

Abstract
Motivation: Bioimaging techniques rapidly develop toward higher resolution and dimension. The increase in dimension is achieved by different techniques such as multitag fluorescence imaging, Matrix Assisted Laser Desorption / Ionization (MALDI) imaging or Raman imaging, which record for each pixel an N-dimensional intensity array, representing local abundances of molecules, residues or interaction patterns. The analysis of such multivariate bioimages (MBIs) calls for new approaches to support users in the analysis of both feature domains: space (i.e. sample morphology) and molecular colocation or interaction. In this article, we present our approach WHIDE (Web-based Hyperbolic Image Data Explorer) that combines principles from computational learning, dimension reduction and visualization in a free web application.
Results: We applied WHIDE to a set of MBI recorded using the multitag fluorescence imaging Toponome Imaging System. The MBI show field of view in tissue sections from a colon cancer study and we compare tissue from normal/healthy colon with tissue classified as tumor. Our results show, that WHIDE efficiently reduces the complexity of the data by mapping each of the pixels to a cluster, referred to as Molecular Co-Expression Phenotypes and provides a structural basis for a sophisticated multimodal visualization, which combines topology preserving pseudocoloring with information visualization. The wide range of WHIDE's applicability is demonstrated with examples from toponome imaging, high content screens and MALDI imaging (shown in the Supplementary Material).
Availability and implementation: The WHIDE tool can be accessed via the BioIMAX website http://ani.cebitec.uni-bielefeld.de/BioIMAX/; Login: whidetestuser; Password: whidetest.
Supplementary information: Supplementary data are available at Bioinformatics online.
Contact: tim.nattkemper@uni-bielefeld.de
1 INTRODUCTION
Bioimage informatics has been established as a new branch in the tree of bioinformatics' fields of research in the last 10 years. The term bioimage comprises all kinds of images generated for biological samples in a biological or biomedical research context using a large diversity of imaging techniques. The techniques range from standard ones such as bright field imaging or phase contrast to advanced technologies that enable recording many molecular variables for each resolvable volume unit. The latter group of technologies can also be referred to as multivariate bioimages (MBIs; Herold et al., 2011). MBI belong to the so called high-content imaging techniques which apply high resolution imaging in time and/or space and/or variables to close those open gaps in systems biology which cannot be bridged by standard, i.e. non-spatial omics techniques (Megason and Fraser, 2007; Starkuviene and Pepperkok, 2007). While these can in principle resolve the almost complete molecular composition in a sample on different levels (genomics, transciptomics, proteomics and metabolomics) they have to leave out the spatial domain. In contrast to that, bioimaging approaches, which usually work with a lower level of molecular resolution, can relate molecular information to spatial features such as morphology.
Typical examples for MBI are Matrix Assisted Laser Desorption / Ionization (MALDI) imaging (Cornett et al., 2007), vibrational spectroscopy/Raman microscopy (van Manen et al., 2005) or MultiEpitope-Ligand Cartography (MELC)/Toponome Imaging System (TIS) (Schubert et al., 2006). The first two techniques measure molecular features and interactions in localized spectra, arranged in a pixel grid. The interpretation of the obtained images aims at the identification of pixel groups that share particular or similar spectral features (e.g. Alexandrov et al., 2010) where as the final identification of molecules and a semantic interpretation remains an unsolved problems for most applications. In contrast to that, MELC/TIS (for the sake of compactness we will refer to this technique with TIS) imaging aims at the imaging of a selected set of N proteins using a library of N fluorescent labeled antibodies, lectins or other specific ligands (referred to as tags, in general) in combination with a cyclic protocol of staining, fluorescence imaging and soft bleaching. To unfold the full potential of all these kinds of MBI, new algorithms and software are needed that allow researchers to visually explore the data and to identify the hidden regularities. In this article, we will focus on images recorded using the TIS technology, however our method is definitely applicable to other MBI data recorded with a different multitag technology or MALDI images as well.
For one selected field of view (FOV) in the sample, TIS records one multivariate image T(s) which consists of a set of N aligned images g(s)a(x, y)a=1,…,N (with x, y as pixel coordinates) with s (s=1,…, S) describing the ID of the TIS image/FOV and g(s)a denoting the fluorescence gray value image for tag a. In practice, a number of S TIS runs with one library of N>10 tags are applied to record a set of S datasets. With gx,y=(g1, g2,…, gN)x,y we will refer to the N gray values for the respective N tags assigned to one pixel (x, y) in a TIS image T(s). To align the N fluorescence images in one TIS image, phase contrast images are recorded in each cycle and used as a reference.
One TIS image or a set of S TIS images resembles a high-dimensional complex data structure that encodes hidden relationships between colocation of proteins and the spatial distribution pattern, which is also referred to as the toponome (Schubert et al., 2006). While on the one hand, the gain in molecular information through toponome data may undoubtedly have the potential to lead to a new understanding of functional molecular networks, the analysis of TIS data represents a new challenging problem with a large number of open issues for bioimage informatics on the other hand. It is evident that by visual inspection of each one of the N single gray value images, colocation of proteins can hardly be identified. Likewise, iteratively superimposing three out of the N images or even all images to obtain RGB fusion images is not feasible for protein network identification since an observer would need to analyze a number of N!/(3!(N−3)!) visualizations and link the results obtained for each image triplet, which is impossible for human observers.
One straightforward way to reduce the complexity of the data is to apply a threshold to each image. Schubert et al. (2006) applied such a method for pixel-wise extraction of binary colocation and anti-colocation vectors, termed combinatorial molecular phenotypes (CMPs), by manually thresholding each image ga(s) for a combinatorial analysis. Random colors are subsequently assigned to each of the n detected CMPs to construct so called toponome maps which encode the spatial location of each CMP with its individual color. Although the concept of binary CMPs has the advantage of a fundamental reduction of data complexity and a clear interpretation on the level of a single CMP, thresholding each image by manual human interaction features several disadvantages. It is quite time consuming and requires a high level of expertise to set reasonable thresholds. Slight modifications of the threshold can lead to different CMP lists, potentially affecting the interpretation of the data. Furthermore, thresholding discards information inherent in the data, so analyzing non-binarized gray value images may be better suited to track protein locations in the cell (Friedenberger et al., 2007). However, the CMP concept has successfully been applied in several studies (Bhattacharya et al., 2010; Bonnekoh et al., 2006; Eyerich et al., 2009; Ruetze et al., 2010), for example revealing proteins controlling the molecular networks of tumor cell lines, or finding CMPs to distinguish between healthy patients, patients with psoriasis and patients with atopic dermatitis. But even regardless of the aforementioned thresholding issue, we believe that the CMP-based visualization concept should be reconsidered as follows. From a visualization point of view, mapping the CMP to random colors follows the idea to treat CMP as nominal variables. On the one hand, this perspective on a colocation pattern is well motivated since similar patterns (CMPs) can constitute different functions (similarity may be quantified using the Hamming distance for binary patterns). But on the other hand, one should also bear in mind that similar patterns may also belong to the same functional group or to the same hierarchically organized network. Another drawback of using random colors for CMPs is that the morphological structure in a random color map can be hard to interpret since the colorful map can overburden the cognitive skills of a user. So an alternative visualization concept is definitely needed, that maps similar patterns to similar colors. In other words, one needs a pseudocoloring that preserves the topology of the N-dimensional fluorescence colocation feature space. In summary, a new method for visual data mining TIS images is needed that features he following. First it has to provide an overview on the entire image using a pseudocolor visualization. Second, it has to support the identification and display of relevant gray value-based protein colocation patterns, referred to as MCEPs (Molecular Co-Expression Phenotypes). Third, the perception of similarities and contrasts in the expressed MCEPs must be possible. Fourth, filtering and zooming must be supported in both domains, tissue morphology and protein colocation.
In this article, we present the visual data mining tool WHIDE (Web-based Hyperbolic Image Data Explorer), which offers the four functions listed above. The idea behind WHIDE is to identify MCEP in TIS images using a special variant of the self-organizing map, the hierarchical hyperbolic self-organizing map (H2SOM), in combination with state-of-the-art internet browser technology and information visualization concepts. Compared with standard SOMs, hyperbolic SOMs have the potential to achieve much better low-dimensional embeddings, since they offer more space due to the effect, that in a hyperbolic plane the area of a circle grows asymptotically exponential with its radius (see Supplementary Material for details). This feature has been identified as a solution to the so called focus and context problem in information visualization (Ware, 2004) by other researchers as well, like in the famous hyperbolic tree browser (Lamping et al., 1995). The tool is integrated in our full-web-based online bioimage analysis platform BioIMAX (BioImage Mining, Analysis and eXploration; Loyek et al., 2011) which uses state-of-the-art web graphics tool kits to realize an online bioimage analysis workbench as a Rich Internet Application (RIA) (see access details given above and details given in the Supplementary Material).
2 APPROACH
WHIDE combines principles from machine learning, scientific visualization and information visualization that shows to be very effective to analyze both aspects of TIS images: space and colocation. H2SOM clustering (Ontrup and Ritter, 2006) is applied to identify MCEPs as cluster prototypes which are organized on a regular 2D grid, following the SOM topology preservation principle. Each MCEP is displayed as a graphical icon called CIPRA (Combinatorial Intensity PRofile Archetype), showing the individual colocation signal characteristics. Using the grid position and the CIPRA icons we are able to render a graphical display of one or two TIS images in dynamic pseudocolor which can be interactively explored in a web browser tool.
We show, how WHIDE is applied to a set of four TIS images {T(c1), T(c2), T(n1), T(n2)}. The images were taken using tissue sections from one colon cancer patient and the four visual fields were selected. Two visual fields were selected in tissue that was classified as normal according to histopathological analysis and two TIS images were recorded (T(n1), T(n2)). The other two images were recorded in tissue classified as cancerous and two TIS images were recorded (T(c1), T(c2)). For all images, the following library of 11 tags (MUC1, Ep-CAM, DAPI, CD166, CD44, CD36, CD29, Ki-67, CK20, CK19 and CD133) was applied yielding N=11 fluorescence images per TIS image. In the Supplementary Material A, we show the 11 fluorescence image from one TIS image T(n1) plus one phase contrast image. Each image was of size 1056×1026 with pixel resolution of 206 × 206 nm/pixel.
3 METHODS
Before a H2SOM is applied, each TIS image is preprocessed in the following manner: first, image registration is applied, i.e. the single images of one TIS image are aligned. To this end, a phase contrast image is recorded within each tag loop so the shifting parameters for the single images can be computed straightforward using the corresponding phase contrast images. Second, each image was preprocessed in three steps: first, a median filter was applied to eliminate outliers. Afterwards, bilateral filtering (Tomasi and Manduchi, 1998) was applied to smoothen homogenous regions while preserving the edge information. The gray values in each image of a stack were scaled to [0; 1] using a tanh() squashing function which also introduces a slight contrast enhancement to the images. The original gray values were replaced computing ga(x, y)=tanh(0.5·E(ga)·ga(x, y)), with E(ga) as the average gray value of image ga. Now, for each pixel the N gray values are written to a colocation feature vector x=(x1, x2,…, xN)1. The feature vectors from one image (or from a selected group of images) resemble a training set Γ=x(ξ)ξ=1,…,nt with nt as the number of training items. We refer to the set of all colocation features from all four images with Γ∪.
The training set Γ∪ is used to train a H2SOM of nr rings and a branching factor of b. The foundations of the H2SOM are explained in the Supplementary Material. To train a H2SOM with nr rings, the training is divided into nr epochs (i.e. one epoch per ring) of length L(r). In each epoch a new ring of nodes is initialized by adding b new branches with child nodes to each parent node. The first ring contains eight nodes which are trained using the SOM training algorithm: In each step, a training example x(ξ) is selected and the prototype vectors {u(k)k=1,…,7} are searched for the best matching unit (BMU) u(κ), with 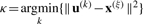 and the learning rule
and the learning rule
is applied to the nodes. The parameters e(t) and σ(t) are monotonically decreasing functions. After the first epoch is completed, each node is expanded by b child nodes and a new epoch starts applying a beam search for the BMU (see Supplementary Material B). This process is repeated until all nr rings of nodes are adapted. A Poincaré projection is applied to map the H2SOM grid to the unit disc. To manipulate the projection direction, the Möbius transform is applied (details are given in the Supplementary Material B).
To assess the quality of the H2SOM projection, we applied the approach proposed by Venna and Kaski (2001) and computed the trustworthiness Tn and the continuity Cn of the H2SOM projection. The two terms empirically determine the projection quality by quantifying for each MCEP, how wide its n most similar MCEPs are scattered across the grid (Cn) and how many non-similar, i.e. false MCEPs have been wrongly mapped into the vicinity in the grid (see the Supplementary Material B for details please).
3.1 CIPRA glyphs
Although clustering greatly aids in finding groupings inherent in the data, the success and efficiency of knowledge discovery mainly depends on suitable, linked visualizations of the feature domain, i.e. the clusters and prototypes, as well as visualizations of the image domain, i.e. the topological ordering of the data items.
First we will focus on visualizations of the feature domain. Second a pseudocoloring technique will be described. The interactive combination of the two techniques showed to be a powerful approach to the analysis of TIS data.
To visualize the feature domain we render a graphical display for each MCEP cluster and we refer to this as the CIPRA glyph of the cluster. The general reason to visualize the MCEPs, is that by focusing on the clustering result, i.e. the CIPRAs, the data complexity is significantly reduced. The main protein colocation characteristics of the data can be visually explored in one rapid knowledge discovery attempt without the need of analyzing single images gs. If interesting CIPRAs are found, the associated data items can be analyzed in a subsequent step following the Ben Schneiderman visualization mantra of ‘Overview first, zoom in and filter, details on demand’ (Schneiderman, 1996). However, a suitable CIPRA visualization is not as straightforward as it seems. A simple strategy for the display of multivariate data such as CIPRAs is an extension of the scatter plot to a generalized drafter's plot (Chambers et al., 1983), also referred to as scatter plot matrix. Here, scatter plots for all possible pairs of features are displayed. A related technique, termed dimensional stacking (LeBlanc et al., 1990), embeds one coordinate system into another and bins the data. These techniques are a straightforward extension of lower-dimensional displays, but are often hard to interpret with increasing dimensionality. This holds especially if a combination of more than two features contribute to an interesting pattern, as it is likely the case in protein colocation studies. Another popular way to display multivariate data are glyph or icon displays. According to Colin Ware ‘A glyph is a graphical object designed to convey multiple data values’ (Ware, 2004), p.145). Each data feature is mapped to a different graphical attribute of the glyph such as size, shape or color. For example Chernoff faces (Chernoff, 1973), star glyphs (Chambers et al., 1983), color icons (Levkovitz, 1991), or stick figures (Pickett and Grinstein, 1988) belong to these types of displays.
The CIPRA glyph combines visualization aspects known from bar charts and star glyphs and is to some extend inspired by the sequence logo display, which represents patterns in nucleotide or amino acid sequences (Schneider and Stephens, 1990). In a sequence logo, for each position of a set of aligned sequences, e.g. nucleotide sequences, the four nucleotides are arranged on top of each other sorted according to their frequency at that position. The character height represents the frequency of the according nucleotide. Through this visualization, a rapid identification of prominent sequence patterns can be achieved as high frequent nucleotides can directly be ‘read’ from the logo. To construct a glyph for one CIPRA uk (k=1,…, K), a horizontal box is drawn for each data feature (Fig. 1). The height, as well as the length, of each box is scaled according to the feature's value. To increase differentiation between neighboring boxes, they are alternating colored black and white. This follows C. Ware's suggestion for star glyphs or whisker plots to increase the number of dimensions by changing length and width of the bars as well as using different luminance levels. To allow for a fast identification of prominent proteins, the protein names are directly incorporated into the visualization. To this end, the associated protein name is written in each bar and scaled in height and length analog to the bar itself. With this strategy, prominent protein co-localization can easily be identified by ‘reading’ the CIPRA analog to the reading of a sequence logo. The color background of the glyph is determined by the position zk in the (Hue, Saturation)-color scale disc (see the following Section 3.2). Figure 1 gives an overview of the construction of the CIPRA display (top) and shows six CIPRA examples that have been computed for one TIS image with N=22. One can see, that the three blue CIPRA glyphs share a large number of features but differ in some features as well (like high/low values for DAPI and CD166). With changing color the differences in the CIPRAs grow as well.
Fig. 1.

The CIPRA glyph: for each H2SOM node, the prototype coefficients uak are read (1) and for each protein a bar is plotted in alternating black/white (2). The length and width of one bar k is scaled so it is proportional to uak (3). The background color of the glyph is chosen depending on the grid coordinates of the prototype in relation to the HS color scale plate (4). In the bottom row five more examples for CIPRA glyphs are shown.
In the display of a CIPRA additional information about the corresponding cluster is shown. In the upper right of a CIPRA display, the size of the corresponding cluster in relation to the entire number of projected pixels is shown as a percentage. If WHIDE is applied to two or more images, one can expect a cluster of one MCEP prototype to include feature vectors from more than one TIS image. This information may be important to users since it could point to differences in MCEP abundances in different samples, which can be an interesting feature resulting from different dynamics of molecular networks. Thus, the information about the composition of each cluster is encoded in a MCEP's CIPRA as well by a graphical line symbol, which encodes the different percentages as line segments. In a bookmarked CIPRA, a mouse over provides the numerical information.
3.2 H2SOM pseudocolor map
The CIPRA glyphs are used to display colocation features of pixel groups, i.e. it shows features of the N-dimensional colocation space. However, as outlined above, the morphological features need to be explored as well. Thus, WHIDE uses the H2SOM training result to visualize a TIS image in pseudocolor. To this end, the Poincaré projection is applied to map the node coordinates of the H2SOM prototypes {u(k)} to coordinates {zk} in a unit disc (see Supplementary Material B for full details). These new coordinates are then used to pick up colors in a circular color scale with radius R=1. In this work, we choose the basic plate of a HSV (Hue, Saturation, Value) color cone as a color scale disc, i.e. the color hue changes with the angle α and the saturation changes with the radius R. One may argue, that isoluminant color scales should be preferred to avoid tendencies for a human observer to perceive contrast of different intensities dependent on the particular color scale region. However, we found that isoluminant color scales have strong negative effect on a human observers ability to resolve smaller structural features. Thus, we use the (Hue, Saturation)-disc and allow the user to rotate the H2SOM projection on the disc to individually choose, which clusters are to be displayed in blueish (lower contrast sensitivity for humans) or in reddish (higher sensitivity) colors.
3.3 Implementation and Web Application
The H2SOM learning and the WHIDE visualization are implemented as modules of the BioIMAX platform and can be applied by all registered users. The H2SOM learning and mapping is realized in a client–server architecture as described in (Langenkämper et al., 2011).
To enable the previously described continuous visual exploration of complex datasets and benefit from the tight integration with the BioIMAX infrastructure, WHIDE was designed as a RIA. RIAs resemble classic desktop applications with regards to the richness of the user interface and computational power, but are more independent from hardware or system limitations and require no extra installation procedures or setup routines. This is achived by executing most of the application's computation, presentation and interaction in a client-side browser plugin, thereby leveraging the local hardware resources and reducing client–server traffic.
The open-source RIA framework choosen for the implementation of WHIDE is Flex.2 It is already employed by the BioIMAX platform, which enables easy access to the H2SOM mapping results, and deploys consistently on most systems due to the high penetration rate of the Adobe Flash Player, which is the properitary browser plugin used for its client-side execution. Furthermore, Flex offers a good selection of predefined but extensible user interface components, e.g. the CIPRA glyph is build upon the standard charting components.
WHIDE has only a short inital communication phase with the server-side of the BioIMAX platform to retrieve the necessary H2SOM mappings as well as image data. All H2SOM mapping data is transfered in a compressed and space optimized file in JSON3 format for fast transfer and parsing. After that the tool needs no further server connections and runs solely on the client-side. Depending on the number of rings in the H2SOM result and the amount of concurrently viewed TIS images the tool may take a while to construct all data structures needed for fast data look-up and interface manipulation. This approach is necessary because all available data are needed right from the start to enable the user to switch rapidly between a coarse overview and focus of arbitrary details.
Computation of interaction relevant data on the server-side would result in high client–server traffic and notable delays in the visualization, hampering the desired free and continuous exploration.
4 RESULTS
The dataset was built by extracting the multidimensional (N = 11) intensity values for each pixel (| Γ∪| = 1 083 456). A H2SOM was initialized with a branching factor of 8 and 3 rings (not counting the central node) yielding a total number of 160 nodes. The H2SOM was trained in 30· P steps following the training algorithm described in Section 3. Training took 4 h, after training for each TIS image a BMU index image was computed, mapping each pixel to the index of the BMU in the H2SOM, which took < 1 min per image. The trustworthiness and the continuity indices were computed at start and stop of each training period and plotted (see Supplementary Material B for details). One can see, how these indices increase over time and the H2SOM approaches a stable state which seems to show no drastic topologic distortions such as wrong folds. Using the WHIDE tool the results have been visually analyzed regarding different aspects. First, the topology preservation is qualitatively analyzed by moving the mouse cursor along the border of color disc. One can observe the continuous changes in the MCEP patterns while the color changes. Some example CIPRAs are shown as bookmarks in Figure 2. One can see, that with changing color (from blue to green to yellow to red) some markers go up (such as DAPI) and some are going down and up again (such as EpCAM or CD133) or vice versa (such as KI-67). The color mapping did not show any strong distortions, such as CIPRAs with similar colors but different colocation pattern. The second interesting aspect was how the WHIDE tool reacts to strong noise in the data. In image T(n2), a strong noise signal can be observed for the CD29 marker showing a large star-shaped group of fully saturated pixels. Such noise can be observed from time to time in TIS imaging and from a data mining point of view these signals form false outlier data clouds in the high-dimensional colocation signal space. The right image of T(n2) in Figure 2 shows this case and some magenta/blue spike of the noise pattern can be observed in the right half of the image. However, the pattern does not have an influence on the global color mapping, since both cases, T(n1) and T(n2), show equivalent color mappings of their morphology and their MCEP patterns. Third, we investigated WHIDE's potential to reveal differences in MCEP statistics and spatial distribution for cancer and normal tissue. To this end, we apply the special WHIDE feature of a continuous interactive tuning of the color mapping. The color mapping is changed in two ways: the color disc can be rotated as shown in Figure 3, where we rotate the color disc, so some regions are drawn in yellow, which are visible in T(c1) and T(c2) as a small number of cellular/sub-cellular objects, marked with white ellipsoids. The motivation to move these regions to yellow is that human observers can perceive more color details in the green–yellow–red interval of the color spectrum than in the bluish region. So the observer might discriminate more colors, i.e. different MCEPs for these regions now. In addition, the Möbius transform is applied to move the nodes from the yellow region toward the center, thereby, squeezing the opposing nodes all into the blue region of the color scale disc (see Fig. 3, a lower row on the right). Please note, that the colors of the bookmarked CIPRAs are adapted accordingly. This transformation has two important consequences: the majority of MCEPs are drawn blue with a low color contrast (so the human observer does not perceive many structural features) and the color contrast for a comparably small subregion of the 11-dimensional colocation feature space, spanned by the rest of the MCEPs is strongly enhanced. The selected individual MCEPs of the selected regions can now be distinguished more easily and analyzed in detail. This way, we enable a kind of a zoom in an N-dimensional space which is interactive and continuously, so the user does not loose the context.
Fig. 2.

The WHIDE result for two TIS images from normal tissue T(n1), T(n2) is shown as a screenshot from the WHIDE tool in the BioIMAX system. On the right, the color disc is shown with H2SOM nodes displayed as square icons at positions computed with a Poincaré projection. The size of the squares encodes the size of the clusters. Moving the mouse over one square activates the display of its CIPRA. Alternatively, CIPRA displays can be activated in the image. At the bottom of the screen, the history of selected CIPRAs is shown as bookmarks. In the upper left of each image display one fluorescence image is shown for an overview and using the sliders below the user can change the opacity of the pseudocolor map and the zoom as it is demonstrated in the lower screenshot. This way, the user can modify the display to relate the found clusters, i.e. MCEPs to individual fluorescence signals for a detailed analysis.
Fig. 3.

The H2SOM architecture provides the structural basis for a synchronized interactive dynamic pseudocoloring of TIS images. In the upper row, the four TIS images T(c1), T(c2), T(n1) and T(n2) from left to right. The bottom row shows a small set of selected bookmarked CIPRAs. On the right, the color disc is shown with its control buttons below. To change the coloring, the user can combine two functions. First, using the rotate-button, the user can turn the color disc so that of the H2SOM grid which is of less interest is mapped to the blue area (since human observers are more sensitive to non-blue colors). Second, the user can use the arrow buttons to change the Möbius projection, i.e. to move H2SOM nodes toward the center and squeeze the opposing nodes into a small cloud. In this example, the nodes from the upper right are moved to enhance the color contrast for a chosen region of interest in one image (marked with white ellipsoids).
For comparison we show results obtained with a Principal Component Analysis (PCA). The PCA was performed on the same dataset Γ∪ and the feature vectors were projected onto the eigenvectors of the three largest eigenvalues to map each pixel to three new coordinates (v1, v2, v3) which were used for a RGB pseudocolor mapping for each image (see Supplementary Material C). While we again made the observation of a difference in colocation feature statistics between normal tissue and cancer tissue, the PCA approach does not feature the structural advances of the H2SOM which allow resolving non-linear features and dynamic interactive manipulation of the colors.
5 DISCUSSION
The WHIDE tool shows significant advantages compared with other approaches to MBI analysis. First, it is able to resolve and embed non-linear data structures. This can be seen by browsing the CIPRAs on the H2SOM visualization on the color disc. Moving the cursor slowly across the discs shows the CIPRAs of neighboring clusters. The CIPRAs show, that similarity in cluster prototypes is reflected by vicinity in the H2SOM grid, i.e. the N-dimensional data topology is preserved regarding local neighborhoods. A second striking feature is the H2SOM visualization using the Möbius transform which allows change of zoom in the N-dimensional feature space by mapping a smaller number of neighboring clusters to a larger area in the color scale. This way, particular groups of MCEPs can be pseudocolored in higher color resolution whereas the rest of the TIS image is colored with a very small part of the color scale, i.e. with low contrast. Another positive feature of the WHIDE approach is the reduction of the TIS data using vector quantization as performed by the H2SOM algorithm which has shown to resolve even small clusters and organize the clusters in a hierarchical structure. If the CIPRA visualization is compared with two classic methods such as bar graphs and star glyphs, it is evident that in the CIPRA display the association of proteins to individual graphical attributes is much easier. Furthermore, besides being able to rapidly identify the dominant proteins, an advantage of the CIPRA display is that only features with high values allocate space, whereas low value features are squeezed. Thereby, space is only allocated proportional to the importance of the protein and the total size of the CIPRA reflects the amount of information provided by the prototype. In some applications, this might not be a desirable feature so that bar graphs, or CIPRAs with constant bar width would be more suited but in our current project this has not been the case yet. Last but not least we must address the issue of preprocessing here although this is not part of the WHIDE tool. The performance and effectiveness of any data mining approach to MBI depends substantially on the preprocessing applied to the data. Maybe the most important preprocessing step is local alignment of the fluorescence images, if volume stacks are recorded alignment must be applied in (x,y,z). If the images are not aligned well, i.e. the image registration failed, the feature vectors extracted for each point display fluorescence values (i.e. molecular signals) from close but different anatomical sites. As a consequence, the H2SOM clustering assigns vectors into false clusters which reflect the misalignment. This would lead to false interpretations and must be avoided. The problem would be even more serious if two or more datasets are analyzed in comparison (like in this study) but the registration fails only in a subset of the data. This could lead to the false assumption that the false clusters are biologically very interesting since they separate this subset of TIS images from the others. Thus, the necessity for an accurate alignment of the data cannot be overstressed. As a consequence we developed a novel registration algorithm which is based on an alignment of square subimages on the phase contrast images (Raza et al., 2012). Another kind of small false signal variations can be noise caused by the imaging chip which can be reduced by filtering (as explained in Section 3). Another, sometimes more critical kind of noise is a locally described over-saturation of imaging elements leading to a nova-like artifact as in the case of this study in the CK19 tag image and in the CD29 tag image as well. We have tested the effect of such kind of distortions to the WHIDE performance and showed, that these do not have a strong influence in the result so masking these areas may not be necessary in many cases. However, we recommend masking such regions and exclude this data from a study.
6 CONCLUSION
Due to advances in machine learning research, present-day internet connection bandwidths and state-of-the-art web graphics technology a new level of MBI analysis is enabled. Web-technology allows a direct connection of researchers to the tools and the result visualizations, independent from their whereabouts and their computer system. Modern RIA technologies allow web-based visualizations to be interactive and dynamic, which are prerequisites for the analysis of MBI data such as TIS. Although, we presented the WHIDE tool in the context of TIS analysis it is evident that the tool is applicable to other MBI data such as MALDI images as well.
Supplementary Material
ACKNOWLEDGEMENTS
MK and SA acknowledge, The Medical and Life Sciences Research Fund and the Warwick University Research and Development Fund for supporting this research. Special thanks go to Sayan Battacharya for efforts in designing the tag library and to Christian Loyek and Julia Herold for their preliminary works on designing the visualizaion framework and the web based bioimage data base. We thank Nasir Rajpoot and his group (Computational Biology and Bioimaging Group, University of Warwick) for providing TIS image alignment which is a pre-processing step of crucial importance to any data mining application in multivariate bioimages. We thank W.Schubert, who introduced us to TIS, and helped us establish a TIS machine at the University of Warwick, and members of his team at ToposNomos and the University of Magdeburg, especially A.Krusche and R.Hillert, who have provided invaluable support when we needed it.
Conflict of Interest: none declared.
Footnotes
1We refer to the feature vector with x to show, that the components differ from the original gray values for the pixel g due to the applied preprocessing.
REFERENCES
- Alexandrov T., et al. Spatial segmentation of imaging mass spectrometry data with edge-preserving image denoising and clustering. J. Proteome Res. 2010;9:6535–6546. doi: 10.1021/pr100734z. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S., et al. Toponome imaging system: in situ protein network mapping in normal and cancerous colon from the same patient reveals more than five-thousand cancer specific protein clusters and their subcellular annotation by using a three symbol code. J. Proteome Res. 2010;9:611225. doi: 10.1021/pr100157p. [DOI] [PubMed] [Google Scholar]
- Bonnekoh B., et al. Profiling lymphocyte subpopulations in peripheral blood under efalizumab treatment of psoriasis by multi epitope ligand cartography (melc) robot microscopy. Eur. J. Dermatol. 2006;16:623–635. [PubMed] [Google Scholar]
- Chambers J.M., et al. Graphical Methods for Data Analysis. Boston, USA: Duxbury Press; 1983. [Google Scholar]
- Chernoff H. The use of faces to represent points in k-dimensional space graphically. J. Am. Stat. Assoc. 1973;68:361–368. [Google Scholar]
- Cornett D., et al. Maldi imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods. 2007;4:828–833. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- Eyerich K., et al. Comparative in situ topoproteome analysis reveals differences in patch test-induced eczema: cytotoxicity-dominated nickel versus pleiotrope pollen reaction. Exp. Dermatol. 2009;19:511–517. doi: 10.1111/j.1600-0625.2009.00980.x. [DOI] [PubMed] [Google Scholar]
- Friedenberger M., et al. Fluorescence detection of protein clusters in individual cells and tissue sections by using toponome imaging system: sample preparation and measuring procedures. Nat. Protoc. 2007;2:2285–2294. doi: 10.1038/nprot.2007.320. [DOI] [PubMed] [Google Scholar]
- Herold J., et al. Data mining in multivariate images. WIREs Data Min. Knowl. Disc. 2011;1:2–13. [Google Scholar]
- Lamping J., et al. Proceedings of the ACM SIGCHI Conference on Human Factors in Computing Systems. New York, USA: ACM; 1995. A focus+content technique based on hyperbolic geometry for viewing large hierarchies; pp. 401–408. [Google Scholar]
- Langenkämper D., et al. Workshop on Microscopic Image Analysis with Applications in Biology (MIAAB) Heidelberg, Germany; 2011. Tical - a web-tool for multivariate image clustering and data topology preserving visualization. [Google Scholar]
- LeBlanc J., et al. VIS '90 Proceedings of the 1st conference on Visualization. CA, USA: '90 IEEE Computer Society Press Los Alamitos; 1990. Exploring n-dimensional databases; pp. 230–237. [Google Scholar]
- Levkovitz H. Visualization '91, Proceedings, IEEE Conference on. San Diego, CA, USA: 1991. Color icons: Merging color and texture perception for integrated visualization of multiple parameters; pp. 164–170. [Google Scholar]
- Loyek C., et al. Bioimax: a web 2.0 approach for easy exploratory and collaborative access to multivariate bioimage data. BMC Bioinformatics. 2011;12:297. doi: 10.1186/1471-2105-12-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megason S., Fraser S. Imaging in systems biology. Cell. 2007;130:784–795. doi: 10.1016/j.cell.2007.08.031. [DOI] [PubMed] [Google Scholar]
- Ontrup J., Ritter H. Large-scale data exploration with the hierarchically growing hyperbolic SOM. Neural Networks. 2006;19:751–761. doi: 10.1016/j.neunet.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Pickett R.M., Grinstein G.G. Iconographic displays for visualizing multidimensional data. Proc. IEEE Conf. Syst. Man Cybern. 1988;1:514–519. [Google Scholar]
- Raza S.A., et al. RAMTaB: robust alignment of multi-tag bioimages. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0030894. e30894. doi:10.1371/journal.pone.0030894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruetze M., et al. In situ localization of epidermal stem cells using a novel multi epitope ligand cartography approach. Integr. Biol. (Camb) 2010;2:241–249. doi: 10.1039/b926147h. [DOI] [PubMed] [Google Scholar]
- Schneiderman B. The eyes have it: a task by data type taxonomy for information visualizations. IEEE Symposium on Visual Languages. 1996:336–343. [Google Scholar]
- Schneider T.D., Stephens R. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert W., et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006;24:1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- Starkuviene V., Pepperkok R. The potential of high-content high-throughput microscopy in drug discovery. Br. J. Pharmacol. 2007;152:62–71. doi: 10.1038/sj.bjp.0707346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi C., Manduchi R. Bilateral filtering for gray and color images. ICCV. 1998:839–846. [Google Scholar]
- van Manen H., et al. Single-cell raman and fluorescence microscopy reveal the association of lipid bodies with phagosomes in leukocytes. PNAS. 2005;102:10159–10164. doi: 10.1073/pnas.0502746102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venna J., Kaski S. Neighborhood preservation in nonlinear projection methods: an experimental study. Artificial Neural Networks-ICANN. 2001:485–491. [Google Scholar]
- Ware C. Information Visualization: Perception for Design. San Francisco, CA, USA: Morgan Kaufmann Publishers Inc.; 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.