

Abstract
Psoriasis (PS) and Crohn disease (CD) have been shown to be epidemiologically, pathologically, and therapeutically connected, but little is known about their shared genetic causes. We performed meta-analyses of five published genome-wide association studies on PS (2,529 cases and 4,955 controls) and CD (2,142 cases and 5,505 controls), followed up 20 loci that showed strongest evidence for shared disease association and, furthermore, tested cross-disease associations for previously reported PS and CD risk alleles in additional 6,115 PS cases, 4,073 CD cases, and 10,100 controls. We identified seven susceptibility loci outside the human leukocyte antigen region (9p24 near JAK2, 10q22 at ZMIZ1, 11q13 near PRDX5, 16p13 near SOCS1, 17q21 at STAT3, 19p13 near FUT2, and 22q11 at YDJC) shared between PS and CD with genome-wide significance (p < 5 × 10−8) and confirmed four already established PS and CD risk loci (IL23R, IL12B, REL, and TYK2). Three of the shared loci are also genome-wide significantly associated with PS alone (10q22 at ZMIZ1, prs1250544 = 3.53 × 10−8, 11q13 near PRDX5, prs694739 = 3.71 × 10−09, 22q11 at YDJC, prs181359 = 8.02 × 10−10). In addition, we identified one susceptibility locus for CD (16p13 near SOCS1, prs4780355 = 4.99 × 10−8). Refinement of association signals identified shared genome-wide significant associations for exonic SNPs at 10q22 (ZMIZ1) and in silico expression quantitative trait locus analyses revealed that the associations at ZMIZ1 and near SOCS1 have a potential functional effect on gene expression. Our results show the usefulness of joint analyses of clinically distinct immune-mediated diseases and enlarge the map of shared genetic risk loci.
Introduction
Psoriasis (PS [MIM 177900]) and Crohn disease (CD [MIM 266600]) are both chronic inflammatory epithelial disorders that are triggered by an activated cellular immune system and have an estimated sibling relative risk (λs) of 4–111,2 and 25–42,3 respectively, and a prevalence of 2%–3% and about 0.1%, respectively, in populations of European ancestry.4,5 PS is a common hyperproliferative disorder of the skin, characterized by red scaly plaques, typically occurring on the elbows, knees, scalp, and lower back.6 In contrast, CD is primarily a gut disorder affecting any aspect of the gastrointestinal tract but with extraintestinal manifestations which might also affect the skin (e.g., erythema nodosum and pyoderma gangraenosum).7 It results from the interaction of environmental factors, including the commensal microflora, with host immune mechanisms in a genetically susceptible host.8 Although PS and CD are clinically distinct diseases, they are observed together more frequently than expected by chance, which could indicate shared genetic factors acting in the etiology of both diseases.9–11 Recently, several genome-wide association studies (GWASs) have successfully been carried out separately for CD and PS12–20 and identified shared susceptibility genes, such as IL23R (MIM 607562), IL12B (MIM 161561), REL (MIM 164910), and TYK2 (MIM 176941), thereby providing further evidence for a genetic overlap of both diseases.13,16,21–23 One of the best characterized risk loci for both CD and PS is IL23R, located in a drug-targetable pathway.24 IL-23, a pro-inflammatory cytokine, is thought to be a key player driving autoimmunity in human disease.25 A recent functional characterization of the amino acid substitution R381Q in IL23R suggests that IL-23-induced Th17 cell effector function is reduced in protective allele carriers and leads to protection against several autoimmune diseases, including PS, CD, and ankylosing spondylitis.26
So far, shared susceptibility loci for CD and PS have been identified by single-disease GWAS for CD or PS separately, and established risk SNPs for one disease are usually tested for association in another disease,27–31 rather than in a combined systematic approach. Combined GWASs were only conducted across clinically related phenotypes, such as CD and ulcerative colitis (UC),32 CD and sarcoidosis (SA)27 or CD and celiac disease (CelD).33 Recently, Zhernakova et al.34 performed a meta-analysis with a similar systematic approach that combined genome-wide genotype data from two autoimmune diseases affecting different organs, namely CelD and rheumatoid arthritis (RA). They identified eight shared risk loci outside the human leukocyte antigen (HLA) region for CelD and RA, four of them previously not known to be associated with either CelD or RA. Based on genome-wide SNP data for CelD and RA, the authors identified an increased probability for CelD risk SNPs to confer also an increase in risk for RA and vice versa. Zhernakova et al.34 postulated criteria for declaring a SNP as being a shared risk factor for two clinically distinct diseases, namely that shared SNPs (1) have to reach genome-wide significance in the combined analysis of the initial GWAS screening stage and the replication stage of the two distinct diseases (pGWAS+Repl < 5 × 10−8) and (2) have to achieve, for each disease separately, nominal significance in the replication stage (pRepl < 0.05) as well as pGWAS+Repl < 10−3 in the combined analysis of screening and replication stage.
We used the criteria proposed by Zhernakova et al.34 in a genome-wide association analysis combining CD and PS to systematically identify shared risk loci associated with both diseases. A two-fold strategy was employed: in a first approach (OVERLAP), we tested established non-HLA CD risk SNPs for association with PS and vice versa, thereby seeking confirmation of whether known risk loci for one disease also play a role in the etiology of the other disease, disregarding the direction of effect. In a second approach (COMBINED), we performed a meta-analysis for the combined phenotype based on genome-wide data sets of both CD and PS in order to increase power for the detection of new shared risk alleles because of an increased sample size. The latter approach allows consideration of same-direction as well as opposing-direction allelic effects of putative shared markers between CD and PS through the use of suitable allele coding. We followed up 20 loci that showed the strongest association in the COMBINED approach and that had not been previously reported as being risk factors for either CD or PS. Follow-up was performed in independent replication panels from Germany, Estonia, Italy, United Kingdom, and the United States (see Table S1, available online).
Subjects and Methods
Study Subjects
We analyzed a collection of different data sets. Figure 1 details the different panels and their use in this study. For discovery, we combined genome-wide case-control data of single-nucleotide polymorphisms (SNP) for psoriasis (PS; panel A) and Crohn disease (CD; panel B) (see Table S1) respectively, conducted genome-wide meta-analyses on PS and CD, respectively, and employed two strategies (COMBINED and OVERLAP, see below) to systematically identify risk loci associated with both diseases. Replication was performed in independent replication panels from Germany (see panels C–E), Estonia (panels C and D), Italy (panel E), United Kingdom (panels C and E), and the United States (panel C) (see Table S1). Written, informed consent was obtained from all study participants and all protocols were approved by the institutional ethical review committees of the participating centers.
Figure 1.
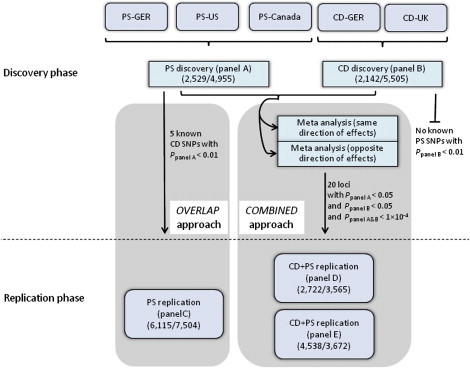
Study Design for the Combined Analysis of CD and PS
For discovery, we conducted a PS and a CD GWAS meta-analysis (panel A and B in Table S1), respectively, and employed two strategies (OVERLAP and COMBINED) to systematically search for shared risk loci. In a first approach (OVERLAP), we tested established non-HLA PS risk SNPs for potential association (p < 0.01) with CD and vice versa. In a second approach (COMBINED), we selected SNPs from 20 loci for being nominally associated in each of the single-disease meta-analyses (ppanel A < 0.05, ppanel B < 0.05) and for being significantly associated in the combined-phenotype association analysis at the 10−4 level (ppanel A&B < 10−4). For replication, follow-up SNP genotyping was performed for CD and PS in independent replication panels (panels C–E in Table S1). The following abbreviations are used: PS-GER, German PS GWAS; PS-US, United States PS GWAS; PS-Canada, Canadian PS GWAS; CD-GER, German CD GWAS; and CD-UK, United Kingdom CD GWAS. For each panel, numbers of cases/controls are displayed in parentheses.
Initial GWAS and German Replication Data
All German CD patients in discovery and replication panels (B and C–E, respectively) were recruited either at the Department of General Internal Medicine, Christian-Albrechts-University, Kiel, and the Charité University Hospital, Berlin; through local outpatient services; or nationwide with the support of the German Crohn and Colitis Foundation. German PS cases in discovery and replication panels (A and C and D, respectively) were recruited either at the Department of Dermatology, Christian-Albrechts-University, Kiel, or the Department of Dermatology and Allergy, Technical University, Munich, or through local outpatient services. Individuals were considered to be affected by PS if chronic plaque or guttate psoriasis lesions covered more than 1% of the total body surface area or if at least two skin, scalp, nail, or joint lesions were clinically diagnosed as psoriasis. The 4,680 German healthy control individuals in discovery and replication panels (A–E) were obtained from the Popgen biobank.35 The additional 3,391 German healthy controls (after quality control measures) in the discovery panels (A and B) were selected from the KORA S3+S4 survey, an independent population-based sample from the general population living in the region of Augsburg, southern Germany.36 Another 674 German healthy controls in the discovery panels (A and B) were selected from ISAAC Phase II study.37 German GWAS controls of the discovery phase were randomly assigned to panels A and B at equal proportions, while ensuring that controls in panel A did not overlap with German GWAS controls used in the independent genome-wide meta-analysis on CD.13 The Collaborative Association Study of Psoriasis (CASP) samples14 consisted of 1,303 PS cases and 1,322 controls after quality control measures and are part of panel A in our study. The data sets used for the analyses described in this manuscript were obtained from the database of Genotype and Phenotype (dbGaP). The genotyping of samples was provided through the Genetic Association Information Network (GAIN).38 The Canadian samples (from Genizon BioSciences and M. Belouchi, unpublished data) consisted of 757 PS cases and 987 controls sampled from the Québec founder population (QFP) after quality control measures. They are part of panel A. Membership in the QFP was defined as having four grandparents with French-Canadian family names who were born in the Province of Québec, Canada, or in adjacent areas in the provinces of New Brunswick and Ontario or in New England or New York state. This criterion assured that all subjects were descendants of French-Canadians living before the 1960s, after which time admixture with non-French-Canadians became more common. CD cases and controls from the United Kingdom were recruited from the 1958 birth cohort and UK National Blood Service for the Welcome Trust Case Controls Consortium (WTCCC) (described in details in WTCCC28). The WTCCC1 CD samples consisted of 1,662 CD cases and 2,860 healthy controls after quality control procedures and entered the analysis as part of panel B.
Additional Replication Data
A number of collaborative data sets were used as replication panels. The Estonian samples used in the OVERLAP and COMBINED approaches (part of panels C and D) were collected at the Department of Dermatology and Venerology and at the Department of Physiology and Centre of Translational Medicine at the University of Tartu.
Additional Estonian samples (part of panel C) used for replication in the OVERLAP approach consisted of samples provided by the population-based biobank of the Estonian Genome Center, University of Tartu. Subjects were recruited by general practitioners (GP) and physicians in the hospitals. Participants in the hospitals were randomly selected from individuals visiting GP offices or hospitals. Diagnosis of PS on the basis of clinical symptoms was posed by a general practitioner and confirmed by a dermatologist. At the moment of recruitment, the controls did not report diagnosis of osteoarthritis, psoriasis, or autoimmune diseases. The United States samples (part of panel C) used for replication in the OVERLAP approach consisted of 2,137 PS cases and 1,903 controls of white European ancestry from the United States. The Italian samples (panel E) used in the COMBINED approach consisted of 688 CD cases and 879 healthy controls that were used in the independent genome-wide meta-analysis on CD.13 The psoriasis data set from the United Kingdom consisted of 2,178 PS cases collected through the Genetic Analysis of Psoriasis Consortium (GAPC) and 2,657 controls from the WTCCC2 common control set, used as the GWAS discovery set described in Strange et al. in 2010.16 Only controls that did not overlap with WTCCC1 controls were used. UK cases and controls entered the analysis as part of panel C and E.
Quality Control and Genome-wide Genotype Imputation
Quality control (QC) was performed for each sample set separately. In each sample set samples with more than 5% missing data were excluded before genotype imputation. We also excluded individuals from each pair of unexpected duplicates or relatives, as well as outlier individuals with average marker heterozygosities of ±5 standard deviation away from the sample mean. The remaining samples were tested for population stratification with the principal components stratification method as implemented in EIGENSTRAT,39 and population outliers were subsequently excluded. SNPs that had more than 5% missing data, a minor allele frequency less than 1% or deviated from Hardy-Weinberg equilibrium (exact p < 10−4 in controls) per sample set were excluded with the PLINK software version 1.07.40 SNP imputation was carried out with the BEAGLE v.3.1.141 software package and 690 HapMap3 reference haplotypes from the CEU, TSI, MEX, and GIH cohorts42 to predict missing autosomal genotypes in silico. We subsequently analyzed only those SNPs that could be imputed with moderate confidence (INFO score r2 > 0.3) and had a minor allele frequency more than 1% in cases or in controls. To take imputation uncertainty into account, phenotypic association was tested for allele dosage data separately for each of the five GWAS data sets in panels A and B through the use of PLINK's logistic regression framework for dosage data. To control potentially confounding effects due to population stratification, we adjusted for the top ten eigenvectors from EIGENSTRAT in the regression analysis. The genomic inflation factor λ is defined as the ratio of the medians of the sample χ2 test statistics and the 1 degree of freedom χ2 distribution (0.455).43 Because the estimated genomic inflation factor λ scales with sample size, it is informative to report the inflation factor for an equivalent study of 1,000 cases and 1,000 controls (λ1000) by rescaling λ.44
Meta-Analyses
Meta-analyses were performed with PLINK's meta-analysis function and with its standard error of odds ratio weighting option (inverse variance weighting), which implicitly deals with imputation uncertainty. For the combined-phenotype analysis, we performed two sorts of meta-analysis in order to detect associations of SNPs with either the same or opposite allelic effects in the two diseases. For the same-effect analysis, the meta-analysis was carried out as usual. For the opposite-effect analysis, first we flipped minor and major alleles of each biallelic SNP in the CD data sets to mimic an opposite-direction effect of the allele in CD and performed a meta-analysis afterward. For both effects models, we considered only those SNPs whose genotypes were available from at least four out of the five GWAS data sets in panels A and B.
Follow-Up Genotyping
Genotyping was carried out with our Sequenom iPlex platform from Sequenom and TaqMan technology from Applied Biosystems. Individuals with more than 3% missing data were removed. SNPs that had more than 3% missing data, a minor allele frequency less than 1% or deviated from Hardy-Weinberg equilibrium (exact p < 10−4 in controls) per sample set were excluded. p values for allele-based tests of phenotypic association for each single-replication sample sets (panels C–E) were calculated with PLINK. PLINK's meta-analysis function was used to obtain p values for the replication data set (pRepl) and for the combined discovery-replication data set (pGWAS+Repl).
Regional Imputation Based on the 1000 Genomes Project Reference
To enable imputation based on the 1000 Genomes project data, SNP positions referring to NCBI build 36 were mapped to build 37. SNP imputation was carried out with the BEAGLE software package v.3.1.141 and 566 EUR (European) haplotypes generated by the 1000 Genomes Project.45 We analyzed only imputed SNPs with moderate imputation confidence (INFO score r2 > 0.3) and a minor allele frequency more than1% in cases or in controls.
Gene Relationships across Implicated Loci Pathway Analysis
The Gene Relationships Across Implicated Loci (GRAIL) software46 quantifies functional similarity between genes by applying established statistical text mining methods to the PubMed database of published scientific abstracts. As input we used the following list of SNPs: rs2201841, rs2082412, rs702873, rs12720356, rs10758669, rs694739, rs281379, rs181359, rs4780355, rs744166, and rs1250544. GRAIL was run with the following settings: HapMap release = HapMap release 22/hg18; HapMap population = CEU (Utah residents with ancestry from northern and western Europe from the Centre d′Étude du Polymorphisme Humain collection); functional data source = PubMed Text (April 2011); and gene size correction = on. GRAIL output results were visualized with VIZ-GRAIL.
Results
Preparation of Single-Disease Meta-Analyses for Discovery Phase by Means of HapMap3 Imputation
The overall study workflow for the combined analysis of CD and PS is displayed in Figure 1. For discovery, we conducted a meta-analysis on PS comprising 2,529 PS cases and 4,955 controls from three previously published GWASs,14,15 all of European descent (panel A in Table S1). SNP data were combined with genotype imputation based on the HapMap3 reference. We subsequently used standard meta-analysis methodology (see Subjects and Methods). In total, 1,121,166 quality-controlled autosomal-imputed SNP markers were available for the analysis on PS. To control for potential population stratification, we adjusted association test statistics by means of principal component analysis (PCA) (see Subjects and Methods). A quantile-quantile (Q-Q) plot of the meta-analysis revealed a marked excess of significant associations in the tail of the distribution (Figure S1A), which is primarily due to thousands of highly significant association signals from the HLA region. Genetic heterogeneity was low; there was an estimated genomic inflation factor of λ1000 = 1.02743,44 (see Subjects and Methods). Results of the meta-analysis on PS are summarized in Figure S2A.
In the same way, we performed a meta-analysis on CD by using 1,034,639 quality-controlled autosomal-imputed markers from a German GWAS13 and a previously published UK GWAS,28 consisting of 2,142 CD cases and 5,505 controls in total (panel B in Table S1). Again, we observed low genomic inflation (λ1000 = 1.032, Figure S1B). Results of the meta-analysis on CD are summarized in Figure S2B.
OVERLAP Approach: Cross-Disease Analysis of Established Risk SNPs
So far, four established GWAS risk loci that are located outside the HLA region and shared between CD and PS have been reported in the literature, namely IL23R, IL12B, REL, and TYK2 (Table 1). Although the markers that showed the strongest association differed between the two diseases at each of the first three loci, we found the same SNP rs12720356 at TYK2 to be associated with both CD and PS. To seek confirmation of whether known risk loci for CD also play a role in the etiology of PS and vice versa, we checked whether markers that were significant (p < 0.01) in our PS meta-analysis were among the 71 established risk SNPs previously implicated in CD13 and whether significant markers (p < 0.01) from our CD meta-analysis were among the 25 established PS risk SNPs.14–20 The four already established shared risk SNPs (Table 1) were excluded. Given the well-established and heterogeneous allelic associations of the HLA region on chromosome 6 with both CD and PS, we also excluded all markers from the extended HLA region (chr6:25-34 Mb). Although none of the known PS SNPs were significantly associated with CD in our analysis, five out of the 71 known CD risk SNPs met our criterion of significance, namely rs10758669 (JAK2 [MIM 147796]), rs694739 (PRDX5 [MIM 606583]), rs281379 (FUT2 [MIM 182100]), rs744166 (STAT3 [MIM 102582]), and rs181359 (YDJC;HGNC 27158). We genotyped these five SNPs by using TaqMan technology in a large independent replication panel comprising 3,937 PS cases and 4,847 controls but also used summary statistics data of the five SNPs from an independent GWAS on PS16 comprising 2,178 PS cases and 2,657 controls. The overall replication panel consisted of 6,115 PS cases and 7,504 controls (panel C, Table S1). We performed single-marker association tests for panel C (pPS-Repl) and conducted a meta-analysis (pPS-GWAS+Repl) by combining association results from the GWAS (pPS-GWAS) and the replication (pPS-Repl) stages (Table 2). All of the CD risk SNPs were also significantly associated with PS at the previously proposed level34 of pPS-Repl < 0.05 and pPS-GWAS+Repl < 10−3 (rs10758669 near JAK2, rs694739 near PRDX5, rs281379 near FUT2, and rs181359 at YDJC, rs744166 at STAT3). These five SNPs have already been reported to show significant association at the genome-wide level (pCD-GWAS+Repl < 5 × 10−8, pCD-Repl < 0.05, and pCD-GWAS+Repl < 10−3) in a very large, independent genome-wide meta-analysis on CD roughly three times the size of this one, that is comprising 6,333 CD cases and 15,056 controls.13 All five SNPs achieved genome-wide significance in the combined analysis of PS discovery panel A, PS replication panel C, and CD discovery data from Franke et al.13 (pCDPS-GWAS+Repl < 5 × 10−8). Furthermore, SNP rs694739, 7.9 kb downstream of PRDX5, as well as SNP rs181359, 53.7 kb downstream of YDJC, reached genome-wide significance for PS only (prs694739 = 3.71 × 10−09 and prs181359 = 8.02 × 10−10). We also observed a highly significant association at the FUT2 locus (prs281379 = 7.86 × 10−08) for PS only.
Table 1.
Established Risk Loci Shared between CD and PS from Published Studies on CD and PS, Respectively
| Locus | Genes of Interesta | Top CD dbSNP IDb | Risk Allele | OR | Top PS dbSNP IDb | Risk Allele | OR | LD between CD and PS SNPs (D′/r2) | Comment |
|---|---|---|---|---|---|---|---|---|---|
| 1p31 | IL23R | rs11209026c | G | 2.66 | rs2201841f | G | 1.13 | 1.0/0.018 | different markers |
| 5q33 | IL12B | rs6556412d | A | 1.18 | rs2082412f | G | 1.44 | 0.715/0.269 | different markers |
| 2p16 | REL | rs10181042e | T | 1.14 | rs702873g | G | 1.12 | 0.036/0.001 | different markers |
| 19p13 | TYK2 | rs12720356e | G | 1.12 | rs12720356g | T | 1.40 | same marker | same marker, opposite direction of effect |
Table 2.
Association Results of OVERLAP Approach from Cross-Disease Comparison of Established Risk Markers
| Chr | SNP | A1 | Locus |
CD GWAS Meta-Analysisa(6,333/15,056) |
PS GWAS (2,529/4,955) |
PS Replication (6,115/7,504) |
PS GWAS and Repl (8,644/12,459) |
CD GWAS Meta-Analysisa+ PS GWAS and Repl (14,977/27,515) |
Status Nowb | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p | OR | p | OR | p | OR | p | OR | p | OR | |||||
| 9 | rs10758669 | C | JAK2 | 1.0 × 10−13 | 1.18 | 2.43 × 10−03 | 1.13 | 2.47 × 10−03 | 1.08 | 2.69 × 10−05 | 1.10 | 1.30 × 10−16 | 1.14 | CD-PS, CD |
| 11 | rs694739 | G | PRDX5 | 3.4 × 10−07 | 0.89 | 1.13 × 10−04 | 0.86 | 6.12 × 10−06 | 0.89 | 3.71 × 10−09 | 0.88 | 2.41 × 10−14 | 0.89 | PS, CD-PS, CD |
| 19 | rs281379 | A | FUT2 | 8.6 × 10−10 | 1.13 | 3.22 × 10−03 | 1.13 | 7.12 × 10−06 | 1.13 | 7.86 × 10−08 | 1.12 | 1.32 × 10−17 | 1.13 | CD-PS, CD |
| 22 | rs181359 | G | YDJC | 6.3 × 10−13 | 0.83 | 4.83 × 10−03 | 0.88 | 3.54 × 10−08 | 0.84 | 8.02 × 10−10 | 0.85 | 1.33 × 10−21 | 0.84 | PS, CD-PS, CD |
| 17 | rs744166c | A | STAT3 | 1.1 × 10−07 | 1.13 | 2.44 × 10−04 | 0.87 | 1.49 × 10−02 | 0.94 | 5.30 × 10−05 | 0.92 | 5.48 × 10−11 | 0.90 | CD-PS, CD |
The following abbreviations are used: Chr, chromosome of marker; SNP, rs ID; A1, minor allele; Locus, one candidate gene in the region; p/OR, p value and corresponding odds ratio with respect to minor allele for the large GWAS meta-analysis of CD,13 GWAS meta-analysis of PS (panel A), PS replication analysis (panel C), combined analysis of PS GWAS meta-analysis (panel A) and PS replication (panel C), and combined analysis of CD GWAS meta-analysis13 and panels A and C. For each panel, numbers of cases/controls are displayed in parentheses.
See Franke et al.13
Status now: new status of association with CD and/or PS. All SNPs are established CD risk SNPs with p < 5 × 10−8 13 that were significant (p < 0.01) in our PS meta-analysis. All SNPs, except for rs744166, showed the same direction of effect for CD and PS. None of the SNPs showed an exact Hardy-Weinberg p value < 0.01 in the PS replication (panel C).
Minor and major alleles of rs744166 were flipped in the CD GWAS meta-analysis in order to calculate the combined-phenotype p value and odds ratio.
COMBINED Approach, Part 1: Meta-Analysis Considering Same-Direction Effects
In order to identify additional shared genetic susceptibility loci in CD and PS, we performed a meta-analysis of the combined phenotype where CD and PS were considered as a single phenotype. The disease-specific meta-analyses (panels A and B) were merged to form a combined-phenotype meta-analysis discovery panel comprising 2,142 CD cases, 2,529 PS cases, and 10,460 healthy controls. In total, 1,123,777 quality-controlled autosomal markers were available for the analysis in at least four out of the five GWAS data sets. As with the OVERLAP approach, we excluded all markers from the extended HLA region (chr6:25-34Mb), leaving 1,116,213 autosomal SNPs for screening of shared risk loci. We observed only low genomic inflation for the same-direction meta-analysis (λ1000 = 1.023; Figure S3A). After exclusion of established loci for PS and CD, the inflation factors further decreased (Figure S3C). To provide proof of principle for our approach, we first examined association signals at the three established shared risk loci with same-direction effects of alleles for CD and PS (see Table 1). We observed highly significant association signals for all three loci (pIL23R = 1.82 × 10−22, pIL12B = 3.32 × 10−7, and pREL = 1.53 × 10−7; Figures S4A–S4C). Subsequently, we selected SNPs for being nominally associated in each of the single-disease meta-analyses (pCD-GWAS < 0.05, pPS-GWAS < 0.05) and for being significantly associated in the combined-phenotype association analysis at the 10−4 level (pCDPS-GWAS < 1 × 10−4). This resulted in 17 SNPs located at 17 distinct loci. Except for the four known shared loci (see Table 1), we did not exclude established risk loci from either CD or PS to maintain the chance of detecting shared risk alleles at these loci. Because one of the 17 SNPs is located at 10q22 (ZMIZ1 [MIM 607159]), which is an established CD risk locus, we added the established CD-associated SNP rs1250550 from this region to the list of follow-up SNPs. We then genotyped these 18 SNPs in an independent panel of 1,713 CD cases, 1,009 PS cases and 3,565 controls (panel D, Table S1D) by using the Sequenom iPlex platform. Association results for all 18 SNPs are shown in Table S2. The strongest association was observed at ZMIZ1 for SNP rs1250544 (pCDPS-GWAS = 1.12 × 10−5 and pCDPS-GWAS+Repl = 2.66 × 10−10; see Table S2 and Figure S5A) and yielded genome-wide significance in the same-effect combined-phenotype analysis of discovery panels A and B and replication panel D. SNP rs1250544 reached also genome-wide significance for PS alone (pPS-GWAS+Repl = 3.90 × 10−8). A robust association with CD, but not with PS, was observed at SOCS1 (MIM 603597) on chromosomal region 16p13 (pCDPS-GWAS = 9.36 × 10−7, pCD-GWAS = 1.47 × 10−3, and pCD-GWAS+Repl = 1.01 × 10−7 for rs4780355; see Table S2 and Figure S5B). We further corroborated both association signals by genotyping SNPs rs1250544 and rs4780355 in additional sample sets from Germany and Italy, comprising 2,360 CD cases and 1,015 healthy controls, but we also used summary statistics data of the two SNPs from the independent GWAS on PS16 comprising 2,178 PS cases and 2,657 controls (panel E in Table S1, the same cases and controls from the United Kingdom, as described in panel C). In the combined analysis of discovery panels A and B and replication panels D and E (Tables 3 and 4), SNP rs4780355 achieved genome-wide significance (pCDPS-GWAS+Repl = 1.37 × 10−13) but also attained genome-wide significance for CD alone (pCD-GWAS+Repl = 4.99 × 10−8).
Table 3.
Association Results of Combined-Phenotype Meta-Analysis Considering Same-Direction Effects of Alleles from COMBINED Approach
| Chr | SNP | A1 | Locus |
CD+PS Discovery GWAS (4,671/10,460) |
PS GWAS (2,529/4,955) |
CD GWAS (2,142/5,505) |
PS Replication (3,187/4,759) |
CD Replication (4,073/2,478) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p | OR | p | OR | p | OR | p | OR | p | OR | ||||
| 10 | rs1250544a | G | ZMIZ1 | 1.12 × 10−05 | 1.13 | 3.85 × 10−05 | 1.18 | 3.31 × 10−02 | 1.09 | 1.94 × 10−04 | 1.14 | 5.28 × 10−07 | 1.22 |
| 10 | rs1250560b | A | ZMIZ1 | 3.13 × 10−06 | 0.87 | 4.31 × 10−06 | 0.82 | 4.87 × 10−02 | 0.92 | 1.06 × 10−03 | 0.87 | 8.06 × 10−10 | 0.79 |
| 10 | rs1250559b | A | ZMIZ1 | 1.53 × 10−07 | 0.87 | 4.58 × 10−06 | 0.82 | 3.46 × 10−02 | 0.91 | 1.16 × 10−03 | 0.87 | 4.10 × 10−10 | 0.79 |
| 16 | rs4780355a | T | SOCS1 | 9.36 × 10−07 | 1.16 | 1.72 × 10−04 | 1.18 | 1.47 × 10−03 | 1.15 | 7.53 × 10−04 | 1.14 | 7.40 × 10−06 | 1.19 |
The following abbreviations are used: Chr: chromosome of marker; SNP: rs ID; A1: minor allele; Locus: one candidate gene in the region; p/OR: p value and corresponding odds ratio with respect to minor allele for the combined-phenotype GWAS meta-analysis of CD and PS (panels A and B), GWAS meta-analysis of PS (panel A), GWAS meta-analysis of CD (panel B), PS replication analysis (as part of panel D), CD replication analysis (part of panel D, panel E). For each panel, numbers of cases/controls are displayed in parentheses. None of the SNPs showed an exact Hardy-Weinberg p value < 0.01 in the PS and CD replication panels (panels D and E).
SNPs were identified via genotype imputation based on the HapMap3 reference and P/OR are given according to that analysis.
SNPs were identified via genotype imputation based on the 1000 Genomes reference and P/OR are given according to that analysis.
Table 4.
Association Results of Combined-Phenotype Meta-Analysis Considering Same-Direction Effects of Alleles from COMBINED Approach
| Chr | SNP | A1 | Locus |
PS GWAS and Repl (5,716/9,714) |
CD GWAS and Repl (6,215/7,983) |
CD+PS GWAS and Repl (11,931/17,697) |
Status Now | |||
|---|---|---|---|---|---|---|---|---|---|---|
| p | OR | p | OR | p | OR | |||||
| 10 | rs1250544a | G | ZMIZ1 | 3.53 × 10−08 | 1.16 | 2.56 × 10−07 | 1.16 | 7.32 × 10−14 | 1.16 | PS, CD-PS, CD |
| 10 | rs1250560b | A | ZMIZ1 | 3.03 × 10−07 | 0.84 | 4.10 × 10−09 | 0.84 | 7.34 × 10−16 | 0.85 | CD-PS, CD |
| 10 | rs1250559b | A | ZMIZ1 | 3.63 × 10−07 | 0.84 | 1.24 × 10−09 | 0.84 | 2.78 × 10−16 | 0.85 | CD-PS, CD |
| 16 | rs4780355a | T | SOCS1 | 5.30 × 10−07 | 1.15 | 4.99 × 10−08 | 1.17 | 1.37 × 10−13 | 1.16 | CD-PS, CD |
For abbreviations used, see Table 3. Combined analysis of PS GWAS meta-analysis (panel A) and PS replication (part of panel D), combined analysis of CD GWAS meta-analysis (panel B) and CD replication (part of panel D, panel E), combined analysis of CD+PS GWAS meta-analysis (panels A and B) and CD+PS replication (panels D and E).
SNPs were identified via genotype imputation based on the HapMap3 reference and p/OR are given according to that analysis.
SNPs were identified via genotype imputation based on the 1000 Genomes reference and p/OR are given according to that analysis.
COMBINED Approach, Part 2: Meta-Analysis Assuming Opposite-Direction Effects
An allele might confer a risk for CD while protecting against PS and vice versa, as is the case for TYK2. Therefore, we also screened our combined-phenotype meta-analysis data (panels A and B) while coding alleles in such a way as to consider the opposite effects of them in the two diseases (see Subjects and Methods). We observed low genomic inflation for the opposite-direction meta-analysis (λ1000 = 1.009, Figure S3B). After excluding established shared loci for PS and CD, the inflation factors further decreased (Figure S3D). In a first step, we checked the known risk SNP rs12720356 (TYK2; see Table 1) for opposite direction of effects. SNP rs12720356 had a p value of 4.09 × 10−5 in the combined analysis of panels A and B (Figure S4D); there was an odds ratio (OR) of 1.29 (95% confidence interval [CI] [1.10,1.51]) for allele A in panel A (pPS-GWAS = 1.39 × 10−3) and of 0.78 (95% CI [0.65,0.94]) in panel B (pCD-GWAS = 1.01 × 10−2). We then selected three SNPs for subsequent genotyping (with Sequenom) and testing in replication panel D (see Table S1D). The selection criteria were the same as for the same-direction effect meta-analysis. However, none of the three SNPs replicated in both diseases at p value < 0.05 (see Table S3).
In Silico Fine-Mapping: Refinement of Association Signals of COMBINED Approach
For refinement of the association signals at ZMIZ1 and SOCS1, we imputed a region of about ±1 Mb around the strongest signals from the discovery panels A and B (see Table S1) by using the EUR reference from the 1000 Genomes Project45 (see Subjects and Methods). In silico fine-mapping of the region around SOCS1 via standard meta-analysis methodology (see Subjects and Methods) confirmed rs4780355 to be highly significant in this region (pGWAS = 4.04 × 10−7). Additionally, another SNP, rs2021511, which is located in the same intron as rs4780355 (2.9 kb downstream of rs4780355) showed the same magnitude of association (pGWAS = 1.58 × 10−7; Figure 2) but was not selected for further replication because of the high linkage disequilibrium (LD) between SNPs rs4780355 and rs2021511 (r2 = 0.934) according to the 1000 Genomes Project EUR reference. Screening of the imputed region of SOCS1 for coding SNPs revealed one missense SNP with p < 10−4 within the TNP2 gene, namely rs11640138. We genotyped this SNP in replication panel D, but it did not replicate in either disease at p value < 0.05.
Figure 2.
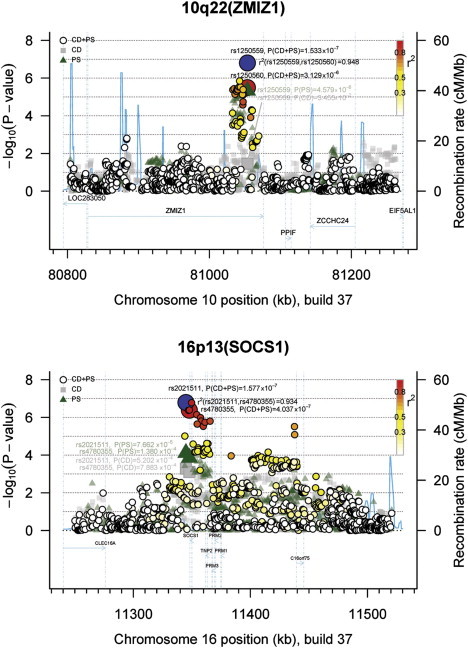
Regional Association Plots of In Silico Fine-Mapping for Newly Detected Shared Risk Loci from COMBINED Approach
Shared risk loci for CD and PS at (A) 10q22 (ZMIZ1) and (B) 16p13 (near SOCS1). eQTL analyses revealed a potential effect of the associations at ZMIZ1 and near SOCS1 on gene expression. p values (−log10p) are depicted with regard to the physical location of markers and are based on imputed genotypes. SNP genotypes were imputed with the EUR reference from 1000 Genomes Project45 (see Subjects and Methods). The following abbreviations are used: blue-filled circle, lead SNP of the combined-phenotype data (panels A and B); other filled circles, analyzed SNPs of the combined-phenotype data (panels A and B) where the fill color corresponds to the strength of linkage disequilibrium (r2) with the lead SNP (for color coding see legend in the upper right corner of each plot); green triangles, analyzed SNPs of the meta-analysis on PS (panel A); gray squares, analyzed SNPs of the meta-analysis on CD (panel B); and blue line, recombination intensity (cM/Mb). Positions and gene annotations are according to NCBI's build 37 (hg19).
In silico fine-mapping of ZMIZ1 with the same methodology narrowed down the association signal to two coding SNPs, namely rs1250559 (pCDPS-GWAS = 1.53 × 10−7, Figure 2) and rs1250560 (pCDPS-GWAS = 3.13 × 10−6). According to the 1000 Genomes Project EUR reference, both SNPs are in near perfect LD (r2 = 0.948). Depending on different splice variants of ZMIZ1, rs1250559 is either intronic or located in the 3-untranslated region (3-UTR), whereas rs1250560 is either an intronic SNP or a missense SNP located in exon 5. The intronic ZMIZ1 SNP rs1250544, which yielded the strongest signal from the initial same-effect combined-phenotype meta-analysis, and the missense SNP rs1250560 are 20.6 kb apart and in moderate LD (r2 = 0.682). In order to substantiate our findings from the in silico analyses, we genotyped both ZMIZ1 SNPs in replication panels D and E (see Table S1). As shown in Tables 3 and 4, both SNPs were associated with PS at the 0.05 level and even showed genome-wide significance with CD (pCD-Repl = 8.06 × 10−10 at rs1250560, pCD-Repl = 4.10 × 10−10 at rs1250559). Interestingly, the association signals of these two SNPs were much stronger in the initial analysis of PS panel A than of CD panel B. The combined analysis of discovery panels A and B and replication panels D and E yielded genome-wide significance for rs1250560 (pCDPS-GWAS+Repl = 7.34 × 10−16) and rs1250559 (pCDPS-GWAS+Repl = 2.78 × 10−16), both of which are of higher significance than was observed for rs1250544 (pCDPS-GWAS+Repl = 7.32 × 10−14) (Tables 3 and 4).
Effect on Gene Expression
We subsequently assessed a potential functional effect of the four SNPs showing association for both CD and PS with the same direction of effects, namely rs1250544, rs1250559, rs1250560 (ZMIZ1), and rs4780355 (near SOCS1). To this end, we investigated the correlation of SNP genotypes with gene expression levels by means of in silico expression quantitative trait locus (eQTL) analysis by using the mRNAbySNPBrowser software.47 This program utilizes genotype data from 408,273 SNPs and gene expression data from Epstein-Barr-virus-transformed lymphoblastoid cell lines that were collected from 400 children and measured with the Affymetrix HG-U133 Plus 2.0 chip. Significant evidence (uncorrected pExpression < 10−4) for causing differential expression of ZMIZ1 was observed for SNP rs1250546 (prs1250546 = 8.10 × 10−5), which is in high LD with our lead SNP rs1250544 (r2 = 0.829). Also, we found an even stronger evidence for association between expression of C16ORF75 (MIM 612426), which is located 90 kb upstream of SOCS1, and SNP rs243323 (prs243323 = 1.10 × 10−8). This SNP is also in high LD with our lead SNP rs4780355 (r2 = 0.931). Both proxy SNPs rs1250546 and rs243323 were also significantly associated in our same-effect combined-phenotype analysis of discovery panels A and B (pGWAS = 7.15 × 10−5 for rs1250546, pGWAS = 1.80 × 10−6 for rs243323). This in silico eQTL analysis supports the notion that our four reported SNPs might affect the expression of ZMIZ1 and C16ORF75. The full list of significant associations between SNP genotypes and gene expression levels is shown in Table S4.
Discussion
In a large combined sample set of 6,215 CD cases, 8,644 PS cases and 20,560 healthy controls, we have identified seven non-HLA susceptibility loci shared between CD and PS (9p24 near JAK2, 10q22 at ZMIZ1, 11q13 near PRDX5, 16p13 near SOCS1, 19p13 near FUT2, 17q21 at STAT3, 22q11 at YDJC). These loci, except for SOCS1, were already known to play a role in CD etiology, but were of unknown significance for PS13 (see also Table S5 for associations with other diseases). Notably, three of these loci showed genome-wide significance when tested for association with PS alone (10q22 at ZMIZ1, 11q13 near PRDX5, and 22q11 at YDJC). Furthermore, we revealed a risk locus for CD (16p13 near SOCS1). The identified shared risk loci point to functionally very interesting genes that might play a role in the pathogenesis of both CD and PS. The gene ZMIZ1 (also known as hZIMP10 or TRAFIP10) encodes for the protein zinc finger MIZ type 1, which is a member of the protein inhibitor of activated STAT (PIAS) family. The protein regulates the activity of several transcription factors such as the androgen receptor, Smad3/4, and p53; regulates TGF-β/SMAD signaling; and is induced by retinoic acid.48 FUT2 encodes α-(1,2)fucosyltransferase (FUT2), a physiological trait that regulates expression of the Lewis human blood group of antigens on the surface of epithelial cells and in body fluids. Genetic variants in FUT2 have been implicated in susceptibility to infections with Norovirus49 and Helicobacter pylori.50 PRDX5 encodes Peroxiredoxin-5, which belongs to the peroxiredoxin family of antioxidant enzymes that reduce hydrogen peroxide and alkyl hydroperoxides and might play a protective role during inflammatory processes. SOCS1 encodes the suppressor of cytokine signaling 1 (SOCS1), a protein that is member of the STAT-induced STAT inhibitor (SSI), also known as suppressor of cytokine signaling (SOCS) family. SOCS1 is a cytokine-inducible negative regulator of cytokine signaling.51,52 Cytokines such as IL2, IL3, erythropoietin, and interferon-gamma can induce expression of SOCS1.53 Moreover, a potential functional effect of the associations at ZMIZ1 and near SOCS1 on gene expression was found by an in silico eQTL analysis.
The present study has increased the number of known shared CD and PS susceptibility loci to eleven (IL12B, IL23R, REL, TYK2, JAK2, ZMIZ1, PRDX5, SOCS1, STAT3, FUT2, and YDJC). To quantify the degree of relatedness between genes within the eleven loci, we used a published statistical genomics method, namely GRAIL (gene relationships across implicated loci),46 that applies statistical text mining to PubMed abstracts (see Subjects and Methods and Figure 3). GRAIL highlights a number of nonrandom and evidence-based connections between the genes within the nine loci that might indicate overlap in the pathways acting in the etiology of CD and PS. Multiple genes (IL12B, IL23R, TYK2, JAK2, SOCS1, and STAT3) are involved in IL23/Th17 signaling and play a critical role in the principal signaling mechanism for a wide array of cytokines and growth factors. It is noteworthy that genes CDC37 ([MIM 605065] 21.7 kb downstream of the established shared risk locus at TYK2) and STIP1 ([MIM 605063] 113.5 kb upstream of the identified shared risk at PRDX5) were found by GRAIL to be significantly connected. CDC37 and STIP1 encode CDC37 and STI1, respectively, two of several auxiliary proteins that associate with the heat-shock protein 90 (HSP90) molecular chaperone and thus are collectively referred to as HSP90 cochaperones.54 HSP90 itself is an abundant, evolutionarily conserved molecular chaperone that acts mainly as a cofactor for the folding of polyproteins into functional, stable, mature proteins, and it physically associates with JAK1 and probably JAK2,55 demonstrating that JAK1/2 are client proteins of HSP90. A study in mice and in patient samples suggested that HSP90 inhibitors might help treat JAK2-dependent myeloproliferative neoplasms (MPNs).56 Moreover, inhibition of HSP90 was found to block Nod2-mediated activation of the transcription factor NF-κB and reduce NALP3-mediated gout-like inflammation in mice,57 and mutations in the gene encoding NALP3, a member of the Nod-like receptor (NLR) protein family, are associated with several autoinflammatory disorders.58,59 Our hypothesis that CDC37 and STIP1 are potential joint risk factors for CD and PS is substantiated by an association peak within CDC37 in our same-effect combined-phenotype analysis of discovery panels A and B (pGWAS = 1.61 × 10−3 for rs11879191, Figure S6).
Figure 3.
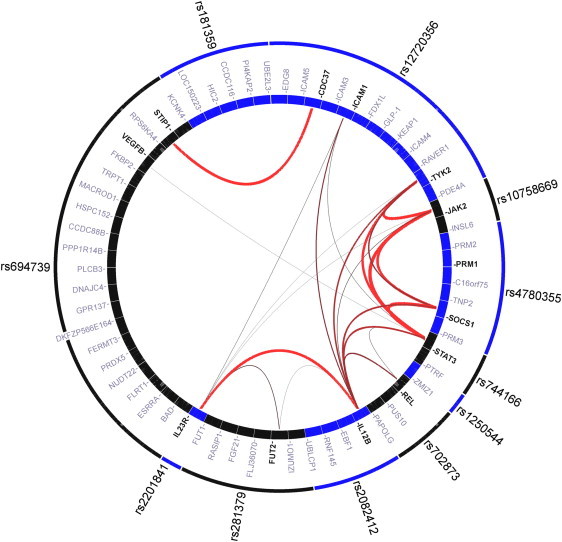
Gene Relationships across the 11 Shared Risk Loci of CD and PS Identified by GRAIL Analysis
GRAIL46 is a statistical text-mining approach to quantify the degree of relatedness among genes in genomic disease regions. It estimates the statistical significance of the number of observed relationships with a null model in which relationships between the genes occur by random chance. A significance score ptext, which is adjusted for multiple hypothesis testing, represents the output GRAIL score. ptext values approximately estimate type-I error rates. Outer circle: lead SNPs from shared risk loci of both diseases; each box represents a SNP. Inner circle: genes of the genomic regions around lead SNPs that were identified based on LD properties; each box represents a gene; genes that were scored at ptext < 0.05 are significantly linked to genes in the other disease regions and are indicated in bold type. Lines: the lines between genes represent significant connections, with the thickness and redness of the lines being inversely proportional to the probability that a text-based connection would be seen by chance.
It is worth noting that we used a two tier strategy to identify shared disease risk loci: Both approaches turned out to be effective and complementary tools for gaining insights into the postulated shared pathogenesis of CD and PS. Application of only a single strategy would have decreased the number of identified loci. Although the OVERLAP approach represents a simple and cost-effective strategy (cross-disease comparison of known risk SNPs), the COMBINED approach provides the power to identify shared susceptibility loci even if association signals are heterogeneous between diseases, that is the particular SNP showing the smallest p value at the considered locus, as was the case, for example, for the identified risk locus at ZMIZ1. This heterogeneity of most strongly associated SNPs could be due to interactions with other genetic variants or environmental factors, to differences in the distribution or effect size of causal alleles, or to the fact the identified SNPs show an association signal only because they are in LD with the actual causal variant. In particular, the increase of power due to increased sample sizes makes the COMBINED approach a potentially powerful tool to detect shared risk loci that might be missed in disease-specific GWASs that are often underpowered because of their comparatively smaller sample sizes.
Because we did not search for loci harboring association signals with different and independent SNPs in terms of LD associated with CD and PS, there is room for improvement. For instance, in a simple rank approach with regard to single-marker association p values, different disease-associated markers for the same locus could be determined when they rank high with regard to their p value in association scans of CD and PS, respectively. This would allow detecting shared susceptibility loci even if association signals are heterogeneous between diseases. An approach to meet the challenge of the heterogeneity of genetic effects of the same markers between different diseases was proposed by Morris et al.60 The authors developed a test of association within a multinomial regression framework and demonstrated the improved power of their multinomial regression-based analysis over existing methods.
It is likely that future studies will identify additional shared disease loci for CD and PS by further increasing the sample size of analyzed case-control panels or, for example, by applying the suggested rank approach. Evidence for a shared etiological basis among several autoimmune and inflammatory diseases is growing. For Crohn disease, for example, Lees and colleagues currently reported that 51 of the known 71 loci overlap with more than 23 distinct diseases, comprising also several nonautoimmune conditions.61 Given the success of this study, we expect the same for the investigation of further combinations of such diseases for shared risk factors.
Acknowledgments
We thank all individuals with psoriasis or CD, their families, control individuals and clinicians for their participation in this project. We thank the WTCCC consortium for the access to the CD case/control data. We acknowledge the cooperation of Genizon Biosciences. We wish to thank Tanja Wesse, Tanja Henke and Susan Ehlers for expert technical help. We acknowledge EGCUT and Estonian Biocentre personnel, especially Ms. M. Hass and Mr. V. Soo. A list of funding sources is included in the Supplemental Data.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
BEAGLE, http://faculty.washington.edu/browning/beagle/beagle.html
EIGENSTRAT, http://genepath.med.harvard.edu/∼reich/Software.htm
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PopGen Biobank, http://www.popgen.de
VIZ-GRAIL, http://www.broadinstitute.org/mpg/grail/vizgrail.html
References
- 1.Bhalerao J., Bowcock A.M. The genetics of psoriasis: A complex disorder of the skin and immune system. Hum. Mol. Genet. 1998;7:1537–1545. doi: 10.1093/hmg/7.10.1537. [DOI] [PubMed] [Google Scholar]
- 2.Elder J.T., Nair R.P., Guo S.W., Henseler T., Christophers E., Voorhees J.J. The genetics of psoriasis. Arch. Dermatol. 1994;130:216–224. [PubMed] [Google Scholar]
- 3.Russell R.K., Satsangi J. IBD: A family affair. Best Pract. Res. Clin. Gastroenterol. 2004;18:525–539. doi: 10.1016/j.bpg.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths C.E., Barker J.N. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370:263–271. doi: 10.1016/S0140-6736(07)61128-3. [DOI] [PubMed] [Google Scholar]
- 5.Logan I., Bowlus C.L. The geoepidemiology of autoimmune intestinal diseases. Autoimmun. Rev. 2010;9:A372–A378. doi: 10.1016/j.autrev.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 6.Sagoo G.S., Cork M.J., Patel R., Tazi-Ahnini R. Genome-wide studies of psoriasis susceptibility loci: A review. J. Dermatol. Sci. 2004;35:171–179. doi: 10.1016/j.jdermsci.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Najarian D.J., Gottlieb A.B. Connections between psoriasis and Crohn's disease. J. Am. Acad. Dermatol. 2003;48:805–821. doi: 10.1067/mjd.2003.540. quiz 822–804. [DOI] [PubMed] [Google Scholar]
- 8.Khor B., Gardet A., Xavier R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yates V.M., Watkinson G., Kelman A. Further evidence for an association between psoriasis, Crohn's disease and ulcerative colitis. Br. J. Dermatol. 1982;106:323–330. doi: 10.1111/j.1365-2133.1982.tb01731.x. [DOI] [PubMed] [Google Scholar]
- 10.Bernstein C.N., Wajda A., Blanchard J.F. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: A population-based study. Gastroenterology. 2005;129:827–836. doi: 10.1053/j.gastro.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 11.Weng X., Liu L., Barcellos L.F., Allison J.E., Herrinton L.J. Clustering of inflammatory bowel disease with immune mediated diseases among members of a northern california-managed care organization. Am. J. Gastroenterol. 2007;102:1429–1435. doi: 10.1111/j.1572-0241.2007.01215.x. [DOI] [PubMed] [Google Scholar]
- 12.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M., NIDDK IBD Genetics Consortium. Belgian-French IBD Consortium. Wellcome Trust Case Control Consortium Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nair R.P., Duffin K.C., Helms C., Ding J., Stuart P.E., Goldgar D., Gudjonsson J.E., Li Y., Tejasvi T., Feng B.J., Collaborative Association Study of Psoriasis Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellinghaus E., Ellinghaus D., Stuart P.E., Nair R.P., Debrus S., Raelson J.V., Belouchi M., Fournier H., Reinhard C., Ding J. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 2010;42:991–995. doi: 10.1038/ng.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strange A., Capon F., Spencer C.C., Knight J., Weale M.E., Allen M.H., Barton A., Band G., Bellenguez C., Bergboer J.G., Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2 A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun L.D., Cheng H., Wang Z.X., Zhang A.P., Wang P.G., Xu J.H., Zhu Q.X., Zhou H.S., Ellinghaus E., Zhang F.R. Association analyses identify six new psoriasis susceptibility loci in the Chinese population. Nat. Genet. 2010;42:1005–1009. doi: 10.1038/ng.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuart P.E., Nair R.P., Ellinghaus E., Ding J., Tejasvi T., Gudjonsson J.E., Li Y., Weidinger S., Eberlein B., Gieger C. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010;42:1000–1004. doi: 10.1038/ng.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hüffmeier U., Uebe S., Ekici A.B., Bowes J., Giardina E., Korendowych E., Juneblad K., Apel M., McManus R., Ho P. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 2010;42:996–999. doi: 10.1038/ng.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X.J., Huang W., Yang S., Sun L.D., Zhang F.Y., Zhu Q.X., Zhang F.R., Zhang C., Du W.H., Pu X.M. Psoriasis genome-wide association study identifies susceptibility variants within LCE gene cluster at 1q21. Nat. Genet. 2009;41:205–210. doi: 10.1038/ng.310. [DOI] [PubMed] [Google Scholar]
- 21.Duerr R.H., Taylor K.D., Brant S.R., Rioux J.D., Silverberg M.S., Daly M.J., Steinhart A.H., Abraham C., Regueiro M., Griffiths A. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cargill M., Schrodi S.J., Chang M., Garcia V.E., Brandon R., Callis K.P., Matsunami N., Ardlie K.G., Civello D., Catanese J.J. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 2007;80:273–290. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nair R.P., Ruether A., Stuart P.E., Jenisch S., Tejasvi T., Hiremagalore R., Schreiber S., Kabelitz D., Lim H.W., Voorhees J.J. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J. Invest. Dermatol. 2008;128:1653–1661. doi: 10.1038/sj.jid.5701255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mannon P.J., Fuss I.J., Mayer L., Elson C.O., Sandborn W.J., Present D., Dolin B., Goodman N., Groden C., Hornung R.L., Anti-IL-12 Crohn's Disease Study Group Anti-interleukin-12 antibody for active Crohn's disease. N. Engl. J. Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 25.Abraham C., Cho J.H. IL-23 and autoimmunity: New insights into the pathogenesis of inflammatory bowel disease. Annu. Rev. Med. 2009;60:97–110. doi: 10.1146/annurev.med.60.051407.123757. [DOI] [PubMed] [Google Scholar]
- 26.Di Meglio P., Di Cesare A., Laggner U., Chu C.C., Napolitano L., Villanova F., Tosi I., Capon F., Trembath R.C., Peris K., Nestle F.O. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE. 2011;6:e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franke A., Fischer A., Nothnagel M., Becker C., Grabe N., Till A., Lu T., Müller-Quernheim J., Wittig M., Hermann A. Genome-wide association analysis in sarcoidosis and Crohn's disease unravels a common susceptibility locus on 10p12.2. Gastroenterology. 2008;135:1207–1215. doi: 10.1053/j.gastro.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 28.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franke A., Balschun T., Karlsen T.H., Hedderich J., May S., Lu T., Schuldt D., Nikolaus S., Rosenstiel P., Krawczak M., Schreiber S. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 30.Wang K., Baldassano R., Zhang H., Qu H.Q., Imielinski M., Kugathasan S., Annese V., Dubinsky M., Rotter J.I., Russell R.K. Comparative genetic analysis of inflammatory bowel disease and type 1 diabetes implicates multiple loci with opposite effects. Hum. Mol. Genet. 2010;19:2059–2067. doi: 10.1093/hmg/ddq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cotsapas C., Voight B.F., Rossin E., Lage K., Neale B.M., Wallace C., Abecasis G.R., Barrett J.C., Behrens T., Cho J., FOCiS Network of Consortia Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imielinski M., Baldassano R.N., Griffiths A., Russell R.K., Annese V., Dubinsky M., Kugathasan S., Bradfield J.P., Walters T.D., Sleiman P., Western Regional Alliance for Pediatric IBD. International IBD Genetics Consortium. NIDDK IBD Genetics Consortium. Belgian-French IBD Consortium. Wellcome Trust Case Control Consortium Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Festen E.A., Goyette P., Green T., Boucher G., Beauchamp C., Trynka G., Dubois P.C., Lagacé C., Stokkers P.C., Hommes D.W. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet. 2011;7:e1001283. doi: 10.1371/journal.pgen.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhernakova A., Stahl E.A., Trynka G., Raychaudhuri S., Festen E.A., Franke L., Westra H.J., Fehrmann R.S., Kurreeman F.A., Thomson B. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krawczak M., Nikolaus S., von Eberstein H., Croucher P.J., El Mokhtari N.E., Schreiber S. PopGen: Population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9:55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- 36.Wichmann H.E., Gieger C., Illig T., MONICA/KORA Study Group KORA-gen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67(Suppl 1):S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 37.Weiland S.K., Björkstén B., Brunekreef B., Cookson W.O., von Mutius E., Strachan D.P., International Study of Asthma and Allergies in Childhood Phase II Study Group Phase II of the International Study of Asthma and Allergies in Childhood (ISAAC II): Rationale and methods. Eur. Respir. J. 2004;24:406–412. doi: 10.1183/09031936.04.00090303. [DOI] [PubMed] [Google Scholar]
- 38.Manolio T.A., Rodriguez L.L., Brooks L., Abecasis G., Ballinger D., Daly M., Donnelly P., Faraone S.V., Frazer K., Gabriel S., GAIN Collaborative Research Group. Collaborative Association Study of Psoriasis. International Multi-Center ADHD Genetics Project. Molecular Genetics of Schizophrenia Collaboration. Bipolar Genome Study. Major Depression Stage 1 Genomewide Association in Population-Based Samples Study. Genetics of Kidneys in Diabetes (GoKinD) Study New models of collaboration in genome-wide association studies: The Genetic Association Information Network. Nat. Genet. 2007;39:1045–1051. doi: 10.1038/ng2127. [DOI] [PubMed] [Google Scholar]
- 39.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 40.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Browning B.L., Browning S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 44.de Bakker P.I., Ferreira M.A., Jia X., Neale B.M., Raychaudhuri S., Voight B.F. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum. Mol. Genet. 2008;17(R2):R122–R128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raychaudhuri S., Plenge R.M., Rossin E.J., Ng A.C., Purcell S.M., Sklar P., Scolnick E.M., Xavier R.J., Altshuler D., Daly M.J., International Schizophrenia Consortium Identifying relationships among genomic disease regions: Predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5:e1000534. doi: 10.1371/journal.pgen.1000534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dixon A.L., Liang L., Moffatt M.F., Chen W., Heath S., Wong K.C., Taylor J., Burnett E., Gut I., Farrall M. A genome-wide association study of global gene expression. Nat. Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 48.Li X., Thyssen G., Beliakoff J., Sun Z. The novel PIAS-like protein hZimp10 enhances Smad transcriptional activity. J. Biol. Chem. 2006;281:23748–23756. doi: 10.1074/jbc.M508365200. [DOI] [PubMed] [Google Scholar]
- 49.Carlsson B., Kindberg E., Buesa J., Rydell G.E., Lidón M.F., Montava R., Abu Mallouh R., Grahn A., Rodríguez-Díaz J., Bellido J. The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 Norovirus infection. PLoS ONE. 2009;4:e5593. doi: 10.1371/journal.pone.0005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ikehara Y., Nishihara S., Yasutomi H., Kitamura T., Matsuo K., Shimizu N., Inada K., Kodera Y., Yamamura Y., Narimatsu H. Polymorphisms of two fucosyltransferase genes (Lewis and Secretor genes) involving type I Lewis antigens are associated with the presence of anti-Helicobacter pylori IgG antibody. Cancer Epidemiol. Biomarkers Prev. 2001;10:971–977. [PubMed] [Google Scholar]
- 51.Starr R., Willson T.A., Viney E.M., Murray L.J., Rayner J.R., Jenkins B.J., Gonda T.J., Alexander W.S., Metcalf D., Nicola N.A., Hilton D.J. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921. doi: 10.1038/43206. [DOI] [PubMed] [Google Scholar]
- 52.Yasukawa H., Sasaki A., Yoshimura A. Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol. 2000;18:143–164. doi: 10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]
- 53.Krebs D.L., Hilton D.J. SOCS: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000;113:2813–2819. doi: 10.1242/jcs.113.16.2813. [DOI] [PubMed] [Google Scholar]
- 54.Abbas-Terki T., Briand P.A., Donzé O., Picard D. The Hsp90 co-chaperones Cdc37 and Sti1 interact physically and genetically. Biol. Chem. 2002;383:1335–1342. doi: 10.1515/BC.2002.152. [DOI] [PubMed] [Google Scholar]
- 55.Shang L., Tomasi T.B. The heat shock protein 90-CDC37 chaperone complex is required for signaling by types I and II interferons. J. Biol. Chem. 2006;281:1876–1884. doi: 10.1074/jbc.M509901200. [DOI] [PubMed] [Google Scholar]
- 56.Marubayashi S., Koppikar P., Taldone T., Abdel-Wahab O., West N., Bhagwat N., Caldas-Lopes E., Ross K.N., Gönen M., Gozman A. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Invest. 2010;120:3578–3593. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayor A., Martinon F., De Smedt T., Pétrilli V., Tschopp J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007;8:497–503. doi: 10.1038/ni1459. [DOI] [PubMed] [Google Scholar]
- 58.Hoffman H.M., Mueller J.L., Broide D.H., Wanderer A.A., Kolodner R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001;29:301–305. doi: 10.1038/ng756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hawkins P.N., Lachmann H.J., Aganna E., McDermott M.F. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–612. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 60.Morris A.P., Lindgren C.M., Zeggini E., Timpson N.J., Frayling T.M., Hattersley A.T., McCarthy M.I. A powerful approach to sub-phenotype analysis in population-based genetic association studies. Genet. Epidemiol. 2010;34:335–343. doi: 10.1002/gepi.20486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lees C.W., Barrett J.C., Parkes M., Satsangi J. New IBD genetics: Common pathways with other diseases. Gut. 2011;60:1739–1753. doi: 10.1136/gut.2009.199679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.