clinical applications. N Engl J Med. 2007;356:444–5. doi: 10.1056/NEJMp068293. [DOI] [PubMed] [Google Scholar]
- 21.Moreau-Gachelin F. Multi-stage Friend murine erythroleukemia: molecular insights into oncogenic cooperation. Retrovirology. 2008;5:99. doi: 10.1186/1742-4690-5-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang G, Ahn YS, Whitcomb CC, Harrington WJ. Development of polycythemia vera and chronic lymphocytic leukemia during the course of refractory idiopathic thrombocytopenic purpura. Cancer. 1984;53:1770–6. doi: 10.1002/1097-0142(19840415)53:8<1770::aid-cncr2820530827>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 23.Hussein K, et al. B-CLL developing in a patient with PV is not affected by V617F mutation of the Janus kinase 2. Eur J Haematol. 2006;77:539–41. doi: 10.1111/j.0902-4441.2006.t01-1-EJH2940.x. [DOI] [PubMed] [Google Scholar]
- 24.zur Hausen H, de Villiers EM. Virus target cell conditioning model to explain some epidemiologic characteristics of childhood leukemias and lymphomas. Int J Cancer. 2005;115:1–5. doi: 10.1002/ijc.20905. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell MK, Gregersen PK, Johnson S, Parsons R, Vlahov D. The New York Cancer Project: rationale, organization, design, and baseline characteristics. J Urban Health. 2004;81:301–10. doi: 10.1093/jurban/jth116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghiotto F, et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113:1008–16. doi: 10.1172/JCI19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Damle RN, et al. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood. 2007;110:3352–9. doi: 10.1182/blood-2007-04-083832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rassenti LZ, et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood. 2008;112:1923–30. doi: 10.1182/blood-2007-05-092882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baxter EJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi K, Hoshino H, Ohta Y, Yoshida N, Mishiro S. Very high prevalence of TT virus (TTV) infection in general population of Japan revealed by a new set of PCR primers. Hepatol Res. 1998;12:233–9. [Google Scholar]
- 31.Swerdlow SH, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th edition. Lyon: IARC; 2008. p. 439. [Google Scholar]
- 32.Biagini P. Classification of TTV and related viruses (anelloviruses) Curr Top Microbiol Immunol. 2009;331:21–33. doi: 10.1007/978-3-540-70972-5_2. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto H. History of discoveries and pathogenicity of TT viruses. Curr Top Microbiol Immunol. 2009;331:1–20. doi: 10.1007/978-3-540-70972-5_1. [DOI] [PubMed] [Google Scholar]
- 34.Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 2010;2009;155:133–46. doi: 10.1007/s00705-009-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maggi F, Bendinelli M. Human anelloviruses and the central nervous system. Rev Med Virol. 2010;20:392–407. doi: 10.1002/rmv.668. [DOI] [PubMed] [Google Scholar]
- 36.Itoh Y, et al. Visualization of TT virus particles recovered from the sera and feces of infected humans. Biochem Biophys Res Commun. 2000;279:718–24. doi: 10.1006/bbrc.2000.4013. [DOI] [PubMed] [Google Scholar]
- 37.Handa A, Dickstein B, Young NS, Brown KE. Prevalence of the newly described human circovirus, TTV, in United States blood donors. Transfusion. 2000;40:245–51. doi: 10.1046/j.1537-2995.2000.40020245.x. [DOI] [PubMed] [Google Scholar]
- 38.Kakkola L, Hedman K, Qiu J, Pintel D, Soderlund-Venermo M. Replication of and protein synthesis by TT viruses. Curr Top Microbiol Immunol. 2009;331:53–64. doi: 10.1007/978-3-540-70972-5_4. [DOI] [PubMed] [Google Scholar]
- 39.Schat KA. Chicken anemia virus. Curr Top Microbiol Immunol. 2009;331:151–83. doi: 10.1007/978-3-540-70972-5_10. [DOI] [PubMed] [Google Scholar]
- 40.Wiestner A, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood. 2003;101:4944–51. doi: 10.1182/blood-2002-10-3306. [DOI] [PubMed] [Google Scholar]
- 41.Shiramizu B, Yu Q, Hu N, Yanagihara R, Nerurkar VR. Investigation of TT virus in the etiology of pediatric acute lymphoblastic leukemia. Pediatr Hematol Oncol. 2002;19:543–51. doi: 10.1080/08880010290097396. [DOI] [PubMed] [Google Scholar]
- 42.Cacoub P, et al. Transfusion-associated TT virus co-infection in patients with hepatitis C virus is associated with type II mixed cryoglobulinemia but not with B-cell non-Hodgkin lymphoma. Clin Microbiol Infect. 2003;9:39–44. doi: 10.1046/j.1469-0691.2003.00481.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhong S, et al. Gross elevation of TT virus genome load in the peripheral blood mononuclear cells of cancer patients. Ann N Y Acad Sci. 2001;945:84–92. doi: 10.1111/j.1749-6632.2001.tb03868.x. [DOI] [PubMed] [Google Scholar]
- 44.Garbuglia AR, et al. Detection of TT virus in lymph node biopsies of B-cell lymphoma and Hodgkin’s disease, and its association with EBV infection. Int J Immunopathol Pharmacol. 2003;16:109–18. doi: 10.1177/039463200301600204. [DOI] [PubMed] [Google Scholar]
- 45.Leppik L, et al. In vivo and in vitro intragenomic rearrangement of TT viruses. J Virol. 2007;81:9346–56. doi: 10.1128/JVI.00781-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato T, et al. Development of a TT virus DNA quantification system using real-time detection PCR. J Clin Microbiol. 2000;38:94–8. doi: 10.1128/jcm.38.1.94-98.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levine RL, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377–9. doi: 10.1182/blood-2005-05-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108:3128–34. doi: 10.1182/blood-2006-04-017392. [DOI] [PubMed] [Google Scholar]
- 49.Larsen TS, Christensen JH, Hasselbalch HC, Pallisgaard N. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br J Haematol. 2007;136:745–51. doi: 10.1111/j.1365-2141.2007.06497.x. [DOI] [PubMed] [Google Scholar]
- 50.Delhommeau F, et al. Evidence that the JAK2 G1849T (V617F) mutation occurs in a lymphomyeloid progenitor in polycythemia vera and idiopathic myelofibrosis. Blood. 2007;109:71–7. doi: 10.1182/blood-2006-03-007146. [DOI] [PubMed] [Google Scholar]
- 51.Eskazan AE, Salihoglu A, Diz-Kucukkaya R, Hancer VS, Soysal T. Chronic lymphocytic leukemia developing in a patient with Janus kinase 2 V617F mutation positive myeloproliferative neoplasm. Ann Hematol. 2011 2011 May 17; doi: 10.1007/s00277-011-1256-6. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 52.Kodali S, Chen C, Rathnasabapathy C, Wang JC. JAK2 mutation in a patient with CLL with coexistent myeloproliferative neoplasm (MPN) Leuk. Res. 2009;33:e236–9. doi: 10.1016/j.leukres.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 53.Lefranc MP, et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37:D1006–12. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]