

Abstract
The E2F family of transcription factors, in association with pocket protein family members, are important for regulating genes required for cellular proliferation. The most abundant E2F, E2F4, is implicated in maintaining the G0/G1 cell cycle state via transcriptional repression of genes that encode proteins required for S-phase progression. Here, we investigate E2F4's role in bone development using E2f4 germline mutant mice. We find that mutation of E2f4 impairs the formation of several bones that arise through intramembranous or endochondral ossification. The most severe defect occurred in the calvarial bones of the skull where we observed a striking delay in their ossification. In vivo and in vitro analyses established that E2F4 loss did not block the intrinsic differentiation potential of calvarial osteoblast progenitors. However, our data showed that E2f4 mutation elevated proliferation in the developing calvaria in vivo and it increased the endogenous pool of undifferentiated progenitor cells. These data suggest that E2F4 plays an important role in enabling osteoblast progenitors to exit the cell cycle and subsequently differentiate thereby contributing to the commitment of these cells to the bone lineage.
Key words: E2f4, osteoblasts, bone, proliferation, differentiation
Introduction
Members of the E2F family of transcription factors are best known for their role in regulating genes required for cellular proliferation.1,2 To date, ten E2F family members have been identified. These can be subdivided into two groups based on their predisposition to activate or repress transcription of E2F-responsive genes. E2F4 functions primarily as a transcriptional repressor and its nuclear localization is largely dependent on its association with pocket protein family members pRb, p107 and p130.3–5 These complexes recruit histone deacetylases to E2F-responsive gene promoters which directly repress transcription.4,6–8 Upon mitogen-induced phosphorylation of the pocket proteins by activated cyclin dependent kinases, the pocket protein/E2F4 complexes dissociate and E2F4 is exported to the cytoplasm via active nuclear export.1,2,9 This removes E2F4 from promoters, thereby relieving the active repression of E2F-target genes and allowing binding of the activating E2Fs.
While the role of the E2F family of transcription factors in cell cycle progression has been well characterized, we are only beginning to understand the role of E2Fs in development.10 E2F4 is known to affect the differentiation and development of many cell lineages through cell cycle-dependent and cell cycle-independent mechanisms. For example, E2f4 knockout embryos are transiently anemic, and exhibit cell autonomous defects in red blood cell maturation.11–13 Erythrocytes in these embryos show incomplete enucleation and there is an increase in the population of immature erythrocytes.11,12 Mutation of E2f4 often leads to neonatal lethality caused by chronic rhinitis and associated opportunistic bacterial infections.11 The susceptibility to infection is a consequence of disrupted airway epithelium development that results in an absence of ciliated cells and an increase in number of mucin-secreting cells, leading to chronic mucus buildup.14 Although the mechanism underlying the cilial cell defect is unknown, it appears to be independent of deregulated cellular proliferation. E2F4 also contributes to adipocyte differentiation and this seems to occur through both cell cycle-dependent and -independent mechanisms.15,16
A role for E2F4 during chondrocyte or osteoblast differentiation has not been investigated. However, its pocket protein binding partners have been implicated in the differentiation of these cell types.17–20 Specifically, mutation of p107 and p130 in vivo prevents chondrocytes from properly exiting the cell cycle.17,21,22 While loss of pRb inhibits osteoblast differentiation in vitro and in vivo.18–20 Therefore, it is plausible that E2F4 may also function in the differentiation of these cell types and, consequently, bone development. Bone development occurs via two distinct processes.23 In the first process, intramembranous ossification, mesenchymal progenitor cells differentiate directly into osteoblasts, which secrete the calcified extracellular matrix that constitutes bone. This occurs primarily in the flat bones of the calvarium (skull) and the medial clavicles. Most bones in the skeleton form via the second process, endochondral ossification, where condensations of mesenchymal progenitor cells differentiate first into chondrocytes. These chondrocytes proliferate and secrete a cartilage matrix, forming the bone template. Upon terminal differentiation, chondrocytes become hypertrophic and undergo apoptosis, which provides space for osteoblasts to invade and generate the bone matrix. In this study, we show that E2f4-/- embryos exhibit a defect in the ossification of some bones formed through either endochondral or intramembranous ossification and establish that E2f4 is required for appropriate maturation of osteoblasts.
Results
E2f4-/- embryos exhibit defects in bone development.
To determine the effect of E2F4 loss on skeletal development, we examined the skeletons of wildtype and E2f4-/- embryos at e18.5 by staining for calcified bone matrix (Alizarin Red) and cartilage (Alcian Blue). At this stage of development, several bones in the E2f4-/- embryos were less ossified compared to the bones of wildtype littermate controls (Fig. 1). The sternebrae, xiphoid process and presphenoid bone (in the base of the cranial vault), which form through endochondral ossification, had reduced Alizarin Red staining in E2f4-null embryos in comparison with littermate wild-type controls, indicating reduced ossification (Fig. 1A and B). A similar defect is seen in the bones that form through intramembranous ossification; in E2f4-/- embryos, the frontal and parietal bones of the skull exhibit dramatically reduced ossification compared to wildtype embryos (Fig. 1C). We also examined the skeletons of mice at birth (P0). At P0, the skeletal defects were less apparent (Fig. 1). Indeed, many of the affected bones appeared to be ossified appropriately at this timepoint, although the presphenoid bone still showed decreased ossification relative to the controls (Fig. 1B).
Figure 1.
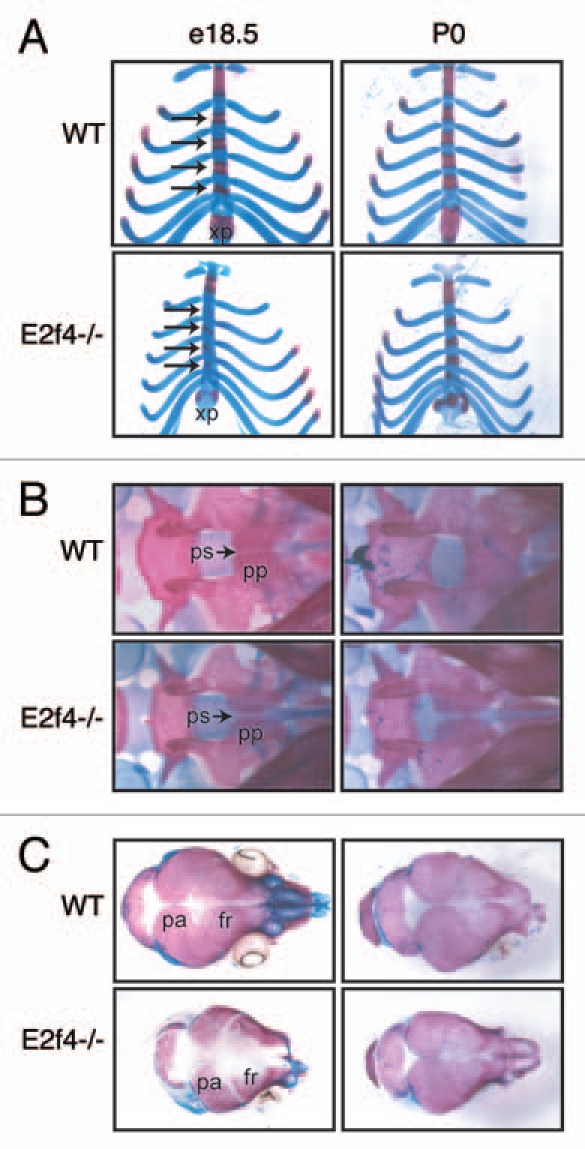
Mutation of E2f4 causes defects in embryonic bone ossification. (A–C) Alizarin Red (bone) and Alcian Blue (cartilage) staining of representative e18.5 and P0 littermate embryos. At e18.5 E2f4-/- embryos exhibit less ossification in (A) the sternebrae (arrows) and xiphoid process, (B) presphenoid bone and (C) cranium (n ≥ 6). By P0 the extent of ossification is similar between wild-type and E2f4-/- animals (n ≥ 4). Abbreviations: xp, xiphoid process; pp, palatine process; ps, presphenoid (with arrow); fr, frontal bone; pa, parietal bone.
To determine if there were defects in the initiation of the ossification process we analyzed embryos at earlier time points. At e13.5, the only bone to become ossified is the clavicle. In E2f4-/- embryos, the clavicle is not ossified as judged by the absence of Alizarin Red staining of this bone in comparison with wildtype littermates (Fig. 2). At e15.5, there is also less Alizarin Red staining in the skulls of E2f4-null embryos compared to wildtype littermates (Fig. 2). In addition, we observed a decrease in the amount of Alcian Blue staining in the basisphenoid and the presphenoid bones at this stage (Fig. 2). This finding suggests that the presphenoid bone ossification defect in e18.5 embryos may, at least in part, be a result of defective or delayed cartilage differentiation. These data indicate that loss of E2f4 affects the normal timing of ossification in specific components of the skeleton but its loss does not block this process.
Figure 2.
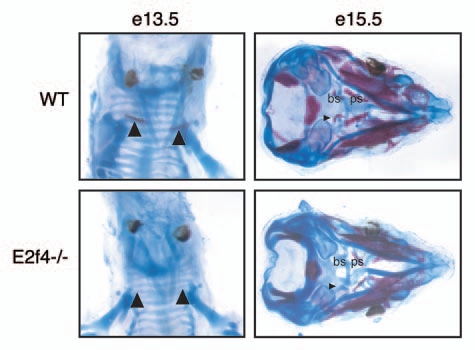
Mutation of E2f4 causes defects in early embryonic cartilage and bone development. Alizarin Red (bone) and Alcian Blue (cartilage) staining of (A) e13.5 and (B) e15.5 embryos. e13.5 E2f4-/- embryos exhibit less ossification in the clavicles, n ≥ 2 (arrowheads). e15.5 E2f4-/- embryos display less ossification of bone (arrowhead) and less deposition of cartilage matrix in the presphenoid and basisphenoid elements, located below the cranial vault, n ≥ 4. Abbreviations: bs, basisphenoid; ps, presphenoid.
Loss of E2f4 affects ossification in vivo.
Bone levels are influenced by three different cell types: osteoclasts, chondrocytes and osteoblasts. Osteoclasts digest bone matrix; thus, we considered the possibility that E2F4 loss delayed bone ossification by increasing osteoclast activity. However, by performing a tartrate resistant acid phosphatase (TRAP) assay, a well-established test for osteoclast activity, we found that there was no detectable difference in osteoclast activity in the frontal calvarial bones of E2f4-null and wildtype embryos (data not shown). Chondrocytes and osteoblasts both play a positive role in bone formation. As we described above, a delay in cartilage development may contribute to the impaired ossification in the E2f4 mutants, raising the possibility of a chondrocyte defect, influencing ossification. However, this could not explain the defect in the intramembranous ossification process seen in the frontal bones of the skull, because this process does not involve a cartilage intermediate. Thus, for this study we focused our attention on osteoblasts because these are required for the differentiation of all of the bones affected by E2f4-deficiency. To eliminate any possible influence of chondrocytes, we used the frontal calvarial bones to assess osteoblast differentiation. First, we examined the expression of alkaline phosphatase (ALP), an early marker of osteoblast differentiation, at e17.5 in coronal sections through the cranium (Fig. 3A). E2f4-/- frontal bones displayed slightly less extentisve ALP activity near the central suture than the wildtype control but a significant difference in the intensity and extent of Alizarin Red staining (Fig. 3A). Thus, these data indicate that E2F4 loss disrupts ossification during or following the generation of ALP positive cells.
Figure 3.
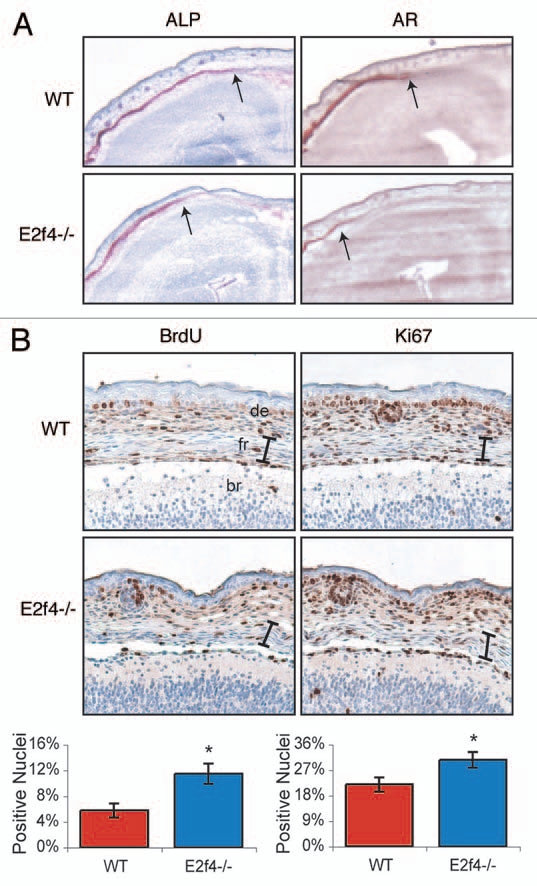
E2f4-deficient frontal bones display decreased levels of ossification and increased levels of proliferating cells. (A) Coronal sections of frontal bones from e17.5 embryos were assessed by histochemical analysis of alkaline phosphatase activity (ALP) or Alizarin Red staining of bone (AR). E2f4-/- frontal bone sections exhibit a slightly decreased extent of ALP and significantly decreased extent and intensity of AR staining in comparison with wildtype embryos (n ≥ 8). 2X magnification shown. Arrows indicate the front of activity or staining, respectively. (B) Immunohistochemical analysis of BrdU incorporation (n ≥ 5) or Ki67 protein expression (n ≥ 3) in coronal sections of frontal bones from e17.5 embryos. The frontal bones (delimited by the bracket) exhibit a greater percentage of nuclei positively staining for both BrdU and Ki67 in E2f4-/- embryos in comparison with wildtype frontal bones. 20X magnification shown. Compiled results from all experiments are quantified below the images; error bars indicate 1 SD, *p < 0.05. Abbreviations: de, dermis; fr, frontal bone; br, brain.
Given that E2F4 is implicated in promoting cell cycle exit, we hypothesized that osteoblast defects in the E2f4 mutants may be associated with a cell cycle defect. Therefore, we examined cell cycle progression in the frontal bones by assessing incorporation of the nucleotide analogue 5-Bromo-2′-deoxyuridine (BrdU) during S-phase, and also expression of Ki67, a proliferation marker. At both e16.5 and e17.5, we observed a statistically significant higher level of BrdU-positive cells in the frontal bones of E2f4-/- embryos than the wildtype littermate controls (Fig. 3B and data not shown). Consistent with this finding, a greater percentage of e17.5 E2f4-/- cells in the frontal bone stained positively for Ki67 in comparison with wildtypes (Fig. 3B). The increased number of cells cycling in the e17.5 E2f4-/- frontal bone was not associated with an apoptotic response, as determined by TUNEL staining (data not shown). Taken together, these data raise the possibility that loss of E2F4 either increases the proliferation rate of cells within the frontal bones or impairs their ability to exit the cell cycle at an appropriate developmental stage and this disrupts the appearance of Alizarin Red positive cells in vivo. Notably, by e18.5, there was no significant difference in the level of proliferation in the frontal bones of wild-type and E2f4 mutant embryos suggesting that this defect is overcome at later stages, (data not shown), correlating with the overtly normal levels of ossification seen at P0 (Fig. 1).
E2f4-/- calvarial preparations undergo ossification to a greater extent than wildtype calvarial preparations in vitro.
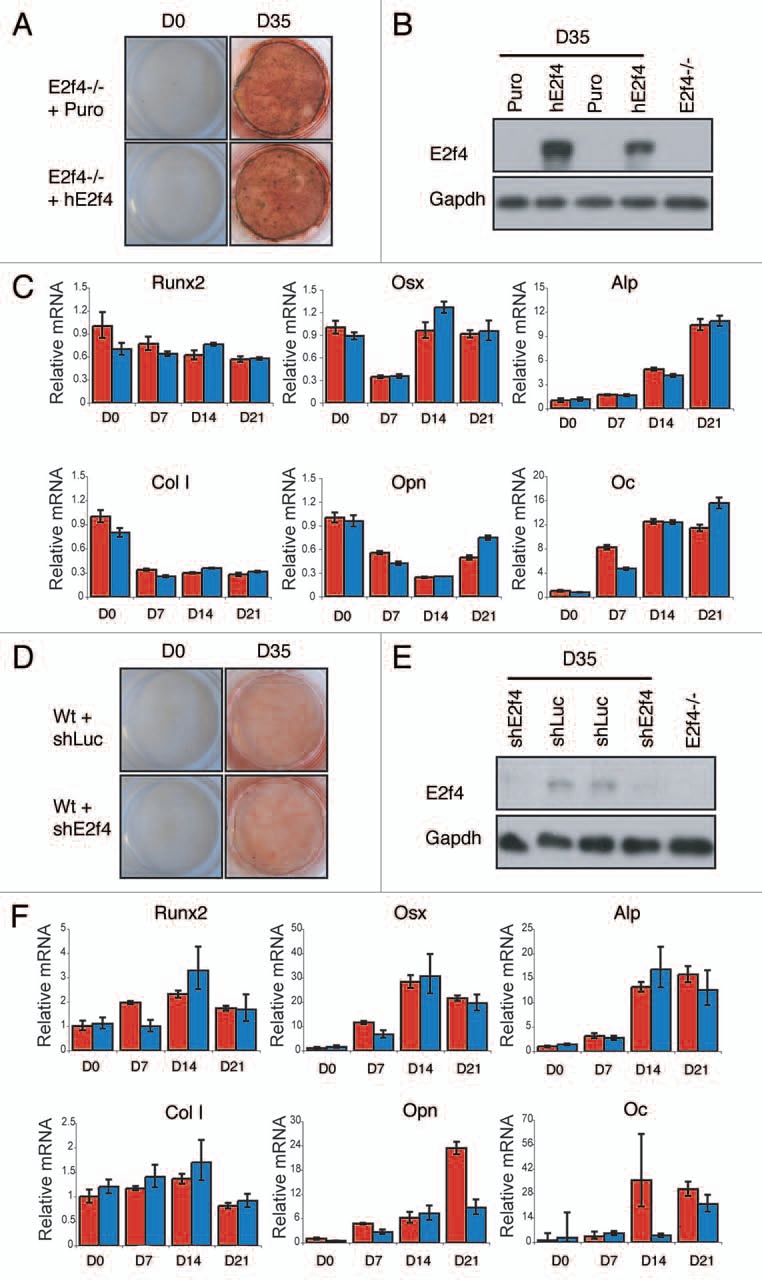
To better understand how loss of E2F4 may affect ossification we used an osteoblast in vitro differentiation assay. Specifically, we generated calvarial cell preparations from e18.5 E2f4-null and wildtype embryos and assessed their ability to secrete calcified bone matrix following induction in vitro. Unexpectedly, we found that the E2f4-deficient calvarial cell populations consistently produced a greater amount of calcified bone matrix than their wildtype littermates, as determined by Alizarin Red staining (Fig. 4A). Notably, we first observed calcified bone matrix the same number of days after the initiation of differentiation in both genotypes (data not shown), suggesting that the rate of matrix formation was similar in the wildtype and E2f4 mutant cells. To further explore this, we used quantitative RT-PCR to determine the expression levels of several osteoblast markers during the differentiation time course. For these studies, we examined Runx2 and Osterix (Osx), two transcription factors that are required for osteoblast differentiation, two early differentiation markers, alkaline phosphatase (ALP) and Collagen1 (Col1), one early/mid-marker, Osteopontin (OPN) and one late-marker, Osteocalcin (OC).24–26 These experiments showed that these genes were induced in both the E2f4-deficient cells and the wildtype controls with a comparable time course (Fig. 4B), indicating that E2F4 loss does not alter the kinetics of the differentiation process. However, the cells generated from E2f4 mutant embryos had significantly higher levels of OSX, OPN and at times, ALP compared to wildtype preparations (Fig. 4B), consistent with the increased amount of calcified matrix produced (Fig. 4A). These results demonstrate that E2f4-/- calvarial preparations express increased mRNA levels of several osteoblast differentiation markers upon induction of ossification compared to wildtype cells and accordingly generate a greater amount of calcified extracellular matrix.
Figure 4.

E2f4-/- calvarial cell populations generate more calcified matrix than wildtype calvarial cell populations, but do not display altered proliferation or cell cycle exit phenotypes. (A) ossification of primary calvarial cells was determined by Alizarin Red staining of secreted calcium deposits after 0 and 35 days of induction. E2f4-/- calvarial cells secrete a greater amount of calcium deposits than wildtype osteoblasts (n ≥ 8). (B) Quantitative RT-PCR measurements of bone marker mRNA levels from wildtype (red bars) and E2f4-/- (blue bars) calvarial cells during induction. E2f4-/- calvarial cells express higher levels of Osterix, Osteopontin and, at times, Alkaline Phosphatase mRNAs compared to wildtype cells. Ubiquitin was used as an internal control to normalize for RNA levels within each sample. Each time point is an average of three replicates. Columns, results from a representative littermate pair; error bars represent 1 SD, a representative experiment is shown (n ≥ 3). (C) Indirect immunofluorescence analysis of proliferation in calvarial cells. Wildtype and E2f4-/- calvarial cells were cultured with BrdU for 24 hours at the indicated time points during induction in vitro. DNA within nuclei is stained with DAPI (blue) and incorporated BrdU labeled by indirect immunofluorescence (green). 20X magnification shown. (D) Quantitation of proliferation, a minimum of 250 cells was analyzed per sample from four separate experiments and the percentage of BrdU positive cells calculated in each case and averaged; error bars indicate 1 SD. Wildtype (red bars) and E2f4-/- (blue bars) calvarial cell cultures display similar percentages of BrdU positive cells indicating similar levels of proliferation and cell cycle exit during the induction time course.
It is well established that cell density correlates positively with differentiation in vitro.27,28 Thus, we hypothesized that the enhanced differentiation properties of the E2f4-/- calvarial preparations might reflect an increased proliferation rate and/or a defect in their ability to exit the cell cycle and hence a higher cell density in culture. To address this possibility, we assessed the number of cells in S-phase in the wildtype and E2f4-/- calvarial cells during induction by analyzing BrdU incorporation during a 24-hour pulse. Contrary to our hypothesis, the percentages of BrdU-positive cells at each stage of the induction time-course was not statistically different between the wild-type and E2f4-/- calvarium derived cell populations (Fig. 4C and D). Moreover, in cycling populations, there was also no detectable difference in the percentage of E2f4-/- and wildtype calvarial cells that incorporated BrdU during a two-hour pulse (data not shown). Taken together, these experiments indicate that E2f4-/- calvarial cell preparations have an increased ability to generate calcified bone matrix in vitro and that this occurs without a detectable change in their proliferative capacity during either asynchronous proliferation, confluence arrest and induction of ossification.
E2f4 deficiency increases the pools of osteoblastic progenitors in vivo.
Given that E2f4 loss in vivo resulted in less ossification of various bones, we were surprised to find that E2f4-/- calvarial cell preparations have an increased potential to generate calcified bone matrix in vitro and displayed no cell cycle defect. We considered two possible hypotheses to reconcile the in vitro and in vivo phenotypes. E2F4 could play a direct role in inhibiting bone ossification that is apparent in vitro but this is obscured in vivo because E2F4 is also required for cell cycle exit. Alternatively, the increased differentiation in vitro could reflect an accumulation of the population of progenitor cells in the E2f4 mutant calvaria because their differentiation was impaired in vivo by their reduced ability to exit the cell cycle at the appropriate timepoint or another unrelated mechanism. This differentiation defect could be relieved in the in vitro induction assay. Thus, we conducted experiments that would specifically test both hypotheses.
To determine whether E2F4 plays a direct role in the induction of ossification, we used two complementary approaches. Initially, we tested the effect of restoring E2F4 in the deficient cultures by isolating E2f4-deficient calvarial cells, infecting them with either a control retrovirus or one expressing the human E2F4 cDNA and then comparing their ability to differentiate. We found that, although cells infected with human E2F4 robustly expressed E2F4 (Fig. 5B), there was no significant difference in the amount of calcium deposits secreted by these cells and those infected with empty virus as judged by Alizarin Red staining (Fig. 5A). In addition we did not observe any consistent difference in the expression of bone differentiation markers between E2f4 mutant cells with or without ectopic E2F4 expression (Fig. 5C). It seemed plausible that the E2f4 mutant cells had past a critical point in the ossification process such that the re-introduction of E2F4 was unable to reverse the defect. Therefore, we performed the converse experiment in which we isolated wildtype calvarial cells, infected them with retroviruses carrying either a hairpin against E2f4 or a control hairpin against luciferase and induced them to differentiate in vitro. Consistent with the add-back experiment, we saw no significant difference in the extent of calcified matrix deposition between either cell population despite a strong reduction of E2F4 protein levels in the knockdown cells (Fig. 5D and E). There was also no consistent difference in the expression level of several genes involved in bone differentiation between these cultures (Fig. 5F). Taken together, these experiments suggest that E2f4 loss does not modulate the intrinsic ability of calvarial osteoblasts to deposit calcified matrix in this in vitro differentiation assay.
Figure 5.
E2F4 does not modulate the induction of calcified matrix in vitro. E2f4-/- calvarial cells were stably infected with hE2F4 or an empty virus and induced to undergo bone deposition. (A) Alizarin Red staining was conducted after 0 and 35 days. No significant difference in the amount of Alizarin red staining is observed between E2f4-/- cells overexpressing hE2F4 or E2f4-/- control cells (n ≥ 6). (B) hE2F4 protein expression is maintained throughout the 35 day duration of a differentiation time course experiment as judged by western blotting. Gapdh levels in the same samples are shown as a loading control. (C) Quantitative RT-PCR (performed as described in Fig. 3B) showed no consistant difference in the expression of osteoblast markers between control cells (red bars) and cells overexpressing E2F4 (blue bars). Wildtype calvarial cells were stably infected with a hairpin against E2f4 or luciferase and induced to generate bone matrix. (D) Alizarin Red staining of secreted bone calcium deposits was conducted after 0 and 35 days. No difference is observed between wildtype cells expressing a hairpin directed against E2f4 in comparison with those expressing the luciferase control hairpin (n ≥ 6). (e) Stable infection of the E2f4 hairpin significantly reduces the level of E2F4 protein through the 35-day duration of the experiment as judged by western blotting. Gapdh levels in the same samples are shown as a loading control. (F) Quantitative RT-PCR showed no consistent in the expression of osteoblast markers between control cells (red bars) and cells with knock down of E2F4 (blue bars).
Our alternate hypothesis was that the increased in vitro differentiation potential of the E2f4-/- calvarial preparations, relative to the wildtype controls, reflects the presence of more progenitor oesteoblast cells in the E2f4 mutant calvaria due to the delayed osteoblast differentiation in vivo. To test this hypothesis, we performed an ALP-progenitor assay in which wildtype and E2f4-/- calvarial cells were plated at very low density directly into differentiation media. Colonies that stain positive for alkaline phosphatase are indicative of early osteoblast progenitor cells. Notably, starting at day 7, the E2f4 mutant calvaria derived cultures had a higher number of alkaline phosphatase-expressing colonies than the wildtype controls (Fig. 6A). To quantify this difference, we performed a limiting dilution assay and determined the number of alkaline phosphatase-positive colonies one week later. This showed that the frequency of ALP-positive colony generating cells (early osteoblast progenitors) is 1 in 21 for E2f4-/- cells versus 1 in 50 for wildtype controls (Fig. 6B). Taken together, our data strongly suggest that E2F4 loss impairs bone formation in vivo by disrupting the ability of osteoblastic progenitors to exit the cell cycle and undergo ossification at the appropriate point in development, thereby increasing the relative levels of osteoblast progenitors. These cells are inducible in vitro, accounting for the higher amounts of calcified matrix seen in E2f4 mutant versus wildtype calvarial cell cultures after induction.
Figure 6.
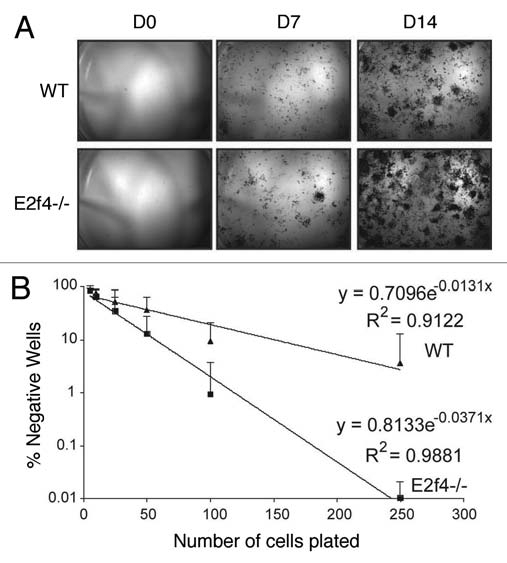
E2f4-/- calvarial preparations contain more ALp-progenitor osteoblasts than wildtype preparations. (A) Alkaline phosphatase (ALP) staining of calvarial cells. Cells isolated from calvaria were sparsely plated in induction media and stained for ALP activity at the indicated time points. E2f4-/- embryo derived calvarial cells produce more alkaline phosphatase-positive colonies, likely derived from osteoblast progenitors, than wildtype embryo derived cells (n ≥ 5). (B) Limiting dilution assay of wildtype and E2f4-/- calvarial cells. Cells were plated directly into induction media and alkaline-phosphatase-positive colony number was assessed 7 days later. The graph shows an average of 9 independent experiments, error bars indicate one standard deviation. E2f4-/- calvarial cultures exhibited a higher clonal frequency of alkaline phosphatase-positive colonies in comparison with wildtype derived cultures.
Discussion
E2F4 is considered to be primarily a repressive E2F that functions during G0/G1 to maintain growth arrest and aid cell cycle exit. E2F4 can bind to all three pocket proteins, pRb, p107 and p130 in vivo.29 Interestingly, all of these pocket proteins have been implicated in bone development. Deletion of p107 and p130 in the mouse causes an increase in chondrocyte proliferation in vivo, affecting the development of long bones,17,21,22 whereas pRb loss perturbs osteoblast differentiation in vitro and in vivo.18–20 Therefore, we hypothesized that E2F4 may have a role in bone development. Our analysis of germline mutant mice showed that E2F4 plays a key role in establishing the appropriate timing of ossification in specific skeletal elements. Further analysis of the calvarial defect shows that this correlates with increased proliferation/impaired cell cycle exit as opposed to a more direct role in the differentiation process. Specifically, we find that E2F4 loss delays ossification in vivo at an early stage, as judged by Alizarin Red staining. Accordingly, in vitro differentiation assays indicate that the E2f4-deficient calvaria retain a higher level of osteoblast progenitors than the wildtype controls. Moreover, both add-back and knockdown experiments argue against a requirement for E2f4 in the actual ossification process.
It is formally possible that the defective cell cycle exit could reflect a non-cell autonomous role for E2f4. For example, E2f4 knockout mice are anemic during embryogenesis,11 and thus the anemic state of the embryo could lead to inadequate gas exchange in tissues. In this situation, many cellular processes could be adversely affected, causing a general delay in the development of embryonic tissues. However, we favor the notion that E2f4 is required to promote cell cycle exit in early calvarial cells through its role as a transcriptional repressor of E2F-responsive genes.
E2F4 is known to maintain cell cycle arrest through its association with pocket protein family members and recruitment of histone deacetylases to E2F-responsive gene promoters to actively repress transcription. It has been previously reported that Rb-deficient embryos also display defects in bone development. Specifically, Rb inactivation leads to defects in the formation of the cranium, the hyoid bone, the palatine process and the sternum. In addition, Rb-/- calvarial osteoblasts fail to properly exit the cell cycle and, like our E2F4 studies, show an accumulation in the progenitor populations both in vivo and in vitro.19,20 Despite these similarities, there are also notable differences between the E2F4 and pRb models. First, the cell cycle exit defect seems to be stronger in the pRb mutant since, in contrast to E2F4, it persists even in vitro culture.20 Second, although both E2f4- and Rb-deficient embryos have defective bone formation in the cranium and sternum, they display unique defects in the formation of several other bones. These phenotypic differences suggest that E2f4 and Rb play overlapping, but not identical roles in bone development.
We believe that there are two non-mutually exclusive possibilities that could account for the difference in phenotype between E2f4- and Rb- deficient embryos. First, it is possible that this reflects the varying abilities of the other E2F and pocket protein members to compensate for the loss of E2F4 versus pRb in individual tissues. Previous studies showed that E2F4 loss alters the interaction between the pocket proteins and the remaining E2Fs, presumably altering their transcriptional properties.30 Moreover, it is well established that p107 and p130 can compensate for the loss of pRb to varying degrees in different settings.31,32 The relative expression levels of E2F4 and pRb, in normal tissues, and the remaining E2F and pocket proteins, in mutant tissues, would account for the specific spectrum of bone defects observed in E2f4 versus Rb mutant embryos. For example, E2F4 might be the predominant E2F in the presphenoid bone, but play a lesser role, compared to the other E2Fs, in the hyoid bone and palatine process. The alternative possibility is that while both E2F4 and pRb play indirect roles in bone development by promoting cell cycle exit, pRb plays an additional, more direct role in bone development by functioning as a co-activator for Runx2, one of the master regulators of bone development.18,33
Our data indicate that E2F4 loss disrupts both intramembranous and endochondral bone ossification. As endochondral bones ossify via a cartilage intermediate, it is possible that some of the defects in bone development could be attributed to disrupted chondrocyte differentiation. If true, we speculate that this defect would also reflect the inability of these cells to exit the cell cycle. It is interesting to note that both osteoblasts and chondrocytes arise from a common mesenchymal precursor cell. If E2F4 loss impairs cell cycle exit of osteoblast progenitor cells, thereby delaying terminal differentiation into mature osteoblasts, it is also possible that chondrocyte progenitor cells would have the same problem. Although our in vitro data suggest that E2F4 does not directly influence the deposition of calcified matrix or the induction of osteoblast specific differentiation markers, it does not rule out the possibility that E2f4 plays a direct role in the differentiation of earlier progenitor cells, such as osteochondral progenitors or even mesenchymal progenitor cells.
Materials and Methods
Animal maintenance and histological analysis.
The generation of E2f4-/- mice (MMHCC# 01XK7) has been described previously.11 All animal procedures followed protocols approved by the Institute's Committee on Animal Care. Gestation was dated by detection of a vaginal plug. For analysis of proliferation status, pregnant mice were injected with 10 µl/gm body weight of 5 mg/ml 5-Bromo-2′-deoxyuridine (BrdU, Sigma) in phosphate buffered saline (PBS) two hours prior to tissue collection. Collected embryonic tissue was immediately embedded in OCT (Tissue-Tek) or fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Histological sections were cut at 6–8 microns. Enzymatic alkaline phosphatase assays were performed on unfixed frozen sections. Briefly, 0.06 g sodium nitrite was dissolved into 1.5 ml of water and added to 600 µl of 50 mg/ml of new fuchsin (Sigma) in 2 M HCl. This solution was added to 210 ml Tris buffer (pH 9.0). Finally, 1.8 ml of 83.3 mg/ml Naphthol AS-Bi-Phosphate (Sigma) in dimethylformamide (Sigma) was added. Sections were incubated with this solution for 15 minutes, washed in PBS and counterstained with hematoxylin. Alizarin red staining was performed by incubating unfixed frozen sections for 5 minutes in 20 mg/ml alizarin red (Sigma), pH 4.2. Immunohistochemical analyses were performed using antibodies against BrdU (1:50 347580, BD Biosciences) or Ki67 (1:50 550609, BD Biosciences) as described.14 Statistical significance was determined using the two sample Student's t-test with two-tailed distribution and unequal variance.
Skeletal staining.
Embryos were euthanized, eviscerated and skinned. The remaining tissue was fixed in 95% ethanol for 4 days, transferred to acetone for 3 days, and subsequently transferred to staining solution (final volume of 0.015% alcian blue 8GX (Sigma), 0.005% alizarin red S (Sigma) and 5% glacial acetic acid in ethanol) at 37°C for two days and room temperature for a third day. Tissue was cleared in 1% potassium hydroxide for several days and ultimately stored in 80% glycerol.
Calvarial preparations and culture.
Calvarium from e17.5 or e18.5 embryos were removed, treated with several rounds of collagenase/trypsin digests at 37°C, and plated onto 6-well plates. Cells were grown and expanded in αMEM with 10% fetal bovine serum (Hyclone) and the antibiotics penicillin and streptomycin (Cellgro). For differentiation, 2.5 × 105 cells were plated onto 3 cm tissue culture plates. Upon reaching confluence, calvarium derived cells were treated with media supplemented with 50 µg/ml of ascorbic acid (Sigma) and 10 mM β-glycerol-phosphate (Sigma). To assay for calcium deposits, plates were stained with 1% alizarin red S solution (pH 5.0) for 15 minutes. For osteoblast progenitor assays, 450 cells/cm2 were plated directly into induction media. Alkaline phosphatase activity was detected by staining with BCIP/NBT Liquid Substrate System (Sigma) following the manufacturer's instructions. In limiting dilution assays, 5, 10, 25, 100 and 250 cells were plated into 96-well plates containing differentiation media and stained with BCIP/NBT Liquid Substrate System (Sigma) 7 days later. Statistical significance was determined using L-Calc Software (StemCell Technologies).
E2f4 knockdown and overexpression.
E2f4 and control luciferase directed hairpins were cloned into the MSCV-LMP retroviral vector (EAV4679 Open Biosystems). The luciferase hairpin was excised from pPRIME-CMV-GFP-FF3 (Stegmeier et al. 2005) with EcoRI and XhoI and subcloned into MSCV-LMP. The E2f4 hairpin, 5′-CAG AGA TTT AGA AAG ATT T-3′, was cloned into MSCV-LMP as described in the manufacturer's instructions (Open Biosystems). Phoenix cells at 60% confluence were transfected with 2 µg/ml MSCV-LMP. The media was replaced 8 hours later and supernatants were collected at 24 hours and filtered. Supernatants in 10% FBS and 8 µg/ml polybrene were added to calvarial cells. Infected cells were selected with 2.5 µg/ml of puromycin for 2 days. pBabe-hE2F4 was used to overexpress hE2F4. Calvarial cells were infected as described above and then selected with 2.5 µg/ml puromycin for 2 days. Knockdown and overexpression of E2F4 protein was confirmed by quantitative RT-PCR and by western, using monoclonal antibodies that recognize both human and mouse E2F4 [1:10 LLF4.229]. Gapdh antibody, (1:5,000, AM4300, Ambion) was used to detect the levels of Gapdh which was used as a loading control.
Immunofluorescence.
For indirect immunofluorescence analyses of BrdU incorporation in vitro, osteoblasts were plated onto coverslips prior to achieving confluence. BrdU was added to the media (final concentration of 10 µM) and incubated for 24 hours prior to fixation using PBS 4% in paraformaldehyde. Incorporated BrdU was detected using an antibody against BrdU (1:50 347580, BD Biosciences) and a Texas Red-X goat anti-mouse secondary (1:1,000, Invitrogen) using standard procedures. Statistical significance was determined using the Student's t Test.
Quantitative real-time PCR.
RNA was isolated from differentiating cells using the Qiagen RNeasy kit. First-strand cDNA was transcribed from 1 µg of RNA using Superscript III Reverse Transcriptase (Invitrogen) following manufacturer's instructions. Quantitative RT-PCR with 100 ng cDNA was performed using SYBR Green (Applied Biosystems). Reactions were run on the ABI Prism 7000 Sequence Detection System and analyzed using the 7000 SDS software. Primers are listed in Table 1. Statistical significance was determined using the Student's t Test.
Table 1.
Quantitative RT-PCR primer pairs
| Gene | Primer sequence |
| Alkaline Phosphatase | For: TCT CCA GAC CCT GCA ACC TC |
| Rev: CAT CCT GAG CAG ACC TGG TC | |
| Collagen1a1 | For: CGA GTC ACA CCG GAA CTT GG |
| Rev: GCA GGC AGG GCC AAT GTC TA | |
| Cyclin A | For: AGT TTG ATA GAT GCT GAC CC |
| Rev: TAG GTC TGG TGA AGG TCC | |
| Cyclin E | For: TGT TTT TGC AAG ACC CAG ATG A |
| Rev: GGC TGA CTG CTA TCC TCG CT | |
| Osteocalcin | For: CTC TGT CTC TCT GAC CTC ACA G |
| Rev: CAG GTC CTA AAT AGT GAT ACC G | |
| Osteopontin | For: TGC TTT TGC CTG TTT GGC AT |
| Rev: TTC TGT GGC GCA AGG AGA TT | |
| Osterix | For: GCA AGG CTT CGC ATC TGA AA |
| Rev: AAC TTC TTC TCC CGG GTG TGA | |
| Runx2 | For: TGA GAT TTG TGG GCC GGA |
| Rev: TCT GTG CCT TCT TGG TTC CC | |
| Ubiquitin | For: TGG CTA TTA ATT ATT CGG TCT GCA T |
| Rev: GCA AGT GGC TAG AGT GCA GAG TAA |
Acknowledgements
We are grateful to the members of the Lees lab, especially Paul Danielian, for helpful discussion throughout this work. We also thank the KI Histology Core Facility, especially Alicia Caron, for the generation of histological sections. This project was supported by NIH grants awarded to J.A.L. (GM53204, CA121921). E.M. is an NIH Graduate Fellow, S.B. is a David H. Koch and NIH Graduate Fellow, J.A.L. is a Ludwig Scholar at MIT.
Abbreviations
- ALP
alkaline phosphatase
- BrdU
5-bromo-2′-deoxyuridine
- WT
wild-type
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/12108
References
- 1.Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene. 2005;24:2810–2826. doi: 10.1038/sj.onc.1208612. [DOI] [PubMed] [Google Scholar]
- 3.Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6:729–735. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- 4.Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verona R, Moberg K, Estes S, Starz M, Vernon JP, Lees JA. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol Cell Biol. 1997;17:7268–7282. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blais A, Dynlacht BD. E2F-associated chromatin modifiers and cell cycle control. Curr Opin Cell Biol. 2007;19:658–662. doi: 10.1016/j.ceb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev. 2000;14:804–816. [PMC free article] [PubMed] [Google Scholar]
- 8.Wells J, Boyd KE, Fry CJ, Bartley SM, Farnham PJ. Target gene specificity of E2F and pocket protein family members in living cells. Mol Cell Biol. 2000;20:5797–5807. doi: 10.1128/mcb.20.16.5797-5807.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 10.McClellan KA, Slack RS. Specific in vivo roles for E2Fs in differentiation and development. Cell Cycle. 2007;6:2917–2927. doi: 10.4161/cc.6.23.4997. [DOI] [PubMed] [Google Scholar]
- 11.Humbert PO, Rogers C, Ganiatsas S, Landsberg RL, Trimarchi JM, Dandapani S, et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell. 2000;6:281–291. doi: 10.1016/s1097-2765(00)00029-0. [DOI] [PubMed] [Google Scholar]
- 12.Rempel RE, Saenz-Robles MT, Storms R, Morham S, Ishida S, Engel A, et al. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell. 2000;6:293–306. doi: 10.1016/s1097-2765(00)00030-7. [DOI] [PubMed] [Google Scholar]
- 13.Kinross KM, Clark AJ, Iazzolino RM, Humbert PO. E2f4 regulates fetal erythropoiesis through the promotion of cellular proliferation. Blood. 2006;108:886–895. doi: 10.1182/blood-2005-09-008656. [DOI] [PubMed] [Google Scholar]
- 14.Danielian PS, Bender Kim CF, Caron AM, Vasile E, Bronson RT, Lees JA. E2f4 is required for normal development of the airway epithelium. Dev Biol. 2007;305:564–576. doi: 10.1016/j.ydbio.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landsberg RL, Sero JE, Danielian PS, Yuan TL, Lee EY, Lees JA. The role of E2F4 in adipogenesis is independent of its cell cycle regulatory activity. Proc Natl Acad Sci USA. 2003;100:2456–2461. doi: 10.1073/pnas.0138064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell. 2002;3:39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 17.Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, et al. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 18.Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8:303–316. doi: 10.1016/s1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- 19.Gutierrez GM, Kong E, Sabbagh Y, Brown NE, Lee JS, Demay MB, et al. Impaired bone development and increased mesenchymal progenitor cells in calvaria of RB1-/- mice. Proc Natl Acad Sci USA. 2008;105:18402–18427. doi: 10.1073/pnas.0805925105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman SD, Yuan TL, Miller ES, Lee EY, Caron A, Lees JA. The retinoblastoma protein tumor suppressor is important for appropriate osteoblast differentiation and bone development. Mol Cancer Res. 2008;6:1440–1451. doi: 10.1158/1541-7786.MCR-08-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J Cell Biol. 2002;158:741–750. doi: 10.1083/jcb.200205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rossi F, MacLean HE, Yuan W, Francis RO, Semenova E, Lin CS, et al. p107 and p130 Coordinately regulate proliferation, Cbfa1 expression and hypertrophic differentiation during endochondral bone development. Dev Biol. 2002;247:271–285. doi: 10.1006/dbio.2002.0691. [DOI] [PubMed] [Google Scholar]
- 23.Wagner EF, Karsenty G. Genetic control of skeletal development. Curr Opin Genet Dev. 2001;11:527–532. doi: 10.1016/s0959-437x(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 24.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 25.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 26.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 27.Gerber I, ap Gwynn I. Influence of cell isolation, cell culture density and cell nutrition on differentiation of rat calvarial osteoblast-like cells in vitro. Eur Cell Mater. 2001;2:10–20. doi: 10.22203/ecm.v002a02. [DOI] [PubMed] [Google Scholar]
- 28.Purpura KA, Aubin JE, Zandstra PW. Sustained in vitro expansion of bone progenitors is cell density dependent. Stem Cells. 2004;22:39–50. doi: 10.1634/stemcells.22-1-39. [DOI] [PubMed] [Google Scholar]
- 29.Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol. 1996;16:1436–1449. doi: 10.1128/mcb.16.4.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell. 2002;2:463–472. doi: 10.1016/s1535-6108(02)00207-6. [DOI] [PubMed] [Google Scholar]
- 31.Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–228. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 32.Wikenheiser-Brokamp KA. Rb family proteins differentially regulate distinct cell lineages during epithelial development. Development. 2004;131:4299–4310. doi: 10.1242/dev.01232. [DOI] [PubMed] [Google Scholar]
- 33.Luan Y, Yu XP, Xu K, Ding B, Yu J, Huang Y, et al. The retinoblastoma protein is an essential mediator of osteogenesis that links the p204 protein to the Cbfa1 transcription factor thereby increasing its activity. J Biol Chem. 2007;282:16860–16870. doi: 10.1074/jbc.M610943200. [DOI] [PubMed] [Google Scholar]