

Abstract
Introduction
The complement cascade is a critical mediator of the inflammatory response following cerebral ischemia. Recent work has demonstrated that genetic-deficiency of Mannose-binding lectin(MBL) ameliorates reperfusion injury and improves outcome in the acute phase of stroke. The present study sought to further delineate the pathogenic role of MBL in stroke and to examine whether the neuroprotection associated with MBL-deficiency is sustained beyond the acute phase. We hypothesized that genetic MBL deficiency would suppress complement activation and ameliorate reperfusion injury in the acute phase, but that persistent inhibition of complement into the sub-acute phase would serve to abrogate this neuroprotective effect.
Methods
The time-course and localization of post-ischemic cerebral MBL and C3 deposition were characterized using both Western-blot and immunohistochemistry. MBL-a/c null(MBL-KO) mice subjected to transient middle cerebral artery occlusion(MCAO) were then employed to investigate the histologic injury and functional outcome associated with genetic MBL deletion at both 24 hours and 7 days.
Results
MBL-a/c rapidly deposit on ischemic endothelium and trigger downstream complement activation in the acute phase. Genetic deficiency of MBL abrogates C3 cleavage as well as the sub-acute accumulation of mononuclear cells in the ischemic region. Although MBL-KO mice demonstrate significantly improved outcome at 24 hours, the neuroprotective effect associated with genetic MBL deletion is not sustained.
Conclusions
Development of a successful anti-complement neuroprotective strategy will require carefully-tailored inhibition coupled with a greater understanding of the functional effects of complement activation during later phases of stroke recovery.
Keywords: Complement, Stroke, Neuroprotection, Inflammation, Mannose-binding Lectin
Introduction
Inflammation promotes acute neuronal injury following reperfusion of an occluded cerebral blood vessel and serves to limit the therapeutic effectiveness of both pharmacologic and mechanical reperfusion strategies[2, 3]. The complement cascade, a primary component of the ischemic inflammatory response, is central to the initiation and propagation of this cerebral reperfusion injury[4-9]. Both genetic and pharmacologic strategies have been employed to ameliorate experimental cerebral injury in the acute phase by directly inhibiting complement component C3 or by antagonizing the binding of the C3a anaphylatoxin to its cognate receptor[1, 6]. However, investigation into the proximal pathway mechanisms responsible for the initiation of complement activation early following ischemic induction has received comparatively less attention[1].
The lectin pathway of the complement cascade classically begins with MBL-mediated recognition of terminal oligosaccharides presented on the surface of a target cell. This incites a conformational change in the MBL molecule, activating the MBL-associated serine proteases(MASPs), followed by MASP-mediated cleavage of complement components C4 and C2 to form the C3-convertase[10]. Emerging evidence suggests that the lectin pathway is central to the propagation of ischemia-reperfusion injury across a variety of tissues[11, 12]. Additionally, two groups have recently published studies analyzing a role for MBL in stroke. The authors of these studies utilized mice genetically-deficient in MBL(MBL-KO) subjected to middle cerebral artery occlusion(MCAO) to implicate MBL in the pathogenesis of cerebral reperfusion injury[13, 14].
Although this work suggests that MBL contributes to inflammatory cerebral injury in the acute phase of stroke, adequate assessment of a candidate therapeutic target in translational stroke research must include an assessment of delayed functional outcome. This is particularly relevant for anti-inflammatory strategies, as inflammation contributes to the process of post-ischemic repair at delayed time-points[15, 16]. Additionally, complement activation itself has been postulated to play a direct role in stroke recovery, as deposition of a variety of complement components, including MBL, facilitate the clearance of damaged cells and debris in the subacute to chronic phases of neurodegeneration[17]. Furthermore, recent work suggests that complement may promote both basal and ischemia-induced neurogenesis[18]. These data argue that suppression of complement beyond the acute post-ischemic period may actually impair endogenous neurorestorative processes.
In order to further elucidate the role of complement activation in cerebral ischemia/reperfusion, we set out to rigorously characterize the activation of the MBL pathway in reperfused stroke, focusing on both the acute and sub-acute phases of stroke recovery. We began by examining the time-course of complement activation for 7 days following reperfused MCAO in mice. MBL-KO mice subjected to transient MCAO were then employed to evaluate the effect of genetic MBL deficiency on downstream complement cleavage, as well as histologic and functional injury, at both 24 hours and 7 days post-reperfusion. We hypothesized that genetic MBL deficiency would suppress complement activation and ameliorate reperfusion injury in the acute phase, but that persistent inhibition of complement into the sub-acute phase would serve to abrogate this neuroprotective effect.
Materials and Methods
All experiments were approved by the Columbia University Institutional Animal Care & Use Committee. All surgical manipulations, functional evaluation and biochemical assessments were performed by a blinded investigator, and random allocation of animals in each experimental cohort was performed throughout these experiments.
Mice
Adult male MBL-null mice(B6.129S4-Mbl1tm1KataMbl2tm1Kata/J)(8-12 weeks old, 23-26g) were obtained from Jackson Laboratories(Bar Harbor, ME, USA). These mice were backcrossed into C57Bl/6J background for 7 generations, and age/weight matched C57Bl/6J mice were used as wild-type controls. Animals were housed in certified barrier-facilities with free access to food and water.
Murine Model of MCAO
Mice were anesthetized with 1.5% isoflurane delivered in a 70% N2O/30%O2 mixture. MCAO was achieved by advancing a silicon-coated 7-0 nylon monofilament to the MCA origin, with reperfusion achieved by withdrawing the filament after 60 minutes[6, 19]. For all animals, transcranial CBF was measured continuously using laser-Doppler flowmetry beginning pre-occlusion and extending 15 minutes post-reperfusion(Periflux System 5000; Perimed, Stockholm, Sweden)[19, 20]. Animals were included if occlusion suppressed blood flow to <30% of baseline, and if reperfusion resulted in prompt resumption of CBF. Core-temperature was maintained at 37.0°C±0.1°C using a Digi-Sense temperature controller(Cole-Parmer, Vernon Hills, IL) via heat-lamp coupled with a thermistor rectal-probe(Model 400, Yellow Springs Instruments Co., Yellow Springs, OH). Post-operatively, an animal intensive care unit(Lyon Electric Company, Chula Vista, CA) was employed to maintain a temperature of 37°C for a total of 150 minutes. Animals were sacrificed at 0h, 24h, and 7d, and brains were harvested and prepared for histology as detailed below. Delayed functional evaluation was performed in animals undergoing 30 minutes of MCAO. Sham animals underwent surgical exposure and filament insertion without occlusion.
Antibodies and Reagents
Primary antibodies used for Western blot include rat anti-mouse monoclonal C3a antibody(BD-Pharmingen, San Diego, CA)(1:500 dilution); rabbit polyclonal anti-mouse IgM antibody(Novus Biologicals, Littleton, CO)(1:1000); goat anti-mouse MBL-1 antibody(R&D Systems, Minneapolis, MN)(1:1000); goat anti-mouse MBL-2 antibody(R&D Systems, Minneapolis, MN)(1:1000); rat anti-mouse monoclonal CD68(AbD Serotec, Raleigh, NC)(1:2000); rabbit polyclonal anti-mouse Factor-B antibody(Santa Cruz Biotechnology, Santa Cruz, CA)(1:1000); and goat polyclonal anti-mouse C1q-A antibody(Santa Cruz Biotechnology, Santa Cruz, CA)(1:1000).
Primary antibodies used for immunohistochemistry include monoclonal anti-MAP-2(Sigma-Aldrich, St. Louis, MO)(1:600); rat anti-mouse monoclonal complement component C3 antibody(Cedarlane, Burlington, NC)(1:200); goat anti-mouse polyclonal IgM(AbD Serotec, Raleigh, NC)(1:200); rat anti-mouse monoclonal CD68(AbD Serotec, Raleigh, NC)(1:200); goat anti-mouse MBL-1 antibody(R&D Systems, Minneapolis, MN)(1:200); goat anti-mouse MBL-2 antibody(R&D Systems, Minneapolis, MN)(1:200); rabbit polyclonal anti-human von Willebrand Factor(DakoCytomation, Ely, Cambridgeshire, UK)(1:100).
Histologic Analyses
Following transcardiac PBS perfusion, brains were harvested, fixed in 4% paraformaldehyde, and cryoprotected. Brains were frozen in optimal cutting temperature compound(Sakura Finetek US, Torrance, CA, USA)(-80°C) and cut by cryostat into 20μm coronal sections. For immunofluorescence, sections were blocked with the secondary antibody-appropriate serum(10% donkey or goat serum) with PBS containing 0.2% Triton X-100 for 30min at room temperature. Sections were incubated in primary antibodies diluted in PBS containing 0.2% Triton X-100 overnight at 4°C. Sections were then washed and incubated with the corresponding secondary antibodies Alexa Fluor 488, 594, or 647(Invitrogen, Carlsbad, CA) for 1h at room temperature. Neurotrace 640/660 fluorescent Nissl(Invitrogen, Carlsbad, CA) was used for counterstaining. Sections were then mounted on slides in Vectashield(Vector Laboratories, Burlingame, CA) and visualized using a Bio-Rad 2000 confocal laser-scanning device(Bio-Rad, Hercules, CA, USA) with a Nikon E800 microscope(Nikon, San Diego, CA).
Western Blot
For analysis of protein profiles by sodium dodecyl sulfate–polyacrylamide gel electrophoresis(SDS-PAGE), mice were killed and immediately perfused intracardially with 2ml ice-cold saline prior to brain harvest. The ischemic and contralateral non-ischemic brain tissues were excised and homogenized in lysis buffer(20mM Tris-HCl pH 7.5, 1% Triton, 150mM NaCl, 1mM EDTA, Halt™Proteinase Inhibitor Cocktail). The samples were electrophoresed under reduced conditions on 4-12% gradient SDS–PAGE gels. The proteins were transferred onto nitrocellulose membrane and hybridized with specific primary antibodies. After incubation with appropriate horseradish peroxidase-conjugated secondary antibodies(Jackson ImmunoResearch Laboratories, West Grove, PA), the signal was detected using chemiluminescence(Supersignal West Pico Chemiluminescent Substrate; Pierce, Rockford, IL). The relative band intensity was quantified using ImageJ(version 1.43, NIH, http://rsb.info.nih.gov/ij) and results were normalized to both β-actin and contralateral hemisphere expression using the formula: Normalized intensity=[(ipsilateral pixel density/ipsilateral β-actin density)/(contralateral pixel density/contralateral β-actin density).
Infarct Volume Determination
For animals sacrificed at 24h, mice were euthanized and 1mm sections of brain were stained with TTC as previously described[20]. Infarct volumes were calculated by 2 blinded, independent observers and expressed indirectly to control for hemispheric edema[21].
For animals sacrificed at 7d, brains were cryosectioned into coronal sections at 0.5mm intervals as detailed above. Sections were stained with H&E, and infarct volume was quantified using a standard computer-assisted analysis technique(Image Pro Plus 4.5, Media Cybernetics, Bethesda, MD).
Neurofunctional Assessment
Neurological function was assessed at 24 hours using a previously-described 28-point scale which assesses both focal and global functional deficits[22]. For delayed functional evaluation at 7 days, an extensive battery was employed including the Morris Water-Maze[23, 24], open-field[25], and corner-turn tests[24, 26].
Navigational memory and learning was evaluated using the Morris water-maze[23, 24]. For this test, mice were tested in a pool 80cm in diameter filled with opaque white water at a depth of 20cm. The water temperature was maintained at 26°C. The rescue-landing platform(3cm diameter) was submerged 0.5 cm under the water surface in one of four quadrants. Navigational cues were placed on the walls of the pool and a small flag was placed on the landing platform. The testing began on post-operative day three with four consecutive training days. On each of these days, three attempts were given to each mouse to find the platform within 120 seconds. Each mouse was allowed 20 minutes to rest between swimming trials. The training session occurred at the same time of the day throughout the training and testing periods. A mouse was placed in the water in the quadrant opposite to the landing. The time(in seconds) spent by each animal to find the platform was recorded. If the mouse failed to locate and climb on the platform within the allotted time, it was gently placed on the platform for 30 seconds. On postoperative day 7, mice were placed in the same swimming pool without the landing platform for 60 seconds (probe trial). The time spent in the quadrant where the landing had been previously situated was recorded in a masked fashion. Each animal was afforded only one attempt.
The open-field test was performed on the day of sacrifice to assess spontaneous locomotor activity and exploratory behavior[25]. Mouse behavior was monitored in the open-field apparatus consisting of a square plastic chamber(50×50×25cm). Animals were allowed to start from one of the four corners, selected randomly by the experimenter. Animal location and animal path length data was collected for 10 min after a 2-minute habituation. All movements of each animal in the open-field apparatus were traced and analyzed with commercially-available software(Smart Junior, Harvard Apparatus, Holliston, Massachusetts, US).
Sensorimotor impairment was analyzed using the corner-turn test[24]. For this test, the mouse was allowed to walk down a corridor into a 30° corner. The mouse’s choice of turn direction was noted. The number of right and left turns out of 10 attempts was recorded. The laterality index(LI) and normalized LI were calculated for each mouse according to the formula: LI=(number of right turns-number of left turns)/(total number of turns)[26]. The LI for the day before surgery(LIBS) and each of the post-surgery days was calculated and normalized using the formula:Normalized LI=(LI+2)/(LIBS+2).
Statistical Analysis
All data are presented as mean±standard error of the mean(SEM). Between-group differences were calculated using two-tailed Student’s t-test or Analysis of variance, with Mann-Whitney and Kruskal-Wallis tests for non-parametric data, as appropriate. All values were considered significant when P<0.05. Statistical analyses were performed using commercially-available software(SPSS Statistics 17.0, SPSS Inc., Chicago, IL).
Results
MBL-a/c deposit in association with IgM at the time-point of reperfusion, prior to activation of either the classical or alternative pathways
Western blots for MBL-a/c and IgM were performed in wild-type mice subjected to sham-surgery, as well as animals subjected to MCAO and sacrificed at 0h, 24 hours, and 7 days. This revealed that MBL-a protein expression increases acutely in the ischemic hemisphere following cerebral ischemia/reperfusion, with a peak at 24h, and subsequent decrease by 7 days(sham: 0.82±0.16, 0h: 2.71±0.79, 24h: 3.61±0.70, 7d: 2.61±0.42; p<0.05; n=4-5/time-point)(Figure 1A). MBL-c demonstrates similar increases by the time of reperfusion(0h), also peaking at 24 hours and decreasing by 7 days(sham: 0.92±0.28, 0h: 3.97±1.46, 24h: 6.47±0.95; 7d: 4.21±1.15; p<0.05, n=4-5/time-point)(Figure 1B). Deposition of IgM occurs rapidly in a similar fashion, but peaks at 7 days(sham: 0.95±0.17, 0h: 4.75±2.24, 24h: 3.54±1.17, 7d: 6.92±1.01; p<0.05)(Figure 1C). Thus, MBL-a/c and IgM protein deposition occur along a similar time-course in the ischemic hemisphere.
Figure 1. Cerebral ischemia/reperfusion results in a rapid induction of MBL-a, MBL-c, and IgM antigen deposition in the ischemic hemisphere.
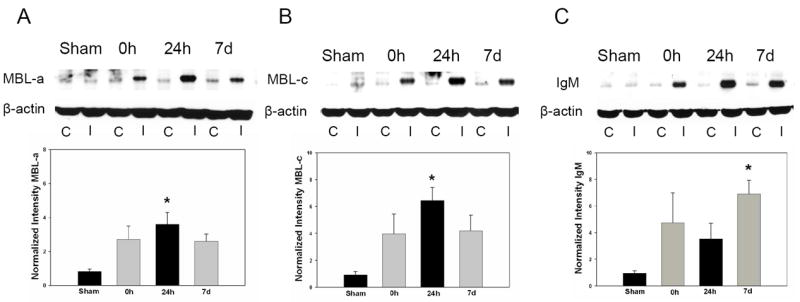
Representative Western blots for MBL-a, MBL-c, and IgM were performed in wild-type mice subjected subjected to sham surgery and following 0h, 24h, or 7 days of reperfusion. C= Contralateral hemisphere, I= Ipsilateral hemisphere. Densitometric analyses represent a composite of multiple independent replicates(n=4-5/time-point). (A) MBL-a protein expression increases acutely in the ischemic hemisphere following cerebral ischemia/reperfusion, with a >3-fold increase in MBL-a at 0h relative to sham animals and a peak at 24h(*p<0.05 vs. sham). (B) MBL-c demonstrates a similar(>6-fold) increase by the point of reperfusion, with a similar peak following 24 hours(*p<0.05 vs. sham). (C) Deposition of IgM was also observed to occur rapidly along a similar time-course, but with a peak at 7 days(*p<0.05 vs. sham).
Immunohistochemistry was also utilized to assess the cellular localization of MBL-a/c and IgM antigen. At 0h post-reperfusion, MBL-a co-localizes with both IgM and C3 on the endothelial surface of ischemic microvessels(Figure 2A). MBL-c demonstrates a similar pattern of early endothelial deposition, including co-localization with IgM and C3 at the same time-point(Figure 2B). The endothelial localization of the MBL-a/c antigen was confirmed by confocal microscopy(Figure 2C, D). In all samples, MBL-a/c expression at the 0h time-point was confined to the ischemic territory, and was not observed in non-ischemic ipsilateral territories or in contralateral brain regions(data not shown).
Figure 2. MBL-a and MBL-c deposit in association with IgM and C3 on ischemic microvessels at 0h post-reperfusion.
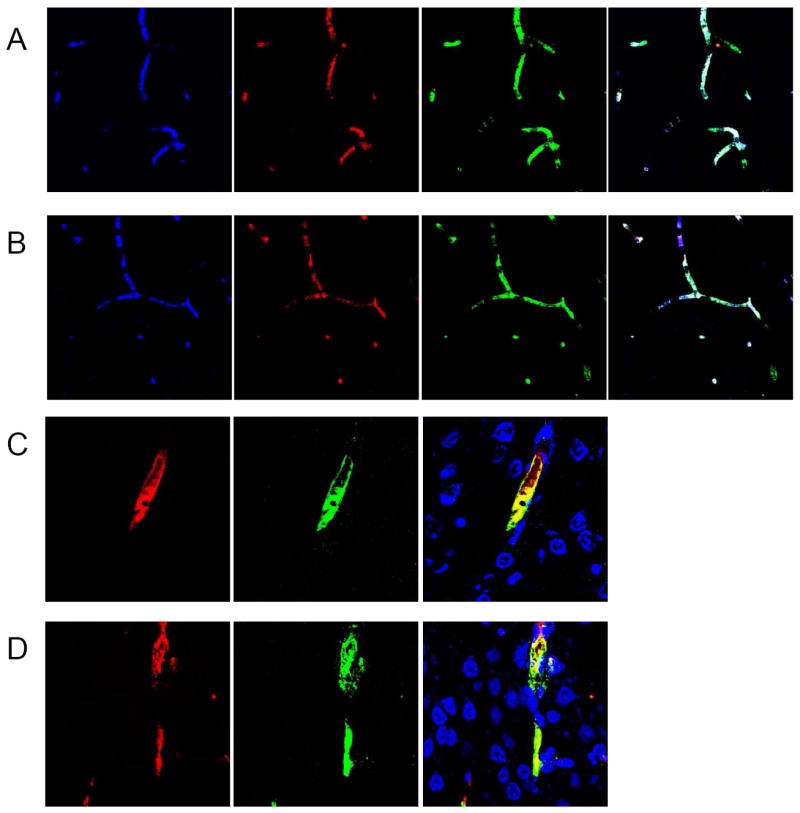
(A) Immunostaining for MBL-a(blue), C3(red), IgM(green), and triple-stain at 0h reveals that MBL-a protein associates with IgM as well as downstream complement components on structures resembling microvessels 0h following reperfusion. (B) At 0h, similar immunostaining for MBL-c(blue), C3(red), IgM(green), and triple-stain reveals that MBL-c deposits with IgM and downstream complement components in a similar pattern at an early time-point(0h) following reperfusion. (C) Immunohistochemistry performed for MBL-a(red) and endothelial marker(vWF-green) on frozen sections obtained from animals sacrificed at 0h post-reperfusion demonstrate unambiguous co-localization(yellow), confirming endothelial localization for MBL-a. (D) Immunohistochemistry performed for MBL-c(red) and endothelial marker(vWF-green) at the same time-point also demonstrate unambiguous endothelial co-localization(yellow).
At 24 hours, MBL-a co-localizes with both IgM and C3 on the surface of cells with neuronal morphology in the ischemic region(Figure 3A). The neuronal identity of these cells was confirmed by co-staining with MAP-2(Figure 3C). By contrast, MBL-c remained associated with C3 in the peri-vascular region, but did not bind to neurons(Figure 3B, Figure 3D).
Figure 3. MBL-a deposits on ischemic neurons at 24h, whereas MBL-c remains in the perivascular region.
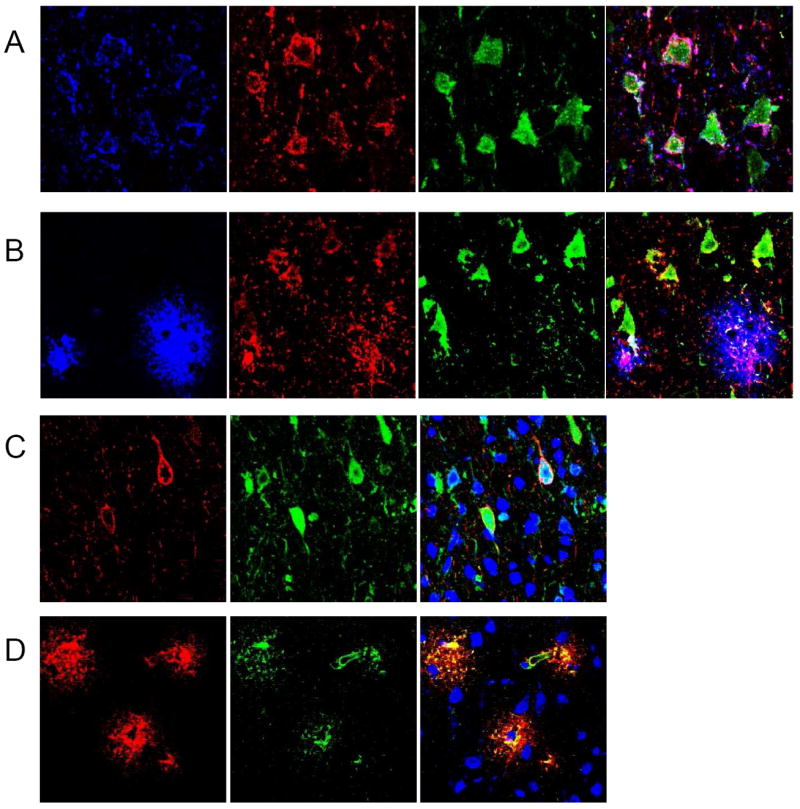
(A) Immunostaining of the ischemic region at 24h post-reperfusion for MBL-a(blue), C3(red), IgM(green), and triple-stain reveals that MBL-a is observed in association with IgM and triggers cleavage of C3 on cells with the morphologic characteristics of neurons. (B) Immunostaining of the ischemic region for MBL-c(blue), C3(red), IgM(green) and triple-stain at 24h post-reperfusion in the same anatomic region reveals that although MBL-c antigen is deposited in association with C3, it is not found on neuronal cells, whereas C3 and IgM co-localize to cells with the morphology of neurons. (C) Immunostaining for MBL-a(red) and MAP-2(green), and double-staining in the same anatomic region at 24h unambiguously confirms the neuronal phenotype of these cells. (D) Further staining in the same region for MBL-a(red), vWF(green), and double-stain suggests that MBL-c antigen remains in and around ischemic microvessels at this time-point. Merged images for all panels are co-stained with Nissl as a nuclear marker (blue).
By comparison, Western blot performed on the same brain samples demonstrates no significant deposition of either factor B(sham: 1.16±0.19, 0h: 1.06±0.28, 24h: 21.65±5.22; p=0.001; n=4/time-point)(Figure 4A) or C1q(sham: 0.99±0.09, 0h: 0.94±0.07, 24h: 2.10±0.35; p=0.006; n=4/time-point)(Figure 4B) at 0h, relative to expression at 24h. Taken together, these data demonstrate that circulating MBL deposits on ischemic endothelium in association with IgM at the time of reperfusion, triggering downstream complement activation prior to activation of the classical and alternative pathways.
Figure 4. Early post-ischemic cerebral complement activation does not occur via the classical or alternative pathways.
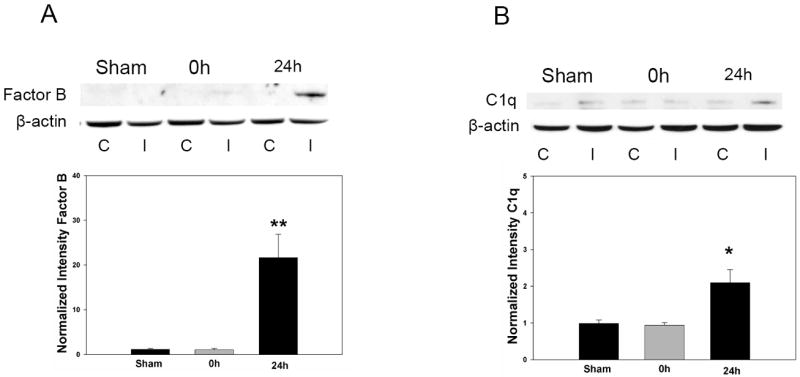
Representative Western blots for Factor B and C1q were performed in wild-type mice (n=4/time-point) subjected to sham surgery or following 0h or 24h of reperfused MCAO. C= Contralateral hemisphere, I= Ipsilateral hemisphere. Densitometric analyses of multiple independent replicates confirms that both alternative(factor B) and classical(C1q) pathway induction is not observed at the same early time point where MBL deposition is prominent. (A) Factor B deposition is significantly increased relative to both sham animals (**P<0.01) and animals sacrificed at 0h(**P<0.01). (B) C1q is also significantly increased at 24h post-ischemia (*P<0.05 relative to sham, and *P<0.05 relative to 0h). Therefore, the MBL pathway is primarily responsible for complement activation at the very early time-point of reperfusion.
Genetic deficiency of MBL suppresses C3 cleavage following reperfused stroke
Western blot for C3a expression was then performed in both MBL-KO and wild-type mice sacrificed at 24 hours and 7 days post-MCAO. This analysis revealed that MBL deletion suppresses ipsilateral C3a expression[24h: 4.78±1.78(n=8) vs. 127.6±24.4(n=7), p=0.0003; 7d: 2.43± 1.3(n=3) vs. 23.6±14.1(n=5), p=0.04](Figure 5A) relative to that of WT mice. Therefore, genetic deletion of MBL attenuates downstream complement activation in the ischemic hemisphere at both acute and subacute time-points.
Figure 5. Genetic deficiency of MBL-a/c suppresses post-ischemic complement activation at 24h and 7 days, as well as decreases CD68 positivity in the ischemic hemisphere at 7 days.
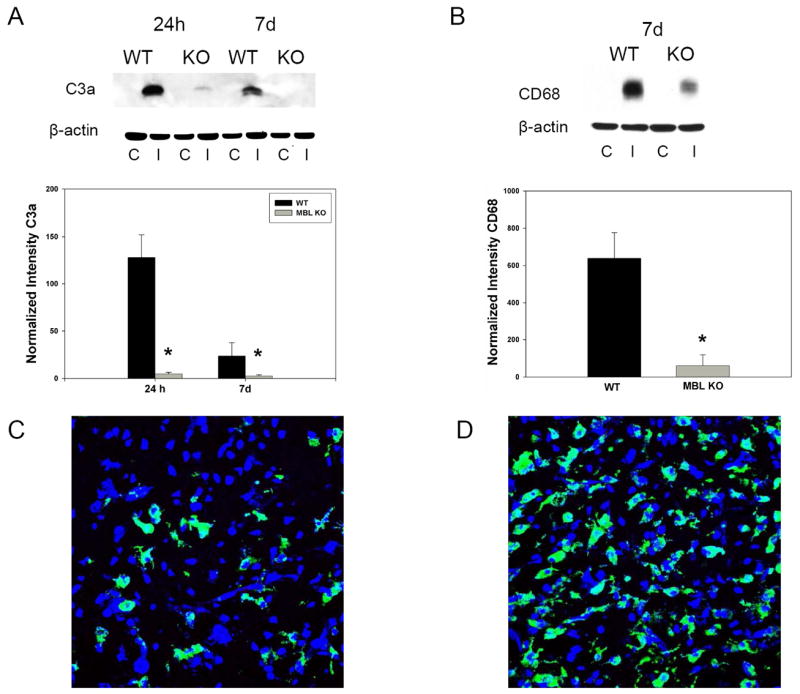
(A) Representative Western blot for C3a in both MBL-KO and wild-type mice reveals that MBL deletion suppresses ipsilateral C3a expression[24h: 4.78±1.78(n=8) vs. 127.6±24.4(n=7), p=0.0003; 7d: 2.43±1.3(n=3) vs. 23.6±14.1(n=5), p=0.04]relative to that of WT mice. C= Contralateral hemisphere, I= Ipsilateral hemisphere. (B) Western blot reveals that MBL-KO mice demonstrate significant reductions in CD68 protein in the ischemic hemisphere at 7 days relative to WT controls [60.37±50.01(n=3) vs. 638.2±138.0(n=5)](*p=0.04). (C) Representative immunostain for CD68(green) and Nissl(blue) used as a nuclear marker in sections obtained from the region of the infarct in a MBL-KO subjected to transient MCAO and sacrificed at 7 days. (D) Immunostaining of the corresponding anatomic region in WT mice reveals a dramatic increase in CD68-positive cells relative to that observed in KO animals.
MBL deletion suppresses inflammatory mononuclear cell infiltrate in the subacute phase of stroke recovery
Mononuclear inflammatory cells(microglia/macrophage) in the ischemic hemisphere were assessed by immunostaining and Western blot for the cell surface protein CD68. At 24h, no significant increase in CD68 immunopositivity was observed by Western Blot, and immunostaining did not reveal significant CD68 antigen in the ischemic territory(data not shown). In contrast, Western blot performed of brain sections obtained from WT and MBL-KO animals sacrificed at 7 days revealed a significant suppression of CD68 immunopositivity in the ischemic hemisphere of KO animals[60.37±50.01(n=3) vs. 638.2±138.0(n=5); p=0.04](Figure 5B). This was supported by immunostaining studies revealing significantly decreased numbers of CD68 positive cells in the region of the infarct in MBL-KO mice(Figure 5C) relative to the corresponding anatomic regions of WT control brain(Figure 5D). Therefore, genetic deletion of MBL suppresses the accumulation of mononuclear inflammatory cells in the ischemic hemisphere in the subacute post-stroke period.
Genetic Deletion of MBL confers robust neuroprotection at 24 hours but not 7 days following MCAO
The functional effect of genetic MBL deletion was assessed by subjecting MBL-KO mice and their wild-type controls to transient MCAO. As variations in cerebrovascular anatomy between strains is known to result in differential susceptibility to cerebral ischemia in mice[27], we stained the circle of Willis by infusing a 1% solution of methylene blue in saline in mice deficient in MBL as well as WT mice (n=3 per cohort)[28]. We did not observe any gross differences in cerebrovascular anatomy between these cohorts of mice (Figure 6).
Figure 6. Comparison of cerebrovascular anatomy in mice deficient in MBL-a/c and wild-type controls.
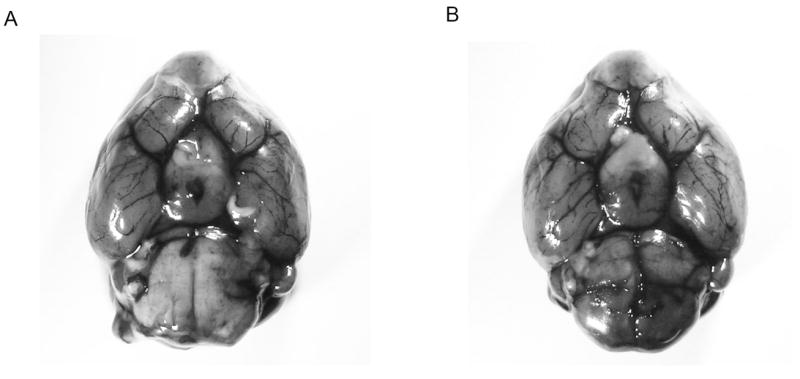
Methylene blue staining of the cerebrovascular anatomy from a ventral perspective reveals no gross anatomic differences in the vascular pattern of the cerebral circulation between (A) MBL-KO and (B) WT mice (representative photograph of n=3 per cohort). Importantly, both genotypes demonstrate bilaterally patent posterior communicating arteries.
At 24 hours, MBL-null mice demonstrated significant improvements in both neurological function[10.2±1.8(n=19) vs. 15.7±1.7(n=18); p=0.01] and infarct volume[39.9±2.5%(n=14) vs. 60.0±2.4%(n=15); p=0.004](Figure 7A). There was no difference in rates of mortality between these cohorts(p=NS). When mice were survived to 7 days, however, infarction volume was not significantly improved in MBL-KO mice[31.4±4.3mm3(n=12) vs. 39.5±6.9mm3(n=10), p=0.54](Figure 7B). The mortality rates at 7 days also did not differ between the cohorts (p=NS). Additionally, no functional benefit for genetic MBL-deletion was observed at 7 days on any functional tests employed[Corner-Turn(Laterality Index): 1.10±0.15(n=12) vs. 1.42±0.22(n=10), p=0.25; Water-Maze(Latency Time, sec): 16.8±3.3(n=11) vs. 15.7±2.0(n=9), p=0.78; Open Field(Total number of crossings): 17.7±3.7(n=12) vs. 22.8±4.7(n=10), p=0.40; Open Field(Ambulatory distance, cm): 1070.2± 126.2(n=12) vs. 1282.6±151.0(n=10), p=0.25](Figure 7C). Thus, the acute neuroprotection afforded by genetic MBL-deficiency is not sustained when assessed at 7 days.
Figure 7. Genetic deletion of MBL confers robust neuroprotection following transient cerebral ischemia, but this effect is not sustained when evaluated at 7 days.
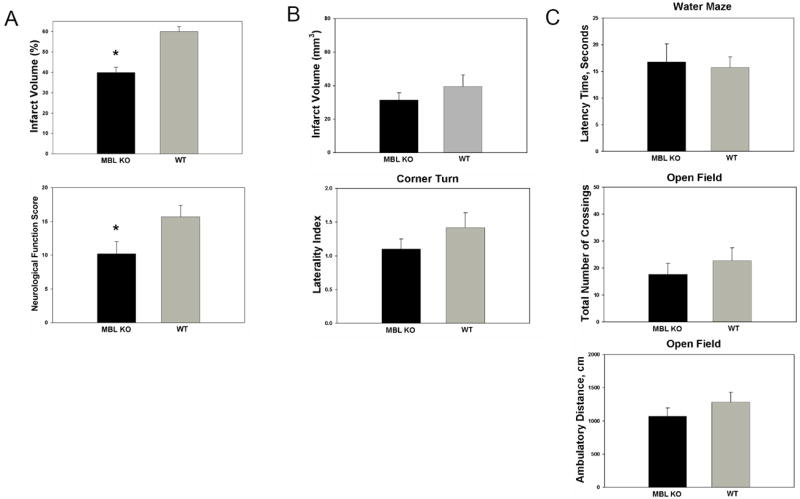
(A) At 24 hours, MBL-null mice demonstrate significant improvement in both infarct volume[39.9±2.5%(n=14) vs. 60.0±2.4%(n=15); p=0.004] and neurological function[10.2±1.8(n=19) vs. 15.7±1.7(n=18); *p=0.01]. (B) At 7 days, infarct volume, determined by analysis of H&E stained sections [31.4±4.3mm3(n=12) vs. 39.5±6.9mm3(n=10), p=0.54] and neurological function, assessed by the corner-turn test[Corner-Turn(Laterality Index): 1.10±0.15(n=12) vs. 1.42±0.22(n=10), p=0.25] were not improved in MBL-KO mice. (C) The early functional improvement associated with genetic MBL-deletion was not sustained on any of the tests employed at 7 days, including the Water-Maze(Latency Time, sec)[16.8±3.3(n=11) vs. 15.7±2.0(n=9), p=0.78], Open-Field(Number of crossings)[17.7±3.7(n=12) vs. 22.8±4.7(n=10), p=0.40], or Open-Field(Ambulatory distance)[1070.2± 126.2(n=12) vs. 1282.6±151.0(n=10), p=0.25] tests.
Discussion
The present study demonstrates that the MBL pathway serves as a critical mechanism of post-ischemic complement activation in the early post-stroke period. Following transient MCAO, MBL-a and MBL-c deposit together with C3 and IgM on ischemic endothelium. By 24 hours, only MBL-a associates with C3 and IgM on neurons in the region of the infarct. In accord with prior work[13], genetic deficiency of MBL suppresses cleavage of C3 and confers robust neuroprotection when assessed at 24h. When outcome was assessed at 7 days, however, this neuroprotective effect was not sustained. Therefore, while the MBL pathway is essential to deleterious complement-mediated ischemic injury in the acute phase of stroke, continued suppression of MBL into the subacute period abrogates this neuroprotection.
Few studies have focused on elucidating the proximal mechanisms responsible for the assembly of the C3-convertase following cerebral ischemia[1, 6, 29]. Multiple independent groups have examined the contribution of the classical pathway to stroke injury and have concluded that complement-mediated post-ischemic injury occurs independent of the classical pathway[1, 30]. In contrast, no study has been published in which selective inhibition of the alternative pathway has been employed in the setting of stroke. Inspired by a series of papers implicating MBL in the pathogenesis of ischemia/reperfusion injury in other organs[31], two competing groups have recently published studies suggesting that MBL-mediated complement activation serves a deleterious role in stroke. First, Cervera et al. directly assessed the role of MBL in stroke using MBL-KO mice subjected to transient MCAO. This study revealed that genetic MBL deletion resulted in smaller infarcts and improved functional outcome when assessed at 48h[13]. More recently, Morrison et al. also found that MBL-KO mice subjected to transient MCAO exhibited significant reductions in striatal injury relative to wild-type controls[14]. The authors of these studies imply that MBL may serve as an effective therapeutic target in stroke.
The present study provides further detail regarding the temporal and spatial sequence of post-ischemic cerebral complement activation. Herein, we demonstrate that MBL is the first complement protein to bind to ischemic cerebral endothelium, where it co-localizes with both IgM and C3 as early as 0h post-reperfusion. This study is also the first to provide evidence that IgM may be involved in the initiation of post-ischemic complement activation in the brain, and that this initial interaction occurs at the ischemic endothelial surface. Whether IgM or MBL directly bind the putative neo-antigens presented by ischemic endothelium, as well as the specific identity of these antigens, however, remains to be elucidated. By comparison, no significant deposition of either Factor B or C1q is observed at the same early time-point, supporting a predominant role for MBL as the proximal mechanism of complement activation in the acute phase of stroke. By 24 hours post-reperfusion, MBL-a convincingly co-localizes with neuronal-specific cellular markers in the ischemic region, while MBL-c remains restricted to the perivascular regions. This suggests that propagation of complement-mediated tissue injury likely involves damage and subsequent breakdown of the blood-brain barrier. Moreover, this differential pattern of staining suggests that MBL-a and MBL-c may serve distinct functions following cerebral ischemia, although this remains unproven.
Our data confirm a robust neuroprotective effect for genetic MBL deletion in the acute post-stroke period. However, as delineated by the STAIR criteria[32], evaluation beyond the acute phase is essential to properly assess the efficacy of a candidate neuroprotective strategy. In fact, in the present study, mice genetically-deficient in MBL do not exhibit improvements in either infarct volume or neurological function when examined at 7 days. This lack of neuroprotection occurs despite a reduction in C3a levels reflective of persistent suppression of downstream complement activation. Previous work by our group and others has demonstrated that C3a expression is associated with worse outcome secondary to increased granulocyte/neutrophil infiltration into the ischemic region[1, 6], and that MBL-inhibition suppresses acute inflammation through a reduction in neutrophil infiltration when assessed at an early time-point[13]. The results of the present study suggest that MBL and downstream complement deposition may play a different role in ischemic injury at later time-points.
At 7 days post-stroke, animals deficient in MBL exhibit a reduction in CD68 positive cells, which represent a predominant class of inflammatory cells present in the subacute infarct[33]. The persistent suppression of post-ischemic inflammation observed in the MBL-KO mice likely contributes to the loss of neuroprotection in the subacute period. For example, inflammatory cells, in particular activated microglia and infiltrating macrophages, are known to promote subacute post-ischemic repair both through phagocytosis of damaged cells as well as through the production of growth factors, such as NGF (Nerve growth factor) and BDNF (Brain-derived neurotrophic factor), and anti-inflammatory cytokines, such as IL-10(Interleukin-10)[34]. Future experiments will directly evaluate the neuroprotective mechanisms associated with MBL suppression in the later phases of stroke. Alternatively, it remains possible that genetic MBL deficiency simply delays the progression of ischemic injury, so that neuroprotection observed with early outcome assessment is not maintained at later time-points. A third possibility is that complement itself, functioning either directly through MBL or through downstream effector components, may serve a beneficial function at later time points following cerebral I/R[35]. For instance, recent work suggests a positive regulatory role for complement in ischemia-induced neurogenesis[18]. Furthermore, complement also functions in the clearance of apoptotic cells, and deposition of MBL as well as other downstream opsonins, such as C3b or C5b, may serve a protective role by clearing apoptotic cells or cellular debris in the subacute period[36]. Clearly, gene-deficient animals do not represent the ideal model to assess the functional effects complement inhibition across time, as they manifest suppressed complement at all times in all cell types. These concepts are supported by recent study by Rahpeymai et al., who found that C3-deficient mice subjected to MCAO exhibited larger infarct volumes when assessed at 7 days, despite a robust neuroprotection observed in these same mice when assessed at 24 hours[1, 18]. Therefore, further experiments utilizing either conditional mutants or rationally-targeted pharmacotherapy must be employed to dissect the multiple possible functional roles for MBL, as well as both its possible deleterious and beneficial effects, across the post-ischemic recovery period.
Conclusion
Careful analysis of the timing and localization of complement activation are critical to the development of effective anti-complement neuroprotective strategies for stroke. The present study delineates the temporal and spatial sequence of MBL deposition and downstream complement cleavage at early time-points following cerebral ischemia. Given the rapid deposition of MBL, pharmacologic strategies of MBL inhibition may prove most successful with pre-ischemic dosing, such as with high-risk patients undergoing neurosurgical or interventional procedures involving cerebral revascularization and/or temporary occlusion. Additionally, the lack of neuroprotection observed when MBL-inhibition is maintained for 7 days suggests that strategies targeting MBL, and complement in general, must be carefully temporally-tailored in order to maximize inhibition of early deleterious inflammation while retaining or even augmenting beneficial inflammatory responses. Despite these caveats, rationally-applied anti-complement therapeutic strategies remain promising in the treatment of post-ischemic cerebral reperfusion injury.
Acknowledgments
This work was supported by a grant from the National Institutes of Health R01 (NS40409 to E.S.C.).
Footnotes
Conflict of Interest Disclosure
The authors have no financial or competing interests to disclose.
References
- 1.Mocco J, Mack WJ, Ducruet AF, Sosunov SA, Sughrue ME, Hassid BG, et al. Complement component C3 mediates inflammatory injury following focal cerebral ischemia. Circ Res. 2006 Jul 21;99(2):209–17. doi: 10.1161/01.RES.0000232544.90675.42. [DOI] [PubMed] [Google Scholar]
- 2.del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ. Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol. 2000 Jan;10(1):95–112. doi: 10.1111/j.1750-3639.2000.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jean WC, Spellman SR, Nussbaum ES, Low WC. Reperfusion injury after focal cerebral ischemia: the role of inflammation and the therapeutic horizon. Neurosurgery. 1998 Dec;43(6):1382–96. doi: 10.1097/00006123-199812000-00076. discussion 96-7. [DOI] [PubMed] [Google Scholar]
- 4.Mocco J, Sughrue ME, Ducruet AF, Komotar RJ, Sosunov SA, Connolly ES., Jr The complement system: a potential target for stroke therapy. Adv Exp Med Biol. 2006;586:189–201. doi: 10.1007/0-387-34134-X_13. [DOI] [PubMed] [Google Scholar]
- 5.Huang J, Kim LJ, Mealey R, Marsh HC, Jr, Zhang Y, Tenner AJ, et al. Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999 Jul 23;285(5427):595–9. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- 6.Ducruet AF, Hassid BG, Mack WJ, Sosunov SA, Otten ML, Fusco DJ, et al. C3a receptor modulation of granulocyte infiltration after murine focal cerebral ischemia is reperfusion dependent. J Cereb Blood Flow Metab. 2008 May;28(5):1048–58. doi: 10.1038/sj.jcbfm.9600608. [DOI] [PubMed] [Google Scholar]
- 7.Kim GH, Mocco J, Hahn DK, Kellner CP, Komotar RJ, Ducruet AF, et al. Protective effect of C5a receptor inhibition after murine reperfused stroke. Neurosurgery. 2008 Jul;63(1):122–5. doi: 10.1227/01.NEU.0000335079.70222.8D. discussion 5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gesuete R, Storini C, Fantin A, Stravalaci M, Zanier ER, Orsini F, et al. Recombinant C1 inhibitor in brain ischemic injury. Ann Neurol. 2009 Sep;66(3):332–42. doi: 10.1002/ana.21740. [DOI] [PubMed] [Google Scholar]
- 9.Arumugam TV, Woodruff TM, Lathia JD, Selvaraj PK, Mattson MP, Taylor SM. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience. 2009 Feb 6;158(3):1074–89. doi: 10.1016/j.neuroscience.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997 Apr 3;386(6624):506–10. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- 11.Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose-binding lectin. Am J Physiol Heart Circ Physiol. 2009 Nov;297(5):H1853–9. doi: 10.1152/ajpheart.00049.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel S, et al. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol. 2006 Oct 1;177(7):4727–34. doi: 10.4049/jimmunol.177.7.4727. [DOI] [PubMed] [Google Scholar]
- 13.Cervera A, Planas AM, Justicia C, Urra X, Jensenius JC, Torres F, et al. Genetically-defined deficiency of mannose-binding lectin is associated with protection after experimental stroke in mice and outcome in human stroke. PLoS One. 5(2):e8433. doi: 10.1371/journal.pone.0008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison H, Frye J, Davis-Gorman G, Funk J, McDonagh P, Stahl G, et al. The Contribution of Mannose Binding Lectin to Reperfusion Injury after Ischemic Stroke. Curr Neurovasc Res. Jan 5; doi: 10.2174/156720211794520260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kriz J. Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol. 2006;18(1-2):145–57. doi: 10.1615/critrevneurobiol.v18.i1-2.150. [DOI] [PubMed] [Google Scholar]
- 16.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006 Jan;147(Suppl 1):S232–40. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, et al. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol. 2004 Sep 1;173(5):3044–50. doi: 10.4049/jimmunol.173.5.3044. [DOI] [PubMed] [Google Scholar]
- 18.Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. Embo J. 2006 Mar 22;25(6):1364–74. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connolly ES, Jr, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996 Mar;38(3):523–31. doi: 10.1097/00006123-199603000-00021. discussion 32. [DOI] [PubMed] [Google Scholar]
- 20.Connolly ES, Jr, Winfree CJ, Springer TA, Naka Y, Liao H, Yan SD, et al. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J Clin Invest. 1996 Jan 1;97(1):209–16. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993 Jan;24(1):117–21. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- 22.Clark WM, Lessov NS, Dixon MP, Eckenstein F. Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurol Res. 1997 Dec;19(6):641–8. doi: 10.1080/01616412.1997.11740874. [DOI] [PubMed] [Google Scholar]
- 23.Ten VS, Bradley-Moore M, Gingrich JA, Stark RI, Pinsky DJ. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav Brain Res. 2003 Oct 17;145(1-2):209–19. doi: 10.1016/s0166-4328(03)00146-3. [DOI] [PubMed] [Google Scholar]
- 24.Rynkowski MA, Kim GH, Garrett MC, Zacharia BE, Otten ML, Sosunov SA, et al. C3a receptor antagonist attenuates brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2009 Jan;29(1):98–107. doi: 10.1038/jcbfm.2008.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kilic E, Kilic U, Bacigaluppi M, Guo Z, Abdallah NB, Wolfer DP, et al. Delayed melatonin administration promotes neuronal survival, neurogenesis and motor recovery, and attenuates hyperactivity and anxiety after mild focal cerebral ischemia in mice. J Pineal Res. 2008 Sep;45(2):142–8. doi: 10.1111/j.1600-079X.2008.00568.x. [DOI] [PubMed] [Google Scholar]
- 26.Bouet V, Freret T, Toutain J, Divoux D, Boulouard M, Schumann-Bard P. Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp Neurol. 2007 Feb;203(2):555–67. doi: 10.1016/j.expneurol.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993 Jul;13(4):683–92. doi: 10.1038/jcbfm.1993.87. [DOI] [PubMed] [Google Scholar]
- 28.Visocchi M, Argiolas L, Meglio M, Cioni B, Basso PD, Rollo M, et al. Spinal cord stimulation and early experimental cerebral spasm: the “functional monitoring” and the “preventing effect”. Acta Neurochir (Wien) 2001;143(2):177–85. doi: 10.1007/s007010170126. [DOI] [PubMed] [Google Scholar]
- 29.Atkinson C, Zhu H, Qiao F, Varela JC, Yu J, Song H, et al. Complement-dependent P-selectin expression and injury following ischemic stroke. J Immunol. 2006 Nov 15;177(10):7266–74. doi: 10.4049/jimmunol.177.10.7266. [DOI] [PubMed] [Google Scholar]
- 30.De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. Am J Pathol. 2004 May;164(5):1857–63. doi: 10.1016/S0002-9440(10)63744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger SP, Daha MR. Emerging role of the mannose-binding lectin-dependent pathway of complement activation in clinical organ transplantation. Curr Opin Organ Transplant. Dec 13; doi: 10.1097/MOT.0b013e3283425509. [DOI] [PubMed] [Google Scholar]
- 32.Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke. 1999 Dec;30(12):2752–8. doi: 10.1161/01.str.30.12.2752. [DOI] [PubMed] [Google Scholar]
- 33.Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009 May;40(5):1849–57. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 34.McCombe PA, Read SJ. Immune and inflammatory responses to stroke: good or bad? Int J Stroke. 2008 Nov;3(4):254–65. doi: 10.1111/j.1747-4949.2008.00222.x. [DOI] [PubMed] [Google Scholar]
- 35.Mastellos D, Lambris JD. Complement: more than a ‘guard’ against invading pathogens? Trends Immunol. 2002 Oct;23(10):485–91. doi: 10.1016/s1471-4906(02)02287-1. [DOI] [PubMed] [Google Scholar]
- 36.Trouw LA, Blom AM, Gasque P. Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol. 2008 Mar;45(5):1199–207. doi: 10.1016/j.molimm.2007.09.008. [DOI] [PubMed] [Google Scholar]