

Abstract
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors are comprised of different combinations of GluA1–GluA4 (also known asGluR1–GluR4 and GluR-A to GluR-D) subunits. The GluA2 subunit is subject to RNA editing by the ADAR2 enzyme, which converts a codon for glutamine (Gln; Q), present in the GluA2 gene, to a codon for arginine (Arg; R) found in the mRNA. AMPA receptors are calcium (Ca2+)-permeable if they contain the unedited GluA2(Q) subunit or if they lack the GluA2 subunit. While most AMPA receptors in the brain contain the edited GluA2(R) subunit and are therefore Ca2+-impermeable, recent evidence suggests that Ca2+-permeable AMPA receptors are important in synaptic plasticity, learning, and disease. Strong evidence supports the notion that Ca2+-permeable AMPA receptors are usually GluA2-lacking AMPA receptors, with little evidence to date for a significant role of unedited GluA2 in normal brain function. However, recent detailed studies suggest that Ca2+-permeable AMPA receptors containing unedited GluA2 do in fact occur in neurons and can contribute to excitotoxic cell loss, even where it was previously thought that there was no unedited GluA2.This review provides an update on the role of GluA2 RNA editing in the healthy and diseased brain and summarizes recent insights into the mechanisms that control this process. We suggest that further studies of the role of unedited GluA2 in normal brain function and disease are warranted, and that GluA2 editing should be considered as a possible contributing factor when Ca2+-permeable AMPA receptors are observed.
Keywords: AMPA receptor, GluA2, GluR2, GluR-B, RNA editing, adenosine deaminases acting on RNA, excitotoxicity
INTRODUCTION
The release of the neurotransmitter glutamate from presynaptic densities activates postsynaptic glutamate-gated ion channels including α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, N-methyl D-aspartate (NMDA) receptors and kainate (KA) receptors. Once activated, the glutamate-gated ion channels flux sodium (Na+) and in some cases will also flux calcium (Ca2+). NMDA receptors are uniformly Ca2+-permeable, with well-recognized roles in synaptic plasticity and disease. Most AMPA receptors and some KA receptors are Ca2+-impermeable, however Ca2+-permeable AMPA receptors (Jia et al., 1996; Lamsa et al., 2007) and KA receptors (Vissel et al., 2001; Lerma, 2003; Sun et al., 2009) also exist at some synapses where they can play a role in synaptic plasticity and disease (Gorter et al., 1997; Pellegrini-Giampietro et al., 1997; Liu and Zukin, 2007). This review focuses on the critical role of glutamine/arginine (Q/R) site RNA editing of the GluA2 subunit in regulating AMPA receptor Ca2+-permeability.
AMPA receptors are tetrameric assemblies of different combinations of four subunits designated GluA1–GluA4 (alternatively known as GluR1–GluR4 and GluR-A to GluR-D; reviewed in Hollmann and Heinemann, 1994; Dingledine et al., 1999; Sobolevsky et al., 2009). The Ca2+-permeability of AMPA receptors varies depending on whether the GluA2 subunit is present within the tetramer. The ability of the GluA2 subunit to regulate Ca2+-permeability of AMPA receptors in turn depends on RNA editing. RNA editing is a post-transcriptional modification that alters a codon encoding glutamine (Gln; Q) to a codon encoding arginine (Arg; R) in the GluA2 mRNA. AMPA receptors are Ca2+-impermeable if they contain the edited GluA2(R) subunit. Conversely, AMPA receptors are Ca2+-permeable if they are GluA2-lacking or if they contain the unedited GluA2(Q) subunit.
Studies to date provide strong evidence that GluA2-lacking AMPA receptors contribute to normal brain function and disease (Pellegrini-Giampietro et al., 1997; Isaac et al., 2007; Wiltgen et al., 2010; Man, 2011). Additional to this, almost no unedited RNA exists in the brain; less than 1% of all RNA on average across the gray matter of the brain encodes unedited GluA2(Q) (Kawahara et al., 2003). Taken together, the evidence has largely been accepted to suggest that unedited GluA2 does not play a major role in brain function and disease. More recently, however, sophisticated studies have revealed a possible role for unedited GluA2 in regulating excitotoxic neuronal cell death in ischemia and ALS (reviewed in Kwak and Kawahara, 2005; Kwak and Weiss, 2006; Liu and Zukin, 2007; Kwak et al., 2010). This is perhaps surprising given many earlier studies previously ruled out a role for unedited GluA2 in ischemia (Akbarian et al., 1995; Rump et al., 1996; Kortenbruck et al., 2001). These recent studies demonstrate that whilst only ~1% GluA2 RNA is unedited on average across the brain, it can vary substantially from cell-to-cell reaching much higher levels in some individual cells. Thus unedited GluA2 RNA is difficult to detect. This raises the possibility of an as-yet undetected role for unedited GluA2 in specific aspects of physiology and in other diseases.
The original discovery of AMPA receptor RNA editing established the critical implications of this type of post-transcriptional modification to genes. As such, RNA editing has now been extensively investigated throughout the mammalian genome (Mattick and Mehler, 2008). In this review, we will explore the role of GluA2 RNA editing and outline new insights into the mechanisms that control this process. We will further discuss the role of GluA2-lacking receptors and GluA2 unedited receptors and suggest that understanding the role of unedited GluA2(Q) containing receptors, and the mechanisms that regulate RNA editing, will become increasingly important for understanding normal brain function and excitotoxic cell death. We propose that, despite the low percentage of unedited GluR2 mRNA present in the brain, it is not yet possible to rule out a critical, and substantial, role of unedited GluA2 in health and disease.
THE GluA2 SUBUNIT OF THE AMPA RECEPTOR PLAYS A KEY ROLE IN NORMAL CENTRAL NERVOUS SYSTEM FUNCTION
Glutamate, the major excitatory neurotransmitter in the central nervous system (CNS), activates AMPA, NMDA, and KA receptors and can potentially cause influx of Ca2+ into the postsynaptic density (Figure 1A). Conventionally, NMDA receptors were considered to be the primary glutamate receptor involved in synaptic plasticity and Ca2+-mediated excitotoxic cell death in the healthy and diseased brain, respectively. However, abundant evidence now suggests that Ca2+-permeable AMPA receptors are also involved in synaptic plasticity (Malinow and Malenka, 2002; Cull-Candy et al., 2006; Isaac et al., 2007; Liu and Zukin, 2007), play a role in memory and learning (Riedel et al., 1999; Wiltgen et al., 2010), and contribute to excitotoxic cell loss in disease (Pellegrini-Giampietro et al., 1997; Tanaka et al., 2000; Kwak and Weiss, 2006).
FIGURE 1.
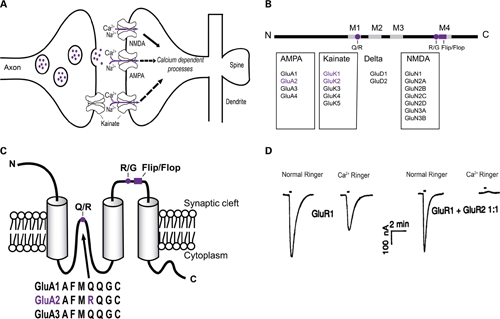
Schematic view of AMPA, NMDA, and KA receptors within the postsynaptic density and the AMPA receptor structure. (A) Glutamate in the synaptic cleft can act on ionotropic receptors including AMPA, NMDA, and KA receptors causing a Ca2+ influx leading to intracellular signaling cascades. (B) Representation of the primary structure of AMPA receptor subunits with N- and C-terminals, four membranes, M1–M4 (gray boxes), two editing sites, Q/R and R/G (violet dots), and alternate spliced flip/flop site (violet box). A summary of glutamate receptors and their subunits are also shown. Subunits shown in violet are subunits that undergo RNA editing at the Q/R site. (C) Topology (secondary structure) of the GluA2 subunit with three transmembrane domains. The Q/R RNA editing site controls Ca2+-permeability of the AMPA receptor site and is located in the putative second membrane domain. The R/G site and flip/flop sites, which control desensitization of the receptor, are located prior to the fourth membrane domain. The protein sequence of GluA2 varies by one codon, arginine (Arg), from GluA1, GluA3, and GluA4, which express a glutamine (Gln). The arginine is post-transcriptionally introduced by an amino acid change in the pre-mRNA of GluA2 by RNA editing. (D) Current response in normal Ringer solution and Ca2+-Ringer solution of oocytes injected with GluA1 cRNA (left) or GluA1+GluA2 cRNA (right). Oocytes containing GluA2 eliminated the inward current, thus showing GluA2 regulates Ca2+-permeability of the AMPA receptor. Reprinted from Hollmann et al. (1991), with permission.
SUBUNIT COMPOSITION DETERMINES AMPA RECEPTOR PROPERTIES AND CHARACTERISTICS
The four subunits of AMPA receptors, GluA1–GluA4, share 68–73% sequence identity. Each subunit consists of a large extracellular N-terminus domain, an intracellular C-domain and three transmembrane domains (M1, M3, and M4; Figure 1B). The putative second membrane domain (M2) consists of a hairpin structure, which changes direction within the membrane and returns to the intracellular side of the cell (Figure 1C). Each subunit exists in two forms created by alternative splicing, termed “flop” and “flip,” which is an interchangeable sequence consisting of 38-amino acids found prior to the fourth membrane domain. Each form is expressed preferentially in different regions of the brain (Hollmann and Heinemann, 1994). The kinetic properties of the AMPA receptors are regulated by the splice variants of the subunits. For example, the flop variants of GluA2–GluA4 desensitize faster than flip variants, but recover more slowly (Sommer et al., 1990). On the other hand, GluA1 flip and flop variants desensitize at equal rates depending on the concentration of glutamate (Mosbacher et al., 1994). Within the hippocampus and cerebral neocortex, the majority of AMPA receptors contain GluA2, which predominantly forms heteromers with GluA1 (Wenthold et al., 1996; Greger et al., 2002). Lower levels of GluA3 and GluA4 are expressed in these areas (Wenthold et al., 1996). In the absence of the GluA2 subunit, AMPA receptors often consist of GluA1/GluA3 hetero-oligomers, which may lead to reduced expression of the AMPA receptor at the synapse (Sans et al., 2003).
THE GluA2 SUBUNIT REGULATES CALCIUM PERMEABILITY AND TRAFFICKING OF AMPA RECEPTORS
The ability of Ca2+ to enter the cell through the AMPA receptor is determined by the GluA2 subunit, which is preferentially incorporated into the receptor (Sans et al., 2003). When AMPA receptors are assembled from combinations of GluA1, GluA3, and GluA4 the receptors are highly permeable to Ca2+. However, when GluA2 is contained within the AMPA receptor, the Ca2+-permeability is profoundly decreased (Figure 1D; Hollmann et al., 1991). Thus, since the vast majority of AMPA receptors are hetero-oligomers consisting of GluA1/GluA2 or GluA2/GluA3 subunits, they are Ca2+-impermeable (Wenthold et al., 1996).
The subunit composition of AMPA receptors is important as it affects not only Ca2+-permeability, but also the trafficking of the receptors (Malinow and Malenka, 2002). AMPA receptors are assembled in the endoplasmic reticulum (ER) and are trafficked to the plasma membrane and their presence at the synapse is in a dynamic equilibrium between insertion (exocytosis) and removal (endocytosis; Malinow and Malenka, 2002; Keifer and Zheng, 2010; Man, 2011). The presence of the GluA2 subunit is important for the stability and trafficking of AMPA receptors within the synapse, though it is not essential (Biou et al., 2008; Panicker et al., 2008). The C-terminus of GluA2 binds with, among other proteins, N-ethylmaleimide-sensitive factor (NSF), an ATPase, which is involved in the insertion of the AMPA receptor into the synapse and synaptic activation of the receptor (reviewed in Bassani et al., 2009). In fact, A synchrony of complex intracellular mechanisms drives AMPA receptor trafficking (Ziff, 2007; Kessels and Malinow, 2009; Jackson and Nicoll, 2011). As we shall discuss, GluA2 RNA editing at the Q/R site can also regulate trafficking (discussed in Section “GluA2 RNA Editing has a Role in AMPA Receptor Trafficking”).
THE IMPORTANCE OF GluA2 IN THE TETRAMERIC ASSEMBLY OF THE AMPA RECEPTOR
As we note above, AMPA receptors are generally considered to be tetramer assemblies of various combinations of GluA1–GluA4 (Rosenmund et al., 1998; Nakagawa, 2010). The GluA1–GluA4 subunits consist of an extracellular amino-terminal domain, which is involved in receptor assembly and trafficking, a ligand binding domain and a transmembrane domain, which form the ion channel (Figure 2A). GluA2 subunit assembly begins with the formation of dimers in which the amino-terminal and transmembrane terminal domain segments are coupled. Sobolevsky et al. (2009) have been able to determine the structure of elements of the GluA2 AMPA receptor ion channel tetramer, using X-ray diffraction data. Their study reveled the receptor has a twofold axis of molecular symmetry and within the ion channel domain there is a fourfold axis of rotational symmetry (Sobolevsky et al., 2009).
FIGURE 2.
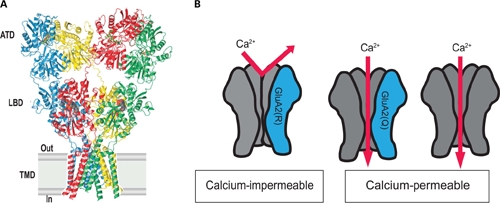
AMPA receptors containing GluA2 are Ca2+-impermeable due to editing at the Q/R site. (A) Current understanding of the quaternary structure of AMPA receptors, thought to be tetrameric assemblies of four AMPA receptor subunits form the ion channel pore. The assembled receptor can comprise any combination of GluA1–GluA4 subunits but usually contains GluA2. Reprinted from Sobolevsky et al. (2009), with permission. (B) AMPA receptors generally contain GluA2 (red) with combinations of GluA1, GluA3, and GluA4 (blue). AMPA receptors containing edited GluA2 are impermeable to Ca2+ (left). AMPA receptors containing unedited GluA2 and AMPA receptors lacking GluA2 are Ca2+-permeable (right).
The crystal structure does not as-yet fully resolve the gate or the Q/R site within the membrane. However it is known that tetrameric assemblies of GluA1–GluA4 subunits form a large pore with a diameter of approximately 0.78 nm (Burnashev et al.,1996). When GluA2 is present within the receptor, the Q/R site is located within the cytoplasmic side of the channel pore, immediately below the transmembrane central cavity (Sobolevsky et al., 2009). Interestingly, the amino acid residue present at the Q/R site does not affect the diameter of the pore. When Gln is present, a negative electrostatic potential is formed with the N-terminus of the M2 loop and thus attracts cations. However, the replacement of the neutral Gln with a large positively charged Arg residue neutralizes this potential and thus prevents the diffusion of divalent cations through the channel (Kuner et al., 2001). It is still unclear, however, how many Arg are required to prevent cation diffusion. This raises the question of how anywhere from one to four Arg’s present at the membrane pore can become selective for Na+ over Ca2+. Further studies are required to determine why the process of RNA editing at only one subunit can dramatically alter the Ca2+-permeability of the receptor, while four GluA2(R) subunits can also have exactly the same effect.
DISCOVERY OF GluA2 RNA EDITING AND ITS NECESSITY FOR AMPA RECEPTOR FUNCTION
The Heinemann group was the first to show that the GluA2 subunit is an essential role determinant of the Ca2+-permeability of AMPA receptors (Hollmann et al., 1991; Hume et al., 1991). The investigators further showed that the effect of GluA2 on AMPA receptor Ca2+-permeability results from a single amino acid present at position 607 in GluA2 mRNA (Figure 1C). Specifically, they discovered that GluA2 mRNA encodes an Arg at position 607, while the equivalent position in GluA1, GluA3, and GluA4 subunit mRNA encodes a Gln. The investigators used site-directed mutagenesis to alter the Arg in GluA2 mRNA to a Gln, as found in the other subunits (Hume et al., 1991). They showed that GluA2(Q) subunits self assembled into functional Ca2+-permeable AMPA receptors while GluA2(R) subunits self assembled into AMPA receptors with low Ca2+-permeability (Hume et al., 1991). Thus, these studies established categorically that the Arg at position 607 of GluA2 is the critical determinant of AMPA receptor Ca2+-permeability.
It was later discovered, somewhat remarkably, by Sommer et al. (1991) in Seeburg’s group, that the Arg at position 607 is in fact not encoded in the DNA but is introduced post-transcriptionally. The investigators found that the codon encoding the critical Arg at position 607 was present in the GluA2 cDNA, however the GluA2 gene DNA sequence at this position encoded a Gln (Sommer et al., 1991). They then showed through a series of elegant studies that the codon for Arg is introduced into the mRNA by RNA editing. Specifically, RNA editing converts an adenosine (A) in the critical CAG codon (encoding Gln) found in the pre-mRNA to an inosine (I), which creates a CIG codon (encoding Arg) in the mRNA (Melcher et al.,1996). Since ribosomes read the inosine as a guanosine (G),the editing effectively alters the CAG codon (encoding Gln) to a CGG codon (encoding Arg). The resulting edited GluA2(R) subunit prevents Ca2+ influx when it is incorporated in the AMPA receptor. Conversely, AMPA receptors containing unedited GluA2(Q) are highly permeable to Ca2+ (summarized in Figure 2B). Unedited GluA2 at the Q/R site can cause AMPA receptor-mediated excitotoxicity, a process we shall discuss later in this review.
Notably, GluA2 RNA editing at the Q/R site occurs in ~99% of cases where GluA2 is present. The editing process is reliant upon an intronic sequence called the editing complementary sequence (ECS), which is located in intron 11 downstream of the editing site (Higuchi et al., 1993). This ECS forms a dsRNA structure with the editing site in the pre-mRNA. The primary RNA editing enzyme, adenosine deaminase acting on RNA (ADAR), identifies this dsRNA structure and alters the CAG codon encoding Gln to a CIG codon encoding Arg, though it is still unclear how this site recognition occurs (Dabiri et al., 1996). Currently, three ADAR family members have been identified (ADAR1–3), in which ADAR2 is the primary modifier of the GluA2Q/R site (Melcher et al., 1996; Bass, 2002). All three ADARs are preferentially expressed in the nervous system. Interestingly, ADAR3 is exclusively expressed in the brain, however its function remains unknown and it may play a regulatory role in RNA editing (Chen et al., 2000).
ADAR2 also edits GluA2 at amino acid position 764, which alters a codon for Arg to a glycine codon (R/G site). The R/G editing site, but not the Q/R site, is also found in GluA3 and GluA4. The R/G site, along with the flip/flop, is involved in desensitization and recovery of the receptor, though is not involved in Ca2+-permeability. Receptors containing GluA2 edited at the R/G site are able to recover from desensitization more rapidly than receptors containing unedited GluA2 at the R/G site. Interestingly, despite this site also being edited by ADAR2, approximately only 90% of RNA is edited at the R/G site of GluA2, and this is lower during development (Lomeli et al., 1994). Both the Q/R site and the R/G site are regulated by ADAR2, yet the extent of editing appears to be regulated differentially between the two sites. It is still unclear as to how and why developmental regulation at the R/G site occurs. However, it appears that currently unidentified mechanisms differentially regulate editing at the Q/R and R/G sites. It remains to be determined if glutamate signaling regulates editing in some way, similar to serotonin regulation of RNA editing of the 5-HT 2C RNA (Burns et al., 1997).
There have been extensive efforts to identify RNA editing sites in the mammalian genome since the discovery of GluA2 RNA editing (Bass, 2002; Mattick and Mehler, 2008). RNA editing was observed early on in the GluK1 and GluK2 subunits of KA receptors, but has so far not been found to occur in NMDA receptors. Recent studies have provided evidence of extensive editing throughout the mammalian genome (Mattick and Mehler, 2008). A summary of editing sites in glutamate receptors is shown in Table 1.
Table 1.
Summary of ionotropic glutamate receptor editing sites and their effects.
| Receptor | Subunit | Site | Editing | Consequence |
|---|---|---|---|---|
| AMPA | GluA2 | Q/R site M2 | A→I | ↓Ca2+-permeability |
| ↓Conductance | ||||
| ↓Spermine block | ||||
| ↓Blockade by toxins | ||||
| GluA2,3,4 | R/G site between M3 and M4 | A→I | ↑Recovery from desensitization | |
| Kainate | GluK1,2 | Q/R site M2 | A→I | ↓Ca2+-permeability |
| ↓Conductance | ||||
| ↓Spermine block | ||||
| ↓Blockade by toxins | ||||
| GluK2 | I/V site M1 | A→I | ↓Ca2+-permeability | |
| Y/C site M1 |
CHARACTERISTICS OF CALCIUM-PERMEABLE AMPA RECEPTORS
AMPA receptors can be Ca2+-permeable because they are GluA2-lacking or because they contain the unedited GluA2(Q) subunit. Ca2+-permeable AMPA receptors can be detected by Ca2+ imaging, but more often than not these receptors are detected using electrophysiology. Unfortunately, however, current techniques are unable to differentiate between GluA2-lacking receptors and AMPA receptors containing unedited GluA2 at the synapse.
AMPA receptors have the same characteristics whether they are GluA2-lacking or whether they contain the unedited GluA2(Q) subunit: First, they are Ca2+-permeable. Second, the AMPA receptors exhibit inward rectification (decreased conductance of AMPA receptors’ synaptic currents’ potentials from positive to 0 mV) when the polyamine, spermine, is diffused into the cell from the recording electrode; this is measured as an altered rectification index. Third, Ca2+-permeable AMPA receptors become selectively sensitive to block by Joro spider toxin (JSTX) and to related drugs such as 1-naphthylacetyl spermine (NAS) and IEM-1460. Sometimes these changes are also associated with AMPA receptor-mediated synaptic transmission being enhanced, which is detected as an increased ratio of AMPA/NMDA receptor-mediated synaptic currents.
While Ca2+ permeable AMPA receptors can be identified by electrophysiology, it is not possible, by any currently available methods to directly distinguish GluA2-lacking AMPA receptors from AMPA receptors containing unedited GluA2 subunits. Unedited GluA2 is, at present, only identified by analyzing RNA from specific cells or brain regions using one of three methods: (1) A “primer extension” assay (Schiffer and Heinemann, 1999; Vissel et al., 2001), (2) DNA sequencing (which measures the ratio of edited G nucleotide peak to unedited A nucleotide peak in total cDNA; Maas et al., 2001), and (3) restriction enzyme based assays, usually using the enzyme BbvI (Kawahara et al., 2004a; Peng et al., 2006).
The only way to infer the presence of functional unedited GluA2 containing AMPA receptors is to combine electrophysiology with an RNA editing assay. In general, this has been achieved by studying neurons with patch clamp electrophysiology to demonstrate the presence of Ca2+-permeable AMPA receptors. Following this, the nucleus is extracted into the patch electrode and the RNA is isolated. Single cell PCR is performed in combination with an editing assay to demonstrate the percentage of edited and unedited GluA2 mRNA within the cell (Peng et al.,2006). If unedited GluA2 mRNA is found to be present concurrently with Ca2+-permeable AMPA receptors it suggests that AMPA receptors contain unedited GluA2 at the synapse. Conversely, if the RNA in a cell does not contain unedited GluA2 then it can be inferred that Ca2+-permeable receptors are GluA2-lacking.
In most studies, however, rarely are editing analyses carried out when Ca2+-permeable AMPA receptors are observed at synapses. Since GluA2 is almost 100% edited in the normal brain, it is generally assumed that unedited GluA2 does not play a significant role in normal brain function. Therefore, while AMPA receptors lacking GluA2 and AMPA receptors containing unedited GluA2 show similar properties, the appearance of Ca2+-permeable AMPA receptors at synapses – usually detected by electrophysiology – is generally interpreted to mean the GluA2-lacking AMPA receptor has been inserted at the synapse, rather than AMPA receptors containing unedited GluA2.
It remains a critical problem that it is difficult to differentiate edited GluA2(R) protein from unedited GluA2(Q) protein at synapses. Many studies have shown that in circumstances where Ca2+-permeable AMPA receptors exist, GluA2 expression is reduced relative to expression of GluA1. This is consistent with the notion that the observed AMPA receptors are Ca2+-permeable because they are GluA2-lacking. However, this does not experimentally rule out that GluA2 unedited receptors may also be present and contribute to Ca2+-permeability in parallel to GluA2-lacking receptors. For example, as we shall discuss, GluA2-lacking receptors have been observed and implicated in the excitotoxic cell loss following brain ischemia (Pellegrini-Giampietro et al., 1992; Calderone et al., 2003). More recently it was found that unedited GluA2 also occurs in ischemia and that this is critical to the excitotoxic neurodegeneration that follows (Peng et al., 2006). Consequently recent data suggests that the Ca2+-permeable receptors following ischemia result at least in part from unedited GluA2. This leaves open a largely unexplored possibility that in other scenarios where Ca2+-permeable receptors are observed, unedited GluA2 may play an as-yet unidentified role, even though previous evidence suggests such receptors are GluA2-lacking.
TO DATE, GluA2-LACKING, BUT NOT UNEDITED GluA2-CONTAINING, AMPA RECEPTORS ARE KNOWN TO PLAY A ROLE IN ENHANCING SYNAPTIC PLASTICITY
We will only briefly touch on the role of GluA2-lacking receptors in this review for the purpose of noting that this form of Ca2+-permeable AMPA receptors are critical in brain function (Cull-Candy et al., 2006; Isaac et al., 2007; Liu and Zukin, 2007; Man, 2011). While there is no known role of unedited GluA2 in normal brain function, there is now thought to be a role for GluA2-lacking AMPA receptors. GluA2-lacking AMPA receptors offer an alternative route to NMDA receptors for Ca2+ flux into cells at synapses and are now recognized to play an important role in synaptic plasticity by triggering LTP (Jia et al., 1996; Harvery et al., 2001; Meng et al., 2003; Lu et al., 2007; Asrar et al., 2009; Wiltgen et al., 2010). Notably however, the properties of AMPA receptor-mediated LTP is different to NMDA receptor-mediated LTP (Lamsa et al., 2007; Kullmann and Lamsa, 2008).
Due to their distinct physiological effects, GluA2-lacking receptors have recently been discovered to be important in learning and memory. Wiltgen et al. (2010) showed that NMDA receptor-independent learning was not impaired in mice with a selective GluA2 KO in the CA1 region of the hippocampus. However, by blocking Ca2+-permeable AMPA receptors during an NMDA receptor-independent learning process, NMDA receptor-independent learning was significantly impaired (Wiltgen et al., 2010). Further, Clem and Barth (2006) demonstrated that evoked activity of a single spared whisker (after removal of the others) increases the trafficking of GluA2-lacking receptors into the barrel column of the spared whisker, suggesting that a sensory experience can increase Ca2+-permeable AMPA receptors and strengthen neocortical excitatory synapses (Clem and Barth, 2006). In addition, GluA2-lacking receptors are increased in the ventral tegmental area of mice following a single injection of cocaine and are thought to contribute to cocaine-evoked plasticity (Bellone and Luscher, 2006), thus contributing to time dependent seeking of cocaine following self administration (Conrad et al., 2008). These studies suggest that GluA2-lacking receptors may be involved in synaptic strengthening and are essential for NMDA receptor-independent learning and processes in the healthy brain.
As noted above, it has generally been concluded that the occurrence of Ca2+-permeable receptors results from the presence of GluA2-lacking receptors, rather than unedited GluA2. It is plausible, however, that like GluA2-lacking receptors, there may indeed be a role for unedited GluA2 in brain processes such as LTP and LTD that has not yet been determined. As we will now show, however, while the role of unedited GluA2 in the healthy brain remains unknown, recent evidence suggests that GluA2 RNA editing is essential for survival and unedited GluA2 plays an important role in neuronal loss in certain diseases.
MICE ENGINEERED WITH GluA2 MUTATIONS REVEAL AMPA RECEPTORS CONTAINING UNEDITED GluA2 HAVE DISTINCT PHYSIOLOGICAL EFFECTS
While GluA2-lacking receptors appear to have a role in synaptic plasticity, unedited GluA2 receptors have a fundamentally different physiological effect. Mice in which GluA2 is ablated (GluA2 KO mice) have Ca2+-permeable AMPA receptors made up of the GluA1, GluA3, and GluA4 subunits. These mice are viable, and live to an old age, suggesting that the presence of Ca2+-permeable AMPA receptors may not be intrinsically excitotoxic. In contrast, mice engineered with reduced RNA editing show a profoundly different phenotype. Brusa et al. (1995) engineered mice in which the ECS is replaced with a loxP sequence (GluA2ΔECS/+ mice). The lack of the ECS in the intron means GluA2 Q/R RNA editing was impaired. This resulted in approximately 25% unedited GluA2 mRNA at the Q/R site compared to age-matched wild-type littermates, which have almost no unedited GluA2 mRNA. In contrast to Ca2+-permeable AMPA receptors in GluA2 KO mice (Jia et al., 1996; Sans et al., 2003; Wiltgen et al., 2010), the presence of an increased number of unedited GluA2 containing AMPA receptors in mice lead to seizures and premature death by several weeks of age (Brusa et al., 1995).
In addition to the GluA2 ΔECS/+ mice engineered by Brusa et al. (1995), Feldmeyer et al. (1999) developed mice with varying degrees of GluA2Q/R RNA editing. These mice included the GluA2ΔECS/+ mouse, and an additional strain in which the loxP-flanked neomycin gene was left in the gene, replacing the ECS site (GluA2neo/neo). The practical result of this was that the most unedited GluA2 RNA was observed in GluA2neo/neo (98% unedited GluA2 mRNA), slightly less was observed in GluA2ΔECS/+ mice (~27% unedited GluA2 mRNA) and less again was observed in GluA2+/neo mice (8.7% unedited GluA2 mRNA). The reduced editing efficiency in each of the mice strains correlated with the Ca2+-permeability of the AMPA receptor and the phenotype. GluA2neo/neo which expressed 98% unedited GluA2(Q) mRNA showed severe dendritic deficits and early death by postnatal day 20. In comparison, GluA2neo/+ mice had 8.7% expression of GluA2(Q), showing less dramatic increases in AMPA receptor Ca2+-permeability and a 20% death rate (Feldmeyer et al., 1999).
Recent studies have indicated that the lethality of the unedited GluA2(Q) subunit may be due to a GluA2-NSF interaction (Mahajan and Ziff, 2007). Thus, alteration to GluA2 RNA editing efficiency has detrimental effects, and the extent of this effect is dependent upon the percentage of unedited GluA2(Q) RNA expressed, highlighting the absolute requirement for GluA2 RNA editing in synaptic function.
Although reducing RNA editing efficiency results in lethality, the unedited GluA2 is not essential for survival. Kask et al. (1998) used gene targeting to generate mice in which the codon for Arg was encoded in the GluA2 gene (GluA2R/R ). These mice are essentially “forced-edited” as the Arg is encoded in the exon and there is no requirement for RNA editing to post-transcriptionally alter GluA2 at the Q/R site, therefore there is no GluA2(Q) present in these mice. The authors noted that these GluA2R/R mice are fundamentally normal with no obvious deficiencies. This suggests that there is no essential functional requirement of the GluA2 Gln allele and it is still unclear as to why RNA editing occurs in the normal brain as opposed to exon coding of the codon for Arg.
As described earlier, ADAR2 is essential for GluA2 RNA editing. Interestingly, mice engineered with ADAR2 ablation (ADAR2-/-; Higuchi et al., 2000) are phenotypically comparable to GluA2 ΔECS/+ mice (Brusa et al., 1995). ADAR2-/- mice show fully unedited GluA2 and display a 30-fold increase in Ca 2+-permeability of AMPA receptors, resulting in lethality at just several weeks of age (Higuchiet al., 2000). In addition, knockdown of hippocampal ADAR2 levels using viral vectors expressing siRNA increased Ca 2+ -permeability of AMPA receptors by approximately 15-fold and increased cell death (Peng et al., 2006).
Notably, when ADAR2-/- mice were crossed with GluA2 R/R mice, the lethal phenotype observed in the ADAR2-/- mice was rescued, and survival rates were significantly increased to several months of age (Higuchi et al., 2000). Therefore, if the GluA2 gene is artificially “forced-edited,” then ADAR2 is no longer essential for survival. This remarkable experiment revealed that ADAR2 is essential for survival because of its role in editing GluA2 at the Q/R site. Thus, although other roles have been explored (Horsch et al., 2011), it appears that RNA editing at the Q/R site of GluA2 is the essential target substrate of ADAR2.
Combined, the aforementioned studies make it clear that alteration to the RNA editing, by modifying the ECS or by ablation of ADAR2 is detrimental to cell survival, leading to the hypothesis that these mechanisms could possibly contribute to excitotoxicity in disease. Perhaps, the most important conclusion that can be drawn from these outstanding studies is that unedited GluA2, when expressed in a neuron, will be expressed at synapses and will have a functional effect.
A POSSIBLE ROLE FOR GluA2 EDITING?
Given its importance for regulating Ca2+ influx, it might be expected that Q/R site RNA editing in GluA2 would be an exquisitely regulated process in the normal CNS. Surprisingly, however, it has been accepted for many years that the AMPA receptor subunit GluA2 in its Q/R site-unedited form is not essential for brain development and function (Higuchi et al., 1993; Kask et al., 1998). Indeed, unedited GluA2 at the Q/R site accounts for < 1% of GluA2 mRNA in the normal brain where GluA2 is present. Despite this, GluA2 Q/R site editing occurs in mammals, amphibians, and some species of fish (Kung et al., 2001), suggesting that GluA2 Q/R site RNA editing is evolutionarily conserved. This raises the significant question as to why a complicated process like RNA editing has evolved to convert a CAG codon encoding Gln in the gene into a CGG codon encoding Arg in the mRNA, as opposed to simply encoding the CGG codon for Arg in the GluA2 gene. A clue to the answer may lie in the observation that the efficiency of RNA editing varies between certain brain regions and may differ between specific cells (Nutt and Kamboj, 1994; Kawahara et al., 2003, 2004a; Peng et al., 2006). This raises the tantalizing possibility that unedited GluA2 RNA may have a role in certain regions in the CNS. Thus, the question still remains as to what role unedited GluA2 plays in the normal functioning brain.
NOVEL INSIGHTS INTO THE IMPORTANCE AND REGULATION OF GluA2Q/R SITE RNA EDITING
It is clear that ADAR2 depletion and reduced editing can cause catastrophic downstream effects. What is not understood, however, is why such a complex RNA editing mechanism occurs in neurons. In addressing this question, it should be taken into consideration that although it is often stated that there is on average ~1% of unedited GluA2 in the adult, it is feasible that editing is temporally and spatially regulated, potentially even varying between individual neurons.
It is now established that ADAR2 expression is regulated, suggesting in turn that GluA2 RNA editing may be regulated. For example, ADAR2 expression is modulated during development and is only expressed within the thalamic nuclei in the fetal brain (Paupard et al., 1999) and expression levels increase gradually over time (Jacobs et al., 2009). In addition to this, studies have shown that human neural progenitor cells (Whitney et al., 2008; Jansson et al., 2011), and malignant gliomas (Maas et al., 2001), are Ca2+-permeable and express unedited GluA2. These studies give indication that unedited GluA2 may play a role in cell function, even though the process is not essential for survival (Higuchi et al., 1993; Kask et al., 1998). Further research is required to understand the need for RNA editing of GluA2 in order to determine if, how, and why it is modulated in the normal brain and in disease.
GluA2 RNA EDITING HAS A ROLE IN AMPA RECEPTOR TRAFFICKING
While it is well established that the GluA2 subunit regulates trafficking (discussed in Section “The GluA2 Subunit Regulates Calcium Permeability and Trafficking of AMPA Receptors”), GluA2 RNA editing has also been shown to be involved in AMPA receptor trafficking. It is known that a significant proportion of edited GluA2 remains in the ER before being trafficked to the synapse (Greger et al., 2002). In addition, the rate of direct insertion of edited GluA2 into the membrane is lower than that of unedited GluA2. This insertion of unedited GluA2, in conjunction with the NSF binding site present on the C-terminus of GluA2, accelerates the insertion into the membrane (Araki et al., 2010). In contrast, edited GluA2 at the Q/R site forms a more stable pool of the subunit in the ER that is stored for later AMPA receptor formation. In addition, GluA2 RNA editing at the Q/R site also plays a role in AMPA receptor tetramerization as edited GluA2 preferentially form dimers (Greger et al., 2003). In the absence of GluA2 Q/R RNA editing, hetero and homotetramers are formed and are rapidly trafficked from the ER to the plasma membrane. This is, however, also dependent upon the editing at the R/G site (Greger et al., 2007). When editing occurs at the Q/R site, but is not present at the R/G site, GluA2 subunits are more likely to tetramerize (Greger et al., 2006). Thus, in the presence of unedited GluA2, Ca2+-permeable AMPA receptors are potentially incorporated into the synapse, permitting an influx of Ca2+ ions, although more studies are required. If so, it is possible that even though there is only ~1% of GluA2 present, there is a disproportionate effect on AMPA receptor Ca2+-permeability.
GluA2 is typically rapidly transported to the synapse when unedited GluA2 forms heterotetramers with GluA3 and interacts with NSF. This could potentially lead to excessive Ca2+ influx in the diseased brain. Mahajan and Ziff (2007) showed that the addition of a peptide, pep2m, blocks the interaction between NSF and the heterotetramer and, thus, reduces the toxicity to the cell in vitro. Though this has yet to be studied in vivo, the results suggest that interfering with the interaction of unedited GluA2 with other proteins may regulate unedited GluA2 mediated excitotoxicity. If true, then interfering with such interactions may act as a viable treatment for diseases associated with low GluA2 editing efficiency (Mahajan and Ziff, 2007).
REGULATION OF GluA2 RNA EDITING
The molecular mechanisms that regulate GluA2 RNA editing are beginning to emerge. Peng et al. (2006) showed that altered cAMP response element-binding (CREB)-transcriptional regulation leads to a reduction in ADAR2 expression. By virally expressing CREB following transient global ischemia, ADAR2 production was restored, thereby increasing GluA2Q/R RNA editing back to levels over 90%. This study was the first to give a mechanism by which GluA2 RNA editing deficiencies occur in the diseased brain. More recently, other transcriptional regulators have been shown to play a role in ADAR2 expression. In the absence of peptidyl-prolyl isomerase NIMA interacting protein 1 (Pin1), which is responsible for phosphorylation of ser/thr-pro motifs, editing efficiency at the Q/R site is reduced (Marcucci et al., 2011). By co-transfecting a plasmid encoding ADAR2 with a GluA2 minigene, an RNA editing level of 100% was achieved. However, when Pin1 was blocked by siRNA, the editing efficiency dramatically fell to 53%. Similar levels of Q/R site editing were seen when co-transfection was conducted with the GluA2 minigene and ADAR2 into immortalized mouse fibroblast cell lines derived from Pin1-/- mice. In addition, the mislocalization of ADAR2 in the absenceof Pin1 also reduces editingatthe R/G site highlighting the necessity of ADAR2 for editing at this site. Further, in the absence of Pin1 the authors observed the mislocalization of ADAR2 into the cytoplasm, rendering it unable to efficiently edit GluA2 (Marcucci et al., 2011). Within the cytoplasm WWP2, which possesses ubiquitin-protein ligase activity, is able to degrade ADAR2 and is therefore a negative regulator of the protein (Marcucci et al., 2011). These sophisticated studies show that there is regulation of ADAR2 and raise significant excitement about the potential for understanding the regulation of RNA editing.
EXCITOTOXICITY IS DETRIMENTAL TO THE EFFICIENCY OF GluA2 RNA EDITING
ADAR2 degradation would be expected to play a role in many diseases where Q/R site editing is compromised. Mahajan et al. (2011) have demonstrated that high concentrations of glutamate can activate processes which induces cleavage of the ADAR2 enzyme in vitro. This glutamate-induced cleavage is both dose- and time-dependent and is reliant upon the activation of NMDA receptors. However, when calpain inhibitors were applied to glutamate-stimulated neurons that virally express ADAR2, no cleavage of the enzyme was observed. Proteasome inhibitors and caspase inhibitors were unable to prevent ADAR2 degradation thus proving calpain mediates ADAR2 cleavage when in the presence of excessive glutamate (Mahajan et al., 2011). Furthermore, calpain inhibitors have been shown to have neuroprotective effects in ischemia (Bartus et al., 1994) and thus suggest a potential therapeutic target for other GluA2 RNA editing deficient disorders. By showing that glutamate, which is deregulated in many diseases, can induce cleavage of ADAR2, Mahajan et al. (2011) has provided insights into a direct mechanism in which neurological catastrophes may potentially cause GluA2 RNA editing deficiencies.
AMPA RECEPTORS ARE INVOLVED IN EXCITOTOXIC NEURONAL DEATH IN DISEASE
DOWNREGULATION OF GluA2 OCCURS IN DISEASE
It has long been known that neuronal death escalates disease progression in ALS and ischemia (reviewed in Doble, 1999). AMPA receptors lacking GluA2 are highly permeable to Ca2+, and many early studies suggested that they might potentially contribute to Ca2+-mediated excitotoxic cell death in disease. Known as the “GluA2 hypothesis” (Pellegrini-Giampietro et al., 1997), this theory suggests that neurological insults potentially decrease the expression of GluA2, resulting in many AMPA receptors that are highly permeable to Ca 2+. This process consequently may contribute to neurodegeneration in disease (Pellegrini-Giampietro et al., 1997; Tanaka et al., 2000).
GluA2 gene downregulation results in formation of AMPA receptors lacking GluA2. Hippocampal cells normally express GluA2, and changes in expression levels may have effects on the Ca2+-permeability of AMPA receptors. It has been shown that 24-72 h following ischemic insult, GluA2 protein is downregulated, particularly in the CA1 region of the hippocampus where cell death is abundant (Pellegrini-Giampietro et al., 1992). The internalization of AMPA receptors following oxygen–glucose deprivation, a model of ischemia, has been shown to lead to a subsequent delivery of Ca2+-permeable AMPA receptors to the synapse (Liu et al., 2006). This process is regulated by the gene silencing transcription factor neuronal repressor element-1 silencing transcription factor (REST; Calderone et al., 2003). Global ischemia increases REST mRNA expression in the CA1 region of the hippocampus, which in turn suppresses GluA2 gene expression. In addition, the suppression of REST expression by the use of antisense oligodeoxynucleotides increased neuronal survival 72 h post oxygen–glucose deprivation (Calderone et al., 2003).
While these Ca2+-permeable AMPA receptors observed in the ischemic brain appear to be in part GluA2-lacking (due to down regulation of GluA2; Pellegrini-Giampietro et al., 1992) recent evidence suggests they may also result from the presence of unedited GluA2 (see GluA2Q/R RNA Editing Deficiencies Occur in Ischemia). These Ca2+-permeable AMPA receptors, when in the diseased state, potentially contribute to excitotoxicity, despite such receptors also being required for maintenance of synaptic plasticity. This is analogous to NMDA receptor-mediated excitotoxicity, whereby a normal Ca2+-permeable receptor that is essential to synaptic plasticity is also critically implicated in excitotoxic cell death (reviewed in Hardingham and Badling, 2003).
MODULATION OF GluA2 RNA EDITING AT THE Q/R SITE HAS IMPLICATIONS FOR SURVIVAL
Through elegant studies, it is now known that modifications to RNA editing at the Q/R site can lead to effects on synaptic and cellular physiology and can lead to neuronal cell loss (Brusa et al., 1995; Kawahara et al., 2004b; Peng et al., 2006). As noted above, GluA2ΔECS/+ mice have altered editing efficiency and thus increased AMPA receptor Ca2+-permeability leading to spontaneous seizures and cell loss within the hippocampus and death by postnatal day 20 (Brusa et al., 1995; Feldmeyer et al., 1999). Whether, by inference, unedited GluA2 plays a role in human epilepsy is not clear. Unedited GluA2 was not found in the hippocampus and temporal cortex of epileptic patients (Kortenbruck et al., 2001). This may be due to the difficulty of detecting unedited RNA in disease (Kawahara et al., 2004a; Peng et al., 2006). Thus, as our current knowledge is limited, further studies using individual cell based assays are required to elucidate the role of GluA2 editing in epilepsy. Similarly, the editing mutant mice raise the possibility, but do not prove that, unedited GluA2 is important in any human disease. The most important point to be taken from the elegant study conducted by Brusa et al. (1995) is that unedited GluA2 can have critical functional effects on cellular physiology.
GluA2Q/R RNA EDITING DEFICIENCIES OCCUR IN AMYOTROPHIC LATERAL SCLEROSIS
As noted above, it has been suggested that Ca2+-permeable AMPA receptors resulting from GluA2 downregulation occurs in various neurological diseases such as ischemia and epilepsy (Pellegrini-Giampietro et al., 1992; Pollard et al., 1993; Gorter et al., 1997). In addition, reduced GluA2 Q/R site RNA editing also results in formation of Ca2+-permeable GluA2(Q)-containing AMPA receptors in ischemia. Historically, numerous attempts were made to identify unedited GluA2 RNA and little, if any, was ever found to be present in both the healthy and diseased brain (Akbarian et al., 1995; Rump et al., 1996; Kortenbruck et al., 2001). This led to the widely held concept that the presence or absence of the GluA2 subunit, rather than RNA editing, is the main process for regulating Ca2+-permeability of AMPA receptors in disease. As such, the role of unedited GluA2 in many neurological disorders has never been investigated on a cellular level as was recently done for ischemia (Peng et al., 2006).
RNA editing deficits have been shown in motor neurons of patients with ALS (Takuma et al., 1999; Kawahara et al., 2004a). No alteration to GluA2 mRNA expression was observed, however, spinal motor neurons showed a GluA2 RNA editing efficiency ranging from 0 to 100% in individual neurons (Kawahara et al., 2004a). Within the same patients, GluA2 RNA editing in Purkinje cells were unaltered, indicating that the deficiency is motor neuron specific. While the work by Kawahara et al. (2004a) does not prove that unedited GluA2 is a cause of cell loss in ALS, the results are highly important when put in the context of the work conducted by Brusa et al. (1995) and Feldmeyer et al. (1999), which showed that the process of RNA editing at the GluA2Q/R site is essential for cell survival.
It is perhaps interesting, however, in mice transgenic for mutant human Cu/Zn-superoxide dismutase (SOD1), a classic model of familial ALS, RNA editing deficiencies at the Q/R site were not observed (Kawahara et al., 2006). This highlights the possibility that mouse models of ALS may not fundamentally represent the human disease pathogenesis, and/or suggests unedited GluA2 mRNA at the Q/R site is not needed for the ALS phenotype. Despite this, studies have shown that crossing SOD1 mice with mice that express Ca2+-permeable AMPA receptors (generated by inserting an asparagine codon at the Q/R site) accelerates motor deterioration and disease onset when compared to age-matched SOD1 mice (Kuner et al., 2005). It is likely that GluA2 RNA editing deficiencies occur in ALS due to the downregulation of ADAR2, which has decreased expression in the spinal cords of patients with sporadic ALS (Kawahara and Kwak, 2005; Hideyama et al., 2010). In fact, the ablation of ADAR2 expression in mice results in a phenotype very similar to ALS patients including the progressive death of motor neurons and a decline in motor function (Hideyama et al., 2010). ADAR2 downregulation in ALS correlates with phosphorylated TDP-43, an RNA binding protein whose mutations are associated with ALS (Aizawa et al., 2010). These studies could suggest that GluA2 RNA editing deficiencies may be an important event in ALS, though the cellular mechanisms that regulate RNA editing deficiencies in ALS remain unsolved.
GluA2Q/R RNA EDITING DEFICIENCIES OCCUR IN ISCHEMIA
While it is evident that GluA2 mRNA downregulation occurs in ischemia (see Downregulation of GluA2 Occurs in Disease), recent evidence has indicated that GluA2 RNA editing deficiencies also occur in ischemia. In a model of transient global ischemia, GluA2 RNA editing efficiency of individual neurons at the Q/R site in the CA1 region of the hippocampus is dramatically decreased (Peng et al., 2006). Using electrophysiology and single cell RT-PCR, the authors correlated the GluA2 RNA editing efficiency of individual neurons to their Ca2+-permeability. RNA editing at the Q/R site showed extremely high variability (7–98%) in editing efficiency following ischemic insult within neurons of the CA1 region of the hippocampus. This editing efficiency correlated closely to the Ca2+-permeability of the neurons. Neither cell death nor GluA2 RNA editing deficiencies were seen in the CA3 region, indicating the CA1 region is most vulnerable to editing changes and ischemic insult. Interestingly, this cellular death closely correlates with the downregulation of ADAR2 (Peng et al., 2006). The authors demonstrated that by virally mediated expression of ADAR2 in vivo, the RNA editing efficiency at the GluA2Q/R site was adequately enhanced to over 95% post the ischemic insult and consequently lead to neuroprotection. These experiments provide substantial evidence that GluA2 RNA editing plays a vital role in mediating excitotoxic neuronal death during ischemia.
Since Ca2+-permeable AMPA receptors appear to occur due to the presence of unedited GluA2 and also from GluA2-lacking receptors in ischemia, the question arises as to which of these two types of AMPA receptors are expressed at the synapse and if only one type, or both, contributes to cell death. Given the astonishing results by Peng et al. (2006), which showed that the over-expression of ADAR2 is able to rescue cell loss following ischemic insult, it is possible that despite GluA2-lacking receptors being present, the unedited GluA2 is critical for cell death, as previously mentioned in Section “Novel Insights into the Importance and Regulation of GluA2Q/R RNA Editing.”
NEUROPROTECTIVE MECHANISMS FOR CALCIUM-MEDIATED EXCITOTOXICITY
Currently, the primary treatments to prevent against Ca2+-mediated neurotoxicity involve NMDA receptor antagonists. The beneficial effects are less than ideal and in addition there are often side effects, possibly due to blocking the physiological actions of glutamate neurotransmission and plasticity in non-injured neurons (reviewed in Parsons et al., 2007). Studies have also suggested, however, that blocking Ca2+-permeable AMPA receptors in animal models of ischemia can protect against cell death in vulnerable neurons. When given intraperitoneal injections of the AMPA receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) following transient forebrain ischemia, cell survival within the CA1 region was significantly increased (Buchan et al., 1991). In addition, 1-naphthyl acetyl spermine (Naspm), which blocks GluA2-lacking and unedited GluA2 containing AMPA receptors, exerts neuroprotective effects. Noh et al. (2005) demonstrated that by administering Naspm to mice 9–40 h postischemic injury, there is increased cell survival within the CA1 region. These studies demonstrate that Ca2+-permeable AMPA receptors play a role in modulating cell death and show antagonists to AMPA can protect against cell loss in ischemia. An important idea, given the evidence in ischemia for example, is whether more effort is needed to block altered RNA editing in order to protect against excitotoxicity in disease.
CONCLUSION
Little consideration has been given to what role the evolutionarily conserved process of GluA2 RNA editing may play in normal brain function. Many early studies suggested that unedited GluA2 exists at very low levels in the brain and that the extent of unedited GluA2 is not altered substantially in most diseases where editing was investigated (Akbarian et al., 1995; Rump et al., 1996; Kortenbruck et al., 2001). Taken together, the evidence has been compelling that unedited GluA2 does not play a major role in brain function. However with some notable exceptions, few recent studies have probed the role of unedited GluA2 in disease (Kawahara et al., 2004a; Peng et al., 2006). We therefore suggest that it is not yet possible to exclude the possibility that unedited GluA2 also contribute to the presence of Ca2+-permeable AMPA receptors in normal brain function, such as synaptic plasticity.
The blockade of Ca2+-permeable AMPA receptors is known to improve cell survival in diseased states, however the extent to which unedited GluA2 contributes to excitotoxic neuron death still needs to be elucidated. Sophisticated approaches recently revealed a possible role for unedited GluA2 in ischemia and ALS, despite previous studies ruling out a role for unedited GluA2 in ischemia (Kawahara et al., 2004a; Peng et al., 2006). By extension, the lack of evidence for extensive unedited GluA2 in other scenarios so far investigated does not rule out a role for unedited GluA2 in normal physiology or disease. Evidence that unedited GluA2 is preferentially trafficked to the cell surface is important because it adds weight to the view that small amounts of unedited GluA2 can have an impact on normal neuronal function (Greger et al., 2002, 2003, 2007). Furthermore, mice engineered with unedited GluA2 to date reveal that unedited GluA2 can have an important effect on cellular physiology and cell survival (Brusa et al., 1995; Feldmeyer et al., 1999). Taken together, it is clear that while the average amount of unedited GluA2 is always very low, it can vary between cells with functional consequences.
In conclusion, we suggest that the presence of unedited GluA2, in addition to GluA2-lacking receptors, should be considered as a possibility in studies where Ca2+-permeable AMPA receptors are described or observed. Numerous studies show that Ca2+-permeable receptors are critical in normal brain function and disease. Studies are essential to determine the extent to which both unedited GluA2 and GluA2-lacking AMPA receptors contribute in these circumstances.
Exciting recent studies have indicated that ADAR2 is downregulated in disease, concomitant with reduced RNA editing. Mechanisms in which ADAR2 expression is regulated are now being investigated and the complex pathways involved in GluA2 RNA editing are being increasingly unraveled. This emerging knowledge may aid in understanding the regulation and functional role of GluA2 Q/R RNA editing. It is possible that such studies in future may reveal that the ~1% unedited GluA2 found in the mammalian brain may have as-yet unknown role in synaptic plasticity and disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Office for Science and Medical Research through their spinal fund, SpinalCure Australia, The Roth Foundation, Bill Gruy, Patricia A. Quick foundation, Nick Kell, Geoffrey Towner, Amadeus Energy Ltd., Walter and Edith Sheldon. We would like to thank members of the Neurodegenerative Disorders Laboratory including Andrea Abdipranoto, LyndseyKonen, Gordon Royle, Sandy Stayte, Richard Tan, and Raphael Zinn for extensive editing of this review. We would also like to thank Sarah Woodworth and Steven Wright for editing and Johanna Stacey and Mauricio Perez for their contribution to the design of figures.
REFERENCES
- Aizawa H., Sawada J., Hideyama T., Yamashita T., Katayama T., Hasebe N., Kimura T., Yahara O., Kwak S. (2010). TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 120 75–84 [DOI] [PubMed] [Google Scholar]
- Akbarian S., Smith M. A., Jones E. G. (1995). Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer’s disease, Huntington’s disease and schizophrenia. Brain Res. 699 297–304 [DOI] [PubMed] [Google Scholar]
- Araki Y., Lin D., Huganir R. L. (2010). Plasma membrane insertion of the AMPA receptor GluA2 subunit is regulated by NSF binding and Q/R editing of the ion pore. Proc. Natl. Acad. Sci. U.S.A. 107 11080–11085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asrar S., Zhou Z., Ren W., Jia Z. (2009). Ca2+ permeable AMPA receptor induced long-term potentiation requires Pi3/MAP kinases but not Ca/CaM-dependent kinase II. PLoS ONE 4 e4339 10.1371/journal.pone.0004339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus R. T., Hayward N. J., Elliott P. J., Sawyer S. D., Baker K. L., Dean R. L., Akiyama A., Straub J. A., Harbe-son S. L., Li Z. (1994). Calpain inhibitor AK295 protects neurons from focal brain ischemia. Effects of postocclusion intra-arterial administration. Stoke 25 2265–2270 [DOI] [PubMed] [Google Scholar]
- Bass B. L. (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71 817–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani S., Valnegri P., Beretta F., Passafaro M. (2009). The GluR2 subunit of AMPA receptors: synaptic role. Neuroscience 158 55–61 [DOI] [PubMed] [Google Scholar]
- Bellone C., Luscher C. (2006). Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat. Neurosci. 9 636–641 [DOI] [PubMed] [Google Scholar]
- Biou V., Bhattacharyya S., Malenka R. C. (2008). Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc. Natl. Acad. Sci. U.S.A. 150 1038–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusa R., Zimmermann F., Koh D. S., Feldmeyer D., Gass P., Seeburg P. H., Sprengel R. (1995). Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270 1677–1680 [DOI] [PubMed] [Google Scholar]
- Buchan A. M., Li H., Cho S., Pulsinelli W. A. (1991). Blockade of the AMPA receptor prevents CA1 hippocampal injury following severe but transient forebrain ischemia in adult rats. Neurosci. Lett. 132 255–258 [DOI] [PubMed] [Google Scholar]
- Burnashev N., Villarroel A., Sakmann B. (1996). Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J. Physiol. 496 165–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns C., Chu H., Rueter S., Hutchin-son L., Canton H., Sanders-Bush E., Emerson R. (1997). Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387 303–308 [DOI] [PubMed] [Google Scholar]
- Calderone A., Jover T., Noh K., Tanaka H., Yokota H., Lin Y., Grooms S., Regis R., Bennet M., Zukin S. (2003). Ischemic insults derepress the gene silencer REST in neurons destined to die. J. Neurosci. 23 2112–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Cho D., Wang Q., Lai F., Carter K., Nishikura K. (2000). A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single-and double-stranded RNA binding domains. RNA 6 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem R. L., Barth A. (2006). Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron 49 663–670 [DOI] [PubMed] [Google Scholar]
- Conrad K. L., Tseng K. Y., Uejima J. L., Reimers J. M., Jun-Heng L. J., Shaham Y., Marinelli M., Wolf M. E. (2008). Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454 118–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S., Kelly L., Far-rant M. (2006). Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr. Opin. Neurobiol. 16 288–297 [DOI] [PubMed] [Google Scholar]
- Dabiri G. A., Lai F., Drakes R. A., Nishikura K. (1996). Editing of the GluR-B ion channel RNA in vitro by recombinant double-stranded RNA adenosine deaminase. EMBO J. 15 34–45 [PMC free article] [PubMed] [Google Scholar]
- Dingledine R., Borges K., Bowie D., Traynelis S. F. (1999). The glutamate receptor ion channels. Pharmacol. Rev. 51 7–61 [PubMed] [Google Scholar]
- Doble A. (1999). The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther. 81 163–221 [DOI] [PubMed] [Google Scholar]
- Feldmeyer D., Kask K., Brusa R., Kornau H. C., Kolhekar R., Rozov A., Burnashev N., Jensen V., Hvalby O., Sprengel R., Seeburg P. H. (1999). Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat. Neuroci. 2 57–64 [DOI] [PubMed] [Google Scholar]
- Gorter J. A., Petrozzino J. J., Aron-ica E. M., Rosenbaum D. M., Opitz T., Bennett M. V. L., Connor J. A., Zukin R. S. (1997). Global ischemia induces downregulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J. Neurosci. 17 6179–6188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger I. H., Akamine P., Khatri L., Ziff E. B. (2006). Developmentally regulated, combinatorial RNA processing modulates AMPA receptor biogenesis. Neuron 51 85–97 [DOI] [PubMed] [Google Scholar]
- Greger I. H., Khatri L., Kong X., Ziff E. B. (2003). AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40 763–774 [DOI] [PubMed] [Google Scholar]
- Greger I. H., Khatri L., Ziff E. B. (2002). RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34 759–772 [DOI] [PubMed] [Google Scholar]
- Greger I. H., Ziff E. B., Penn A. C. (2007). Molecular determinants of AMPA receptor subunit assembly. Trends Neurosci. 30 407–416 [DOI] [PubMed] [Google Scholar]
- Hardingham G. E., Badling H. (2003). The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 26 81–89 [DOI] [PubMed] [Google Scholar]
- Harvery S. C., Koster A., Yu H., Skolnick P., Baumbarger P., Nisenbaum E. S. (2001). AMPA receptor function is altered in GluR2-deficient Mice. J. Mol. Neurosci. 17 35–43 [DOI] [PubMed] [Google Scholar]
- Hideyama T., Yamashita T., Suzuki T., Tsuji S., Higuchi M., Seeburg P. H., Takashashi R., Misawa H., Kwak S. (2010). Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J. Neurosci. 30 11917–11925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M., Maas S., Single F. N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P. H. (2000). Point mutation in the AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406 78–81 [DOI] [PubMed] [Google Scholar]
- Higuchi M., Single F. N., Kohler M., Sommer B., Sprengel R., Seeburg P. H. (1993). RNA editing of AMPA receptor subunit GluR-B: a base paired intron-exon structure determines position and efficiency. Cell 75 1361–1370 [DOI] [PubMed] [Google Scholar]
- Hollmann M., Hartley M., Heine-mann S. (1991). Ca2+ permeability of KA-AMPA – gated glutamate receptor channels depends on sub-unit composition. Science 252 851–853 [DOI] [PubMed] [Google Scholar]
- Hollmann M., Heinemann S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17 31–108 [DOI] [PubMed] [Google Scholar]
- Horsch M., Seeburg P. H., Adler T., Aguilar-Pimentel J. A., Becker L., Calzada-Wack J., Garret L., Gotz A., Hans W., Higuchi M., Holter S. M., Naton B., Prehn C., Puk O., Racz I., Rathkolb B., Rozman J., Schrewe A., Adamski J., Busch D. H., Esposito I., Graw J., Ivandic B., Klingenspor M., Klopstock T., Mempel M., Ollert M., Schulz H., Wolf E., Wurst W., Zimmer A., Gailus-Durner V., Fuchs H., DeAngelis M., Beckers J. (2011). Requirement of the RNA-editing enzyme ADAR2 for normal physiology in mice. J. Biol. Chem. 286 18614–18622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume R. I., Dingledine R., Heinemann S. F. (1991). Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 253 1028–1031 [DOI] [PubMed] [Google Scholar]
- Isaac J. T., Ashby M. C., Mcbain C. J. (2007). The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54 859–871 [DOI] [PubMed] [Google Scholar]
- Jackson A. C., Nicoll R. A. (2011). The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 70 178–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs M. M., Fogg R. L., Emeson R. B., Stanwood G. D. (2009). ADAR1 and ADAR2 expression and editing activity during forebrain development. Dev. Neurosci. 31 223–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson L. C., Wigren H. K., Nordstrom T., Akerman K. E. (2011). Functional α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in differentiating embryonic neural progenitor cells. Neuroreport 22 282–287 [DOI] [PubMed] [Google Scholar]
- Jia Z., Agopyan N., Miu P., Xiong Z., Henderson J., Gerlai R., Taverna F. A., Velumian A., Macdonald J., Carlen P., Abramow-Newerly W., Roder J. (1996). Enhanced LTPinmice deficientinthe AMPA receptor GluR2. Neuron 17 945–956 [DOI] [PubMed] [Google Scholar]
- Kask K., Zamanillo D., Rozov A., Burnashev N., Sprengel R., Seeburg P. H. (1998). The AMPA receptor subunit GluR-B in its Q/R site-unedited form is not essential for brain development and function. Proc. Natl. Acad. Sci. U.S.A. 95 13777–13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y., Ito K., Sun H., Aizawa H., Kanazawa I., Kwak S. (2004a). Glutamate receptors: RNA editing and death of motor neurons. Nature 427 801 [DOI] [PubMed] [Google Scholar]
- Kawahara Y., Ito K., Sun H., Ito M., Kanazawa I., Kwak S. (2004b). Regulation of glutamate receptor RNA editing and ADAR mRNA expression in developing human normal and Down’s syndrome brains. Brain Res. Dev. Brain Res. 148 151–155 [DOI] [PubMed] [Google Scholar]
- Kawahara Y., Ito K., Sun H., Kanazawa I., Kwak S. (2003). Low editing efficiency of GluR2 mRNA is associated with a low relative abundance of ADAR2 mRNA in white matter of normal human brain. Eur. J. Neurosci. 18 23–33 [DOI] [PubMed] [Google Scholar]
- Kawahara Y., Kwak S. (2005). Excitotoxicity and ALS: what is unique about the AMPA receptors expressed on spinal motor neurons? Amyotroph. Lateral Scler. Other Motor Neuron Disord. 6 131–144 [DOI] [PubMed] [Google Scholar]
- Kawahara Y., Sun H., Ito K., Hideyama T., Aoki M., Sobue G., Tsuji S., Kwak S. (2006). Under-editing of GLuR2 mRNA, a neuronal death inducing molecular change in sporadic ALS, does not occur in motor neurons in ALS1 or SBMA. Neurosci. Res. 54 11–14 [DOI] [PubMed] [Google Scholar]
- Keifer J., Zheng Z. (2010). AMPA receptor trafficking and learning. Eur. J. Neurosci. 32 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels H., Malinow R. (2009). Synaptic AMPA receptor plasticity and behaviour. Neuron 61 340–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortenbruck G., Berger E., Speck-mann E. J., Musshoff U. (2001). RNA editing at the Q/R site for the glutamate receptor subunits GLUR2, GLUR5, and GLUR6 in hippocampus and temporal cortex from epileptic patients. Neurobiol. Dis. 8 459–468 [DOI] [PubMed] [Google Scholar]
- Kullmann D. M., Lamsa K. (2008). Roles of distinct glutamate receptors in induction of anti-Hebbian long-term potentiation. J. Physiol. 586 1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R., Groom A. J., Bresink I., Kornau H., Stefovska V., Muller G., Hartmann B., Tschauner K., Waibel S., Ludolph A. C., Ikonomi-dou C., Seeburg P. H., Turski L. (2005). Late-onset motoneuron disease caused by a functionally modified AMPA receptor subunit. Proc. Natl. Acad. Sci. U.S.A. 102 5826–5831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner T., Beck C., Sakmann B., Seeburg P. (2001). Channel-lining residues of the AMPA receptor M2 segment: structural environment of the Q/R site and identification of the selectivity filter. J. Neurosci. 21 4162–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung S. S., Chen Y. C., Lin W. H., Chen C. C., Chow W. Y. (2001). Q/R RNA editing of the AMPA receptor subunit 2 (GRIA2) transcript evolves no later than the appearance of cartilaginous fishes. FEBS Lett. 509 277–281 [DOI] [PubMed] [Google Scholar]
- Kwak S., Hideyama T., Yamashita T., Aizawa H. (2010). AMPA receptor-mediated neuronal death in sporadic ALS. Neuropathology 30 182–188 [DOI] [PubMed] [Google Scholar]
- Kwak S., Kawahara Y. (2005). Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 83 110–120 [DOI] [PubMed] [Google Scholar]
- Kwak S., Weiss J. H. (2006). Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 16 281–287 [DOI] [PubMed] [Google Scholar]
- Lamsa K. P., Heeroma J. H., Somogyi P., Rusakov D. A., Kull-mann D. M. (2007). Anti-Hebbian long-term potentiation in the hip-pocampal feedback inhibitory circuit. Science 315 1262–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerma J. (2003). Roles and rules of kainate receptors in synaptic transmission. Nat. Rev. Neurosci. 4 481–495 [DOI] [PubMed] [Google Scholar]
- Liu B., Liau M., Mielke J. G., Ning K., Chen Y., Li L., El-Hayek Y. H., Gomez E., Zukin R. S., Fehlings M. G., Wan Q. (2006). Ischemic insults directs glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J. Neurosci. 26 5309–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. J., Zukin R. S. (2007). Ca2+-permeable AMPA receptors in synap-tic plasticity and neuronal death. Trends Neurosci. 30 126–134 [DOI] [PubMed] [Google Scholar]
- Lomeli H., Mosbacher J., Melcher T., Hoger T., Geiger J., Kuner T., Monyer H., Higuchi M., Bach A., Seeburg P. (1994). Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266 1709–1713 [DOI] [PubMed] [Google Scholar]
- Lu Y., Allen M., Halt A. R., Weisen-haus M., Dallapiazza R. F., Hall D. D., Usachev Y. M., Mcknight G. S., Hell J. W. (2007). Age-dependentrequirementofAKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26 4879–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S., Patt S., Schrey M., Rich A. (2001). Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. U.S.A. 98 14687–14692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S. S., Thai K. H., Chen K., Ziff E. B. (2011). Exposure of neurons to excitotoxic levels of glu-tamate induces cleavage of the RNA editing enzyme, adenosine deami-nase acting on RNA 2, and loss of GLUR2 editing. Neuroscience 189 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan S. S., Ziff E. B. (2007). Novel toxicity of the unedited GluR2 AMPA receptor subunit dependent on surface trafficking and increased Ca2+-permeability. Mol. Cell. Neurosci. 35 470–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R., Malenka R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25 103–126 [DOI] [PubMed] [Google Scholar]
- Man H. Y. (2011). GluA2-lacking, calcium-permeable AMPA receptors – inducers of plasticity? Curr. Opin. Neurobiol. 21 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci R., Brindle J., Paro S., Casadio A., Hempel S., Morrice N., Bisso A., Keegen L. P., Del Sal G., O’Connell M. A. (2011). Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 30 4211–4222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J., Mehler M. (2008). RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci. 31 227–233 [DOI] [PubMed] [Google Scholar]
- Melcher T., Maas S., Herb A., Sprengel R., Seeburg P. H., Higuchi M. (1996). A mammalian RNA editing enzyme. Nature 379 460–464 [DOI] [PubMed] [Google Scholar]
- Meng Y., Zhang Y., Jia Z. (2003). Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron 39 163–176 [DOI] [PubMed] [Google Scholar]
- Mosbacher J., Schoepfer R., Monyer H., Burnashev N., Seeburg P. H., Ruppersber J. P. (1994). A molecular determinant for submillisecond desensitization in glutamate receptors. Science 266 1059–1062 [DOI] [PubMed] [Google Scholar]
- Nakagawa T. (2010). The biochemistry, ultrastructure, and subunit assembly mechanism of AMPA receptors. Mol. Neurobiol. 42 161–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh K. M., Yokota H., Mashiko T., Castillo P. E., Zukin R. S., Bennet M. V. (2005). Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. U.S.A. 102 12230– 12235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt S. L., Kamboj R. K. (1994). Differential RNA editing efficiency of AMPA receptor GluR-2 in human brain. Neuroreport 5 1679–1683 [DOI] [PubMed] [Google Scholar]
- Panicker S., Brown K., Nicoll R. A. (2008). Synaptic AMPA receptor subunit trafficking is independent of the C terminus in the GluR2-lacking mouse. Proc. Natl. Acad. Sci. U.S.A. 105 1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons C. G., Stoffler A., Danysz W. (2007). Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system – too little activation is bad, too much is even worse. Neuropharmacology 53 699–723 [DOI] [PubMed] [Google Scholar]
- Paupard M.-C., O’Connell M. A., Greber A. P., Zukin R. S. (1999). Patterns of developmental expression of the RNA editing enzyme rADAR2. Neuroscience 95 869–879 [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro D. E., Gorter J. A., Bennet M. V., Zukin R. S. (1997). The GluR2 (GluR-B) hypothesis: Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 20 464–470 [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro D. E., Zukin R. S., Bennet M. V., Cho S., Pulsinelli W. A. (1992). Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc. Natl. Acad. Sci. U.S.A. 89 10499–10503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng P.L., Zhong X., Tu W., Soundara-pandian M. M., Molner P., Zhu D., Lau L., Liu S., Liu F., Lu Y. (2006). ADAR2-dependent RNA editing of AMPA receptor sub-unit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 49 719–733 [DOI] [PubMed] [Google Scholar]
- Pollard H., Heron A., Moreau J., Ben-Ari Y., Khrestchatisky M. (1993). Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience 57 545–554 [DOI] [PubMed] [Google Scholar]
- Riedel G., Micheau J., Lam A. G., Roloff E. L., Martin S. J., Bridge H., De Hoz L., Poeschel B., Mcculloch J., Morris R. G. (1999). Reversible neuralinactivation reveals hippocampal participation in several memory processes. Nature 2 898–905 [DOI] [PubMed] [Google Scholar]
- Rosenmund C., Stern-Bach Y., Stevens C. F. (1998). The tetrameric structure of a glutamate receptor channel. Science 280 1596–1599 [DOI] [PubMed] [Google Scholar]
- Rump A., Sommer C., Gass P., Bele P., Meissner D., Kiessling M. (1996). Editing of GluR2 RNA in the gerbil hippocampus after global cerebral ischemia. J. Cereb. Blood Flow Metab. 16 1362–1365 [DOI] [PubMed] [Google Scholar]
- Sans N., Vissel B., Petralia R. S., Wang Y. X., Chang K., Royle G. A., Wang C. Y., O’Gorman S., Heinemann S. F., Wenthold R. J. (2003). Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J. Neurosci. 23 9367–9373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffer H. H., Heinemann S. F. (1999). A quantitative method to detect RNA editing events. Anal. Biochem. 276 257–260 [DOI] [PubMed] [Google Scholar]
- Sobolevsky A., Rosconi M., Gouaux E. (2009). X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B., Keinanen K., Verdoorn T. A., Wisden W., Burnashev N., Herb A., Kohler M., Takagi T., Sakmann B., Seeburg P. H. (1990). Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science 249 1580–1585 [DOI] [PubMed] [Google Scholar]
- Sommer B., Köhler M., Sprengel R., Seeburg P. H. (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67 11–19 [DOI] [PubMed] [Google Scholar]
- Sun H. Y., Bartley A. F., Dobrunz L. E. (2009). Calcium-permeable presynaptic kainate receptors involved in excitatory short-term facilitation onto somatostatin interneurons during natural stimulus patterns. J. Neurophysiol. 101 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H., Kwak S., Yoshizawa T., Kanazawa I. (1999). Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann. Neurol. 46 806–815 [DOI] [PubMed] [Google Scholar]
- Tanaka H., Grooms S. Y., Bennet M. V., Zukin R. S. (2000). The AMPAR subunit GluR2: still front and center-stage. Brain Res. 886 190–207 [DOI] [PubMed] [Google Scholar]
- Vissel B., Royle G. A., Christie B. R., Schiffer H. H., Ghetti A., Tritto T., Perez-Otano I., Radcliffe R. A., Sea-mans J., Sejnowski T., Wehner J. M., Collins A. C., O’Gorman S., Heinemann S. F. (2001). The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron 29 217–227 [DOI] [PubMed] [Google Scholar]
- Wenthold R. J., Petralia R. S., Bla-hos J. I., Niedzielski A. S. (1996). Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 16 1982–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney N. P., Peng H., Erdmann N. B., Tian C., Monaghan D. T., Zheng J. C. (2008). Calcium-permeable AMPA receptors containing Q/R-unedited GluR2 direct human neural progenitor cell differentiation to neurons. FASEB J. 22 2888–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltgen B. J., Royle G. A., Gray E. E., Abdipranoto A., Thangthaeng N., Jacobs N., Saab F., Tonegawa S., Heinemann S. F., O’Dell T. J., Fanselow M. S., Vissel B. (2010). A role for calcium-permeable AMPA receptors in synaptic plasticity and learning. PLoS ONE 5 e12818 10.1371/journal.pone.0012818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziff E. B. (2007). TARPs and the AMPA receptor trafficking paradox. Neuron 53 627–633 [DOI] [PubMed] [Google Scholar]