

A PacL homologue from L. monocytogenes has been purified and crystallized. The crystals diffracted to 3.2 Å resolution.
Keywords: PacL, Lmo0818, P-type ATPases, transporters, membrane proteins
Abstract
Ca2+-ATPases are members of a large family of membrane proteins that maintain the selective movement of cations across biological membranes. A putative Listeria monocytogenes Ca2+-ATPase (Lmo0818) was crystallized in an unknown functional state. The crystal belonged to space group P212121 and a complete data set was collected to 3.2 Å resolution. The molecular-replacement solution obtained revealed that Lmo0818 is likely to adopt an E2-like state mimicking the phosphorylated intermediate in the functional cycle of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) and a stacked bilayer ‘type I’ packing in the crystal.
1. Introduction
P-type ATPases are integral membrane proteins and a fundamental part of the tightly controlled cellular metal homeostasis. They couple the vectorial transport of charged ions across cellular membranes to ATP hydrolysis. P-type ATPases are often linked to key processes in both prokaryotic and eukaryotic cells, for example in cell signalling (Strehler & Treiman, 2004 ▶), by maintaining Ca2+ levels in the micromolar range or maintaining the membrane potential across the plasma membrane in both mammals and plants (Morth et al., 2011 ▶). P-type ATPases belong to a large family of evolutionarily-related ion pumps that can be found in almost all living organisms (Kühlbrandt, 2004 ▶; Palmgren & Axelsen, 1998 ▶). They share a common domain topology, generally consisting of a transmembrane-spanning domain (TM) with between seven and 12 helices and three cytosolic domains: an actuator domain (A), a phosphorylation domain (P) and a nucleotide-binding domain (N). The Ca2+-ATPases belong to subgroup II of the P-type ATPase family (Palmgren & Axelsen, 1998 ▶). The functional cycle necessary for vectorial transfer is a two-step procedure. The first, high-energy step involves the transfer of the γ-phosphate from an adenosine triphosphate (ATP) molecule to a conserved aspartate residue in the P domain to form the phospho-intermediate. Upon release of the phosphoryl group, the P-type ATPase returns to the ground state and is ready to accept another ATP molecule. The ‘P’ in P-type refers to the intermediate energy level (Pedersen & Carafoli, 1987 ▶; Bublitz et al., 2011 ▶). Ca2+-ATPases can be further divided into two subfamilies: IIa and IIb. Traditionally, the denomination of subfamilies IIa and IIb refers to the intracellular localization of the pumps in mammalian cells: the sarco(endo)plasmic reticulum Ca2+-ATPases (SERCAs) and the plasma membrane Ca2+-ATPases (PMCAs), respectively. However, type IIa Ca2+-ATPases have also been found in the Golgi apparatus (Liang et al., 1997 ▶) and in the plasma and vacuole membrane of some plants (Wimmers et al., 1992 ▶) and type IIb Ca2+-ATPases have also been found in various organelle membranes in the cytoplasm, such as the endoplasmic reticulum (ER; Harper et al., 1998 ▶), vacuole membrane (Malmström et al., 1997 ▶) and chloroplast envelope (Huang et al., 1993 ▶). It is therefore sometimes misleading to discuss type nomenclature based on subcellular location. The importance of Ca2+ regulation has been well established in eukaryotic cells, but less is known about calcium homeostasis in prokaryotes. There is a widespread distribution of open reading frames (ORFs) encoding putative Ca2+-ATPases in the prokaryotic genomes that have been sequenced to date (UniProt Consortium, 2011 ▶). Listeria monocytogenes is a Gram-positive intracellular bacterium and is one of the most virulent foodborne pathogens (Ramaswamy et al., 2007 ▶). Infection, although infrequent, often only manifests itself as uncomplicated mild gastrointestinal symptoms in healthy individuals. However, individuals suffering from immunosuppression and elderly people are considered to be in a high-risk group with a mortality rate as high as 30% (Ramaswamy et al., 2007 ▶). Ca2+ transport has been less studied in bacteria and the first prokaryotic Ca2+-ATPase to be characterized was the pacL gene product from Synechococcus sp. (Berkelman et al., 1994 ▶). The gene locus lmo0818 of L. monocytogenes encodes a PacL homologue, a cation P-type ATPase (Lmo0818). Sequence and structural analysis strongly suggest that Lmo0818 could indeed be a Ca2+-ATPase and thus the second Ca2+-ATPase to be found in L. monocytogenes. The first L. monocytogenes Ca2+-ATPase 1 (LMCA1) has recently been identified and characterized (Faxén et al., 2011 ▶). It has been shown that P-type ATPases in L. monocytogenes are coupled to functions such as virulence and survival at high extracellular Ca2+ concentrations in infected host cells (Rosch et al., 2008 ▶; Francis & Thomas, 1997 ▶). This underlines the importance of studying the molecular mechanisms of bacterial P-type ATPases, which could potentially be potent drug targets for increased control of bacterial pathogens such as L. monocytogenes.
2. Materials and methods
2.1. Cloning, expression and purification
The DNA encoding the L. monocytogenes EGD-e putative Ca2+-ATPase Lmo0818 was cloned into a pET30-derived vector (Novagen) with an N-terminal fusion consisting of a 6×His tag and a tobacco etch virus (TEV) protease cleavage site. Lmo0818 was expressed in Escherichia coli C41 (DE3) cells grown in standard Luria–Bertani (LB) medium. The cells were harvested at 277 K and resuspended in homogenization buffer [50 mM Tris–HCl pH 7.6, 200 mM KCl, 1 µg ml−1 DNase I, 10%(w/v) glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF)]. The cells were lysed using a high-pressure homogenizer (HPH) and cellular debris was removed by centrifugation at 20 000g for 20 min. Membranes were isolated by ultracentrifugation at 235 000g for 90 min at 277 K and the pellet was resuspended in suspension buffer [50 mM Tris–HCl pH 7.6, 10%(w/v) glycerol, 3 mM MgCl2, 50 µM ATP, 1 mM PMSF, 5 mM β-mercaptoethanol (BME)] and adjusted to a total membrane concentration of ∼10 mg ml−1. The membrane fraction was solubilized by adding n-dodecyl β-d-maltoside (DDM) at an approximately 1:1 protein:detergent ratio by weight and solubilization was performed at 277 K overnight with moderate magnetic stirring. Solubilized membranes were ultracentrifuged at 235 000g for 90 min at 277 K. Often, a small light brown pellet was observed; additional detergent-solvation procedures yielded no additional protein from this pellet. The supernatant was removed and subsequently imidazole was added to a final concentration of 30 mM. The solubilized protein was captured on an Ni–NTA Chelating HP column (GE Healthcare) in calibration buffer [50 mM Tris–HCl pH 7.6, 10%(w/v) glycerol, 200 mM KCl, 3.0 mM MgCl2, 50 µM ATP, 30 mM imidazole, 1.0 mM C12E8, 5 mM BME] followed by extensive washing with calibration buffer. The protein was eluted using an imidazole gradient, increasing the concentration from 30 to 500 mM. The His tag was removed by the addition of tobacco-etch virus (TEV) protease (∼0.05 mg ml−1 final concentration) followed by dialysis for 12 h against Ni–NTA dialysis buffer in excess with a minimum of a 50-fold volume [50 mM Tris–HCl pH 7.6, 10%(w/v) glycerol, 200 mM KCl, 3.0 mM MgCl2, 50 µM ATP, 5 mM BME]. Noncleaved Lmo0818 was removed by applying the protein solution to the Ni–NTA column again and collecting the flowthrough. This was followed by a final size-exclusion chromatographic step on a Superdex 200 column (GE Healthcare) in separation buffer [50 mM Tris–HCl pH 7.6, 10%(w/v) glycerol, 100 mM KCl, 3 mM MgCl2, 50 µM ATP, 1 mM C12E8, 1 mM dithiothreitol (DTT)]. The protein was flash-cooled and stored at 193 K in storage buffer [10 mM histidine pH 6, 100 mM KCl, 3 mM MgCl2, 10%(v/v) glycerol, 1 mM DTT]. Protein purity was assessed by SDS–PAGE. Protein concentration was measured by absorption at a wavelength of 280 nm using a calculated extinction coefficient of 0.64 mg−1 ml cm−1.
2.2. Crystallization and data collection
Prior to crystallization, the protein was concentrated to ∼10 mg ml−1 in Vivaspin 20 concentrators (Sartorius Stedim Biotech) with a 10 kDa molecular-weight cutoff and re-lipidated with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti Polar Lipids). The lipid:detergent ratio was determined empirically for each protein batch as described in Gourdon et al. (2011 ▶); the ratio ranges tested were 1:0.2, 1:0.45 and 1:0.7 (µg protein:µg lipid). A thin lipid film was generated by dispensing DOPC dissolved in CHCl3 in a glass tube followed by evaporation of CHCl3 in an argon atmosphere, thus preventing oxidation. Lmo0818 was re-lipidated over 12 h at 277 K with stirring. Insoluble DOPC and aggregated Lmo0818 were removed by centrifugation at 164 000g for 20 min. All crystallization experiments were performed using the hanging-drop vapour-diffusion method (2 + 2 µl drops on glass cover slips equilibrated against 250 µl reservoir solution). Additional DDM (to the C12E8 already present) was added to the protein mixture to a final concentration of 1% prior to the crystallization experiments, which used commercially available screens. The initial crystal hit was obtained in 31%(w/v) polyethylene glycol 2000, 0.2 M MgCl2, 0.1 M sodium cacodylate pH 6.5. Crystals were further optimized by varying the precipitant concentration and the DDM:C12E8 detergent ratio. The secondary-detergent concentration was determined empirically for each protein batch, using a final DDM concentration varying from 0.3 to 1.1%.
Crystals were mounted in LithoLoops (Molecular Dimensions) and cryocooled in liquid nitrogen. A complete data set was collected at 100 K on beamline 14.1 at Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY).
2.3. Structure solution and refinement
Reflections were indexed and integrated with XDS (Kabsch, 2010 ▶). Molecular replacement (MR) was performed using Phaser (RFZ = 4.5, TFZ = 10.4, PAK = 14, LLG = 160; McCoy et al., 2007 ▶); the initial phases were derived from a homology model using MODELLER (Eswar et al., 2007 ▶) with SERCA1A as a template in the E2(MgF3 −) stabilized state (PDB entry 3fgo; Laursen et al., 2009 ▶). Preliminary refinement and rebuilding was performed using the PHENIX software package (Adams et al., 2010 ▶) and Coot (Emsley et al., 2010 ▶).
3. Results and discussion
The crystals of Lmo0818 belonged to space group P212121 and contained one molecule per asymmetric unit, with unit-cell parameters a = 47.85, b = 97.56, c = 276.07 Å. A complete data set consisting of 240 images with 0.5° oscillation per image was collected on beamline 14.1 at BESSY and integrated to a maximum resolution of 3.2 Å (Fig. 1 ▶ a). The data revealed an anisotropic tendency, but this did not pose a major problem at the current resolution. However, such anisotropy could pose a considerable problem at higher resolution. Data statistics are summarized in Table 1 ▶.
Figure 1.
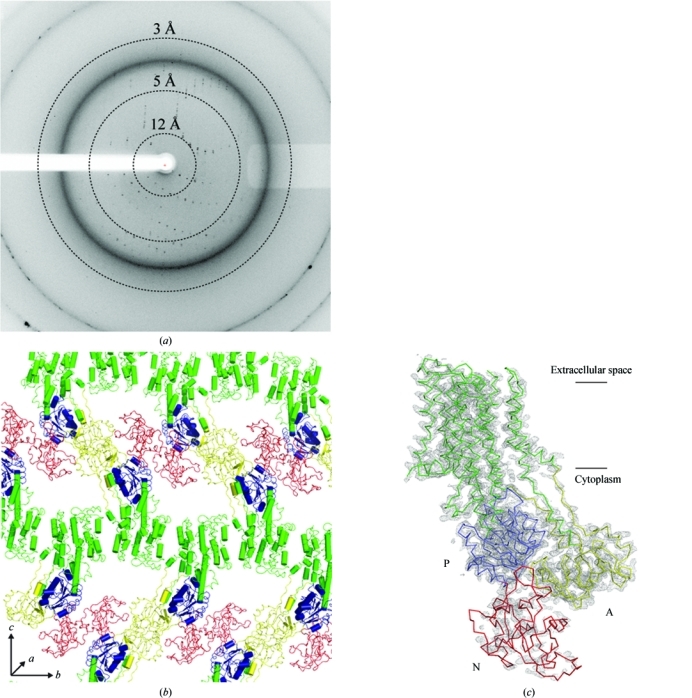
(a) X-ray diffraction image of Lmo0818 crystals. Resolution circles are indicated by dashed lines. (b) Type I packing of Lmo0818. Cytoplasmic domains are colour-coded as follows: A, yellow, P, blue, N, red; the transmembrane domain is shown in green. (c) Preliminary model of Lmo0818 extending to 3.2 Å resolution. Initial phases were estimated using a homology model produced with MODELLER (Eswar et al., 2007 ▶) using SERCA1a in the E2 conformation (PDB entry 3fgo) as a template (Laursen et al., 2009 ▶). A 2F o − F c electron-density map contoured at 1.5σ is shown. Cytoplasmic domains are colour-coded as in (b). Lines indicates the plane of the membrane.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Beamline | 14.1, BESSY |
| Wavelength (Å) | 0.918 |
| Rotation range per image (°) | 0.5 |
| Total rotation range (°) | 120 |
| Resolution range (Å) | 50–3.2 (3.3–3.2) |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 47.85, b = 97.56, c = 276.07 |
| Total No. of measured reflections | 107249 |
| Unique reflections | 21875 |
| Multiplicity | 4.9 |
| Mean I/σ(I) | 10.02 (1.98) |
| Completeness (%) | 98.3 (99.7) |
| Rmeas†‡ (%) | 17.0 (78.7) |
| Rmrgd-F†§ (%) | 24.1 (97.2) |
| Molecules per asymmetric unit | 1¶ |
| Matthews coefficient (Å3 Da−1) | 3.34¶ |
| Solvent content (%) | 63.2¶ |
R meas and R mrgd-F are quality measures of the individual intensity observations and the reduced structure-factor amplitudes, respectively (Diederichs & Karplus, 1997 ▶).
R
meas is defined as 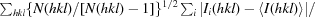
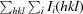 .
.
R
mrgd-F is defined as 
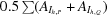 (Diederichs & Karplus, 1997 ▶).
(Diederichs & Karplus, 1997 ▶).
For the most probable solution according to statistical sampling (Kantardjieff & Rupp, 2003 ▶).
The MR solution indicated the presence of one molecule in the asymmetric unit, revealing an E2-like state with ‘type I’ packing of the molecules in the crystal (Ostermeier & Michel, 1997 ▶) with continuous bilayers of transmembrane domains (Fig. 1 ▶ b), as also observed for re-lipidated SERCA1a and LMCA1 (Jidenko et al., 2005 ▶; Andersen et al., 2011 ▶). The MR solution was refined to R = 27.9 and R free = 36.3. The preliminary model consisted of residues 4–266 and 274–869; however, several loops and large parts of the N domain are presently poorly resolved. The poor electron density observed in the N domain can in part be explained by the model N domain of the MR model. The N domain of Lmo0818 is considerably smaller than that of SERCA1a (172 versus 241 residues) and furthermore has low sequence identity (21%). This hampers modelling of the starting model, leading to an initially poor phase estimate. However, with further refinement and manual rebuilding a much better resolved N domain should be within reach. A preliminary model of Lmo0818 including the current electron-density map (2F o − F c) can be seen in Fig. 1 ▶(c). Higher resolution data and experimental phases will undoubtedly facilitate the rebuilding of the N domain to complete the model. Further crystal optimization is in progress.
In comparison to PacL and other known Ca2+-ATPases, the Ca2+-binding sites are highly conserved in Lmo0818 (Fig. 2 ▶ a). Compared with SERCA1a, Ca2+ site II is 100% conserved in terms of Ca2+-coordinating residues, and the structurally important motif involving Leu65, Val304 and Ile307 (SERCA1a numbering), which are important for stabilizing site II, is also conserved. Site I shows a lower degree of conservation; however, the substituted residues show a high similarity to those in SERCA1a (Fig. 2 ▶ b). A functional activity assay is needed for Lmo0818, without which it has not been possible to calculate the exact stoichiometry of transported Ca2+ ions. Owing to the fully conserved Ca2+ site II, it could be suggested that Lmo0818 transports a single Ca2+ only in a similar manner as has been described for LMCA1 and PMCA1 (Faxén et al., 2011 ▶; Niggli et al., 1981 ▶). However, because of the degree of similarity to site I in SERCA1a, the presence of a second Ca2+-binding site in Lmo0818 cannot presently be fully dismissed. Only an activity assay or the structure of a Ca2+-bound state will be able to confirm or reject the presence of a second binding site in Lmo0818.
Figure 2.

(a) Alignment of the calcium-coordinating region in five calcium-transporting P-type ATPases: SERCA1a from rabbit (accession No. ABW96358), Lmo0818 from L. monocytogenes (CAC98896), PacL from Synechococcus elongatus (BAA03906), LMCA1 from L. monocytogenes (Q8Y8Q5) and PMCA1 from human (P20020). Site I coordinating residues are shown in green and site II coordinating residues in blue. Completely conserved residues are indicated in bold. Residues that have lost the ability to coordinate a metal ion to their side-chain chemical group are shown in red. The numbering is according to SERCA1a. (b) SERCA1a Ca2+-binding pocket (PDB entry 1t5s; Sorensen et al., 2004 ▶) showing Ca2+-coordinating residues and structurally important residues. Colour-coding is as in (a).
Acknowledgments
We thank Pascale F. Cossart (Institute Pasteur, Paris, France) for generously providing us with L. monocytogenes genomic DNA and the Swiss Light Source (Paul Scherrer Institute), European Synchrotron Facility and the Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung for providing data-collection facilities and outstanding support. We are grateful to Anna Marie Nielsen for her technical assistance. The work was supported by a Centre of Excellence grant from the Danish National Research Foundation and Lundbeckfonden.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Andersen, J. L., Gourdon, P., Møller, J. V., Morth, J. P. & Nissen, P. (2011). Acta Cryst. F67, 718–722. [DOI] [PMC free article] [PubMed]
- Berkelman, T., Garret-Engele, P. & Hoffman, N. E. (1994). J. Bacteriol. 176, 4430–4436. [DOI] [PMC free article] [PubMed]
- Bublitz, M., Morth, J. P. & Nissen, P. (2011). J. Cell Sci. 124, 2515–2519. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Eswar, N., Webb, B., Marti-Renom, M. A., Madhusudhan, M. S., Eramian, D., Shen, M. Y., Pieper, U. & Sali, A. (2007). Curr. Protoc. Protein Sci., ch. 2, Unit 2.9. [DOI] [PubMed]
- Faxén, K., Andersen, J. L., Gourdon, P., Fedosova, N., Morth, J. P., Nissen, P. & Møller, J. V. (2011). J. Biol. Chem. 286, 1609–1617. [DOI] [PMC free article] [PubMed]
- Francis, M. S. & Thomas, C. J. (1997). Microb. Pathog. 22, 67–78. [DOI] [PubMed]
- Gourdon, P., Andersen, J. L., Hein, K. L., Bublitz, M., Pedersen, B. P., Liu, X., Yatime, L., Nyblom, M., Nielsen, T. T., Olesen, C., Moller, J. V., Nissen, P. & Morth, J. P. (2011). Cryst. Growth Des. 11, 2098–2106.
- Harper, J. F., Hong, B., Hwang, I., Guo, H. Q., Stoddard, R., Huang, J. F., Palmgren, M. G. & Sze, H. (1998). J. Biol. Chem. 273, 1099–1106. [DOI] [PubMed]
- Huang, L., Berkelman, T., Franklin, A. E. & Hoffman, N. E. (1993). Proc. Natl Acad. Sci. USA, 90, 10066–10070. [DOI] [PMC free article] [PubMed]
- Jidenko, M., Nielsen, R. C., Sørensen, T. L., Møller, J. V., le Maire, M., Nissen, P. & Jaxel, C. (2005). Proc. Natl Acad. Sci. USA, 102, 11687–11691. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Kühlbrandt, W. (2004). Nature Rev. Mol. Cell Biol. 5, 282–295. [DOI] [PubMed]
- Laursen, M., Bublitz, M., Moncoq, K., Olesen, C., Møller, J. V., Young, H. S., Nissen, P. & Morth, J. P. (2009). J. Biol. Chem. 284, 13513–13518. [DOI] [PMC free article] [PubMed]
- Liang, F., Cunningham, K. W., Harper, J. F. & Sze, H. (1997). Proc. Natl Acad. Sci. USA, 94, 8579–8584. [DOI] [PMC free article] [PubMed]
- Malmström, S., Askerlund, P. & Palmgren, M. G. (1997). FEBS Lett. 400, 324–328. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Morth, J. P., Pedersen, B. P., Buch-Pedersen, M. J., Andersen, J. P., Vilsen, B., Palmgren, M. G. & Nissen, P. (2011). Nature Rev. Mol. Cell Biol. 12, 60–70. [DOI] [PubMed]
- Niggli, V., Adunyah, E. S., Penniston, J. T. & Carafoli, E. (1981). J. Biol. Chem. 256, 395–401. [PubMed]
- Ostermeier, C. & Michel, H. (1997). Curr. Opin. Struct. Biol. 7, 697–701. [DOI] [PubMed]
- Palmgren, M. G. & Axelsen, K. B. (1998). Biochim. Biophys. Acta, 1365, 37–45. [DOI] [PubMed]
- Pedersen, P. L. & Carafoli, E. (1987). Trends Biochem. Sci. 12, 146–150.
- Ramaswamy, V., Cresence, V. M., Rejitha, J. S., Lekshmi, M. U., Dharsana, K. S., Prasad, S. P. & Vijila, H. M. (2007). J. Microbiol. Immunol. Infect. 40, 4–13. [PubMed]
- Rosch, J. W., Sublett, J., Gao, G., Wang, Y.-D. & Tuomanen, E. I. (2008). Mol. Microbiol. 70, 435–444. [DOI] [PMC free article] [PubMed]
- Sorensen, T. L.-M., Moller, J. V. & Nissen, P. (2004). Science, 304, 1672–1675. [DOI] [PubMed]
- Strehler, E. E. & Treiman, M. (2004). Curr. Mol. Med. 4, 323–335. [DOI] [PubMed]
- UniProt Consortium (2011). Nucleic Acids Res. 39, D214–D219.
- Wimmers, L. E., Ewing, N. N. & Bennett, A. B. (1992). Proc. Natl Acad. Sci. USA, 89, 9205–9209. [DOI] [PMC free article] [PubMed]