

SUMMARY
The continuous monolayer of intestinal epithelial cells (IECs) lining the gut lumen functions as the site of nutrient absorption and as a physical barrier to prevent the translocation of microbes and associated toxic compounds into the peripheral vasculature [1]. IECs also express host defense proteins such as intestinal alkaline phosphatase (IAP), which detoxify bacterial products and prevent intestinal inflammation [2-5]. Our laboratory recently showed that IAP is enriched on vesicles that are released from the tips of IEC microvilli and accumulate in the intestinal lumen [6, 7]. Here, we show that these native ‘lumenal vesicles’ (LVs): (i) contain catalytically active IAP that can dephosphorylate lipopolysaccharide (LPS), (ii) cluster on the surface of native lumenal bacteria, (iii) prevent the adherence of enteropathogenic E. coli (EPEC) to epithelial monolayers, and (iv) limit bacterial population growth. We also find that IECs upregulate LV production in response to EPEC and other Gram-negative pathogens. Together, these results suggest that microvillar vesicle shedding represents a novel mechanism for distributing host defense machinery into the intestinal lumen, and that microvillus-derived LVs modulate epithelial-microbial interactions.
Keywords: actin, myosin, brush border, host defense, intestinal alkaline phosphatase, enteropathogenic E. coli
RESULTS AND DISCUSSION
A defining feature of the IEC apical domain is the array of microvilli known as the brush border, which extends into the intestinal lumen [1]. Within the microvillus, the apical membrane and underlying core actin bundle are linked by myosin-1a (Myo1a), a membrane-binding actin-based motor [8, 9]. Our previous studies suggest that Myo1a applies force to the apical membrane, leading to the accumulation of membrane at microvillar tips and ‘shedding’ of vesicles into the lumen [6, 7]. In mice lacking Myo1a, LVs are reduced in number and exhibit perturbations in morphology and composition [7]. Proteomic studies revealed that native LVs are enriched in IAP [7], a host defense factor that reduces the toxicity of LPS and other bacterial compounds, limits toll-like receptor-4 (TLR4) signaling, and prevents mucosal inflammation [2-5, 10]. The absence of IAP also leads to alterations in gut microbiota [11]. Thus, IAP plays a critical role in preventing mucosal inflammation and maintaining gut homeostasis.
LV-associated IAP dephosphorylates LPS
Given the evidence implicating IAP in gut host defense and homeostasis [2-5, 10, 11], we hypothesized that IAP-enriched LVs produced by microvilli regulate epithelial-microbial interactions. To test this proposal, we first sought to determine if LVs could dephosphorylate the pro-inflammatory bacterial product, LPS. Dephosphorylation of LPS reduces the ability of this compound to activate TLR4 on host cell membranes [2, 10]. Our laboratory previously developed methods for isolating native microvillus-derived LVs from rodent small intestine [7]. Native vesicles isolated with these methods are able to dephosphorylate LPS from Escherichia coli serotype O55:B5 in a concentration dependent manner, which was sensitive to the IAP inhibitor, L-phenylalanine (L-phe) (Fig. 1A) [12]. This activity was not specific to E. coli O55:B5 LPS as assays with other LPS variants also gave rise to robust phosphate release (Fig. 1B, black bars). Purified IAP added at equivalent units of activity (0.1 U IAP = 10 μg LV, data not shown) demonstrated a comparable response (Fig. 1B, gray bars). Kinetic analysis of phosphate release from P. aeruginosa and E. coli LPS (substrates that supported the highest and lowest activities, respectively) revealed that P. aeruginosa LPS gives rise to higher rates of phosphate release at lower substrate concentrations (i.e. exhibits a lower KM; Fig. S1). Thus, LV-associated IAP is catalytically active and can dephosphorylate LPS from a variety of Gram-negative species.
Figure 1. LVs dephosphorylate LPS and interact with native lumenal bacteria.
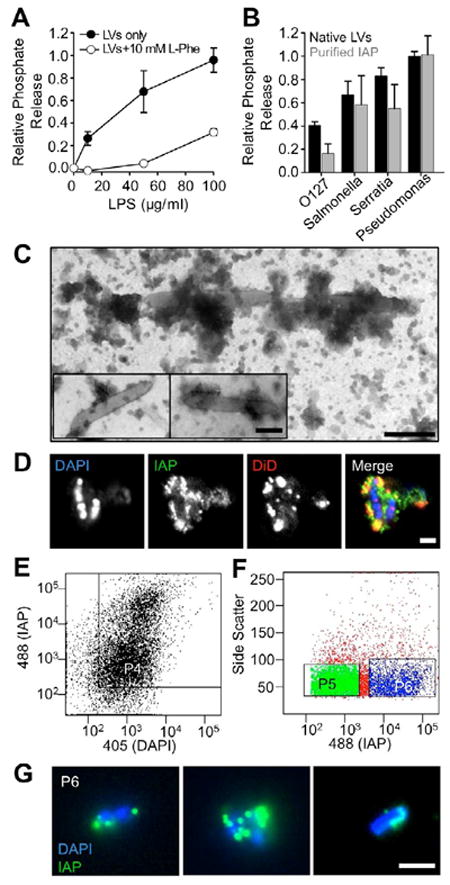
(A) LVs release phosphate from E. coli O55:B5 LPS in a concentration dependent manner; phosphate release is reduced by the IAP competitive inhibitor L-phenylalanine (L-Phe). (B) LVs (200 μg LVs/ml) and purified IAP (2 U/ml) differentially dephosphorylate LPS derived from various Gram-negative bacterial species. Data represent mean ± SEM. (C) TEM imaging reveals vesicle-like particles in close association with native lumenal bacteria from rat LV preparations. Bar, 200 nm. (D) CM imaging reveals IAP enrichment (green) on membranes (red) associated with DAPI-stained bacteria (blue). Bars, 2 μm. (E) Pre-sort input material was analyzed for staining with both anti-IAP (LVs) and DAPI (bacteria) using the P4 gate. (F) P4 material was sorted using gates for IAP (P5, low IAP enrichment) and dual stained material (P6, high IAP enrichment). (G) CM images of sorted P6 material show native LV/microbe complexes (green = IAP, blue = DAPI). Bar, 2 μm.
LVs physically interact with lumenal microbes
Because LVs are able to chemically modify bacterial compounds (Figs. 1A, B; S1), we next sought to determine if these vesicles could interact directly with lumenal microbes. To address this question, we examined resuspended pellets produced during native LV preparations from rat intestinal lumen wash using negative stain transmission electron microscopy (TEM). The resulting images revealed rod-shaped bacteria coated or in contact with clusters of material that resembled small vesicles (Fig. 1C). We labeled the same fraction with an anti-IAP antibody, a membrane dye (DiD), and DAPI (to label bacteria) and imaged samples using confocal microscopy (CM). Consistent with the presence of microvillus-derived LVs, we found that microbe-associated material was highly enriched in IAP and DiD (Fig. 1D).
To analyze minimally processed lumen wash material for the presence of LV/microbe complexes, we also employed a modified form of fluorescence-activated vesicle sorting (FAVS)[7, 13]. Raw lumen wash from rat small intestine was allowed to settle at 1 × g and then passed through a 40 μm filter to remove large particulate matter. Samples were then labeled as in Fig. 1D and applied to the flow cytometer. FAVS analysis revealed that 99% of all DAPI-labeled particles are associated with some level of IAP signal (P4, Fig. 1E). Double-positive particles were further analyzed to select for populations with low (P5, Fig. 1F) and high (P6, Fig. 1F) levels of IAP enrichment. Imaging of post-sort ‘P6’ material revealed numerous examples of rod-shaped bacteria surrounded by IAP-enriched puncta (Fig. 1G). Thus, the LV/microbe complexes observed in conventional lumen wash preparations (Fig. 1C, D) are most likely formed in the lumen and not as a result of centrifugation or other processing.
LVs prevent EPEC adherence to IECs
Because LVs bind directly to lumenal bacteria, microvillar vesicle shedding could serve as a host defense mechanism capable of preventing the adherence of pathogenic bacteria that target the IEC apical surface. To test this idea, we sought to determine if LVs impacted the ability of enteropathogenic E. coli (EPEC, strain E2348/69) to infect human IECs (HT-29 and CACO-2BBE cells) in culture. EPEC uses a type III secretion system to inject factors into host cells that lead to microvillar effacement, which in turn enables intimate adhesion to the apical surface, an event essential for EPEC pathogenesis [14]. We incubated confluent monolayers of HT-29 cells with ~1×105 colony forming units (CFU) EPEC/ml media in the presence or absence of 200 μg LVs/ml media (see Supplemental Experimental Procedures). After a 3 hr incubation, monolayers were stained and then imaged using CM (Fig. 2A, B) or scanning electron microscopy (SEM, Fig. 2C, D). Consistent with a previous report [15], we found that IAP was enriched on the surface of bacteria bound to the cell monolayer, independent of the presence of actin pedestals (arrowheads, Fig. 2A, B). Confocal images were further analyzed by counting: (i) the number of EPEC organisms bound per unit area, independent of stage of attachment (Fig. 2E), and (ii) the number of micro-colonies per unit area (Fig. 2F) [16]. Both indices showed that LVs significantly reduced EPEC attachment to HT-29 monolayers (Fig. 2E, F). Similar results were observed when experiments were carried out with CACO-2BBE cells (not shown). SEM on CACO-2BBE cultures confirmed that EPEC actin pedestal formation was reduced in the presence of LVs (Fig. 2C, D). Neither fixing LVs nor inhibiting LV-associated IAP activity with L-Phe impacted the ability of vesicles to limit EPEC attachment to HT-29s (Fig. 2E, F). Finally, purified IAP had little impact on the number of bacteria bound to the monolayer surface (Fig. 2E, F). Thus, LVs prevent EPEC from intimately attaching to IECs in culture via a mechanism that does not depend on IAP catalytic activity.
Figure 2. LVs prevent EPEC attachment to IECs.
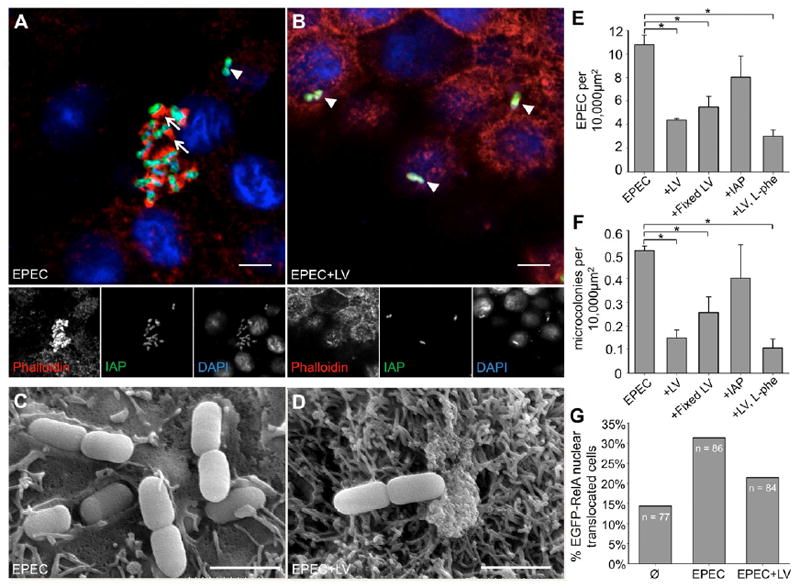
(A, B) CM images of HT-29 monolayers treated with EPEC ± LVs stained for IAP (green), F-actin (phalloidin, red) and bacteria (DAPI, blue), show striking enrichment of IAP surrounding attached bacteria. Most EPEC in non-LV treated samples (A) form micro-colonies and are associated with actin pedestals (arrows), indicating intimate attachment, whereas this rarely occurs in the presence of LVs (B). Arrowheads in A, B denote examples of EPEC superficially attached to the monolayer, indicated by the absence of pedestal formation. Bars, 5 μm. (C, D) Representative SEM images of CACO-2BBE cells demonstrate EPEC intimately associated with the cell surface in the absence (C), but not the presence of LVs (D). Bars, 1.67 μm. (E) The number of EPEC attached to HT-29 cells is significantly reduced in the presence of LVs, LVs fixed in paraformaldehyde, and LVs with IAP activity inhibited by L-Phe, but not in the presence of purified IAP (*p < 0.05). (F) LVs also reduce the number of micro-colonies formed. (G) Nuclear translocation of EGFP-RelA is increased in HT-29 cells incubated with EPEC, an effect which is partially ameliorated by treatment with LVs.
Bacteria and bacterial products activate pro-inflammatory NF-κB signaling [17]. Thus, we sought to determine if LVs were capable of preventing activation of NF-κB signaling in response to EPEC. A hallmark of NF-κB pathway activation is translocation of the transcription factor RelA into the nucleus [4]. We transfected HT-29 cells with EGFP-tagged human RelA [18], incubated cells in serum free media for 3 hrs, and then scored the fraction of transfected cells with nuclear RelA. Under these conditions, 14% of cells expressing the construct demonstrated RelA in the nucleus (Fig. 2G). In cells treated with EPEC, nuclear localization of RelA is observed in 31% of expressing cells, compared with 21% in EPEC-exposed cells treated simultaneously with 200 μg LVs/ml (Fig. 2G). The attenuated response observed here is likely due to the fact that IECs limit expression of cell-surface receptors such as TLR4, to prevent constitutive pro-inflammatory signaling [19]. Together these data indicate that LVs inhibit interactions between adherent pathogenic bacteria and IECs, and limit the downstream NF-κB signaling that would otherwise be triggered by these interactions.
LVs limit bacterial population growth
Direct association with microbes might also enable LVs to impact bacterial viability. To investigate this possibility, liquid cultures with ~2.5×104 CFU EPEC/ml were treated with either 0 or 50 μg LVs/ml and incubated overnight; OD600 was measured at 10 min intervals. While addition of LVs did not prevent initiation of log growth in EPEC (t = 4 - 8 hr), the maximum OD600 achieved in the presence of LVs was significantly reduced (Fig. 3A, black lines). We carried out similar growth assays with bacterial isolates derived from native lumenal material using MacConkey selection [20]. As with EPEC, native isolates entered log phase approximately 4 hr into the assay, but inclusion of 50 μg LVs/ml reduced the maximum OD600 (Fig. 3A, red and blue lines). 16S ribosomal subunit DNA sequence analysis showed that both commensals used in this assay were members of the genus Escherichia, most likely E. coli (not shown). Further experimentation revealed that the impact of LVs on bacterial population growth is not mediated by IAP or its catalytic activity (Supplemental Text 1; Fig. S2A). Moreover, LVs were not reducing the concentration of nutrients available to growing bacteria or otherwise conditioning the culture environment (Supplemental Text 1; Fig. S2B). Adding LVs to stationary phase EPEC cultures also led to a striking decline in OD600 over a similar 16 hr time course (Fig. 3B, orange lines). Comparable results were obtained with stationary EPEC cultures that were first replenished with fresh growth media (Fig. 3B, dark red lines). Finally, CFU analysis of culture end points revealed that LVs most likely reduce OD600 values by accelerating bacterial death (Supplemental Text 2; Fig. 3C). Together, these results indicate that LVs limit the population density of commensal and pathogenic E. coli in a manner independent of the catalytic activity of IAP or other enzymes.
Figure 3. LVs inhibit bacterial population growth.
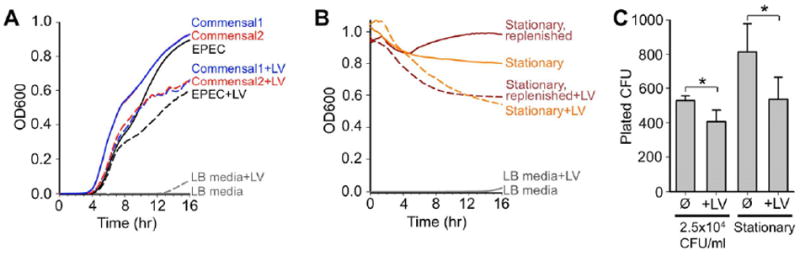
(A) Incubating both EPEC and native intestinal bacterial cultures with 50 μg LVs/ml reduces OD600 achieved at stationary phase (t = 8 - 16 hr) following log growth (t = 4 - 8 hr). Black lines = EPEC, blue and red lines = native E. coli. (B) Stationary phase EPEC cultures incubated with LVs show a reduction in OD600 compared to untreated samples over the course of 16 hrs, both in cultures with nutrient-depleted media (orange lines) or fresh media (dark red lines). Traces represent averaged recordings from multiple experiments (n = 3 - 8). (C) LVs reduce the number of viable bacteria (CFU), as sampled at the end of cultures started with 2.5 × 104 CFU/ml and incubated for 16 hrs or stationary cultures incubated for 16 hrs. *p < 0.05 vs. untreated.
EPEC stimulates LV production
All of our assays with native LVs isolated from rodent small intestine (Figs. 1-3) indicate that LVs likely function as a host defense platform. As many host defense pathways and processes are regulated by bacterial toxins or intact bacteria [21, 22], we sought to determine if LV production was controlled in a similar manner. For these experiments, we developed a culture model of microvillar vesicle shedding using CACO-2BBE cells [23-25]. CACO-2BBE cells polarize, form a well-ordered brush border, and demonstrate robust microvillar vesicle shedding similar to native IECs, as indicated by the accumulation of small vesicles (Fig. S3A) containing LV markers, IAP and annexin A13b [7], in culture media (Fig. 4A). When CACO-2BBE cells were exposed to ~1×106 CFU EPEC/ml for six hours, total cellular levels of IAP increased dramatically (Fig. 4B), consistent with previous work in zebrafish [26]. EPEC exposure also significantly increased the amount IAP-enriched LVs released into culture media (Fig. 4C). Accumulation of IAP in the 100,000 × g pellet likely represents bona fide vesicle shedding, rather than microvillar fragmentation, as no actin is observed in these fractions (Fig. 4C). Additionally, CACO-2BBE cells expressing the Myo1a-TH1 domain (TH1 DN), which acts as a dominant negative inhibiting the function of endogenous Myo1a [27], do not release IAP-enriched vesicles into culture media (Fig. 4C). We also found that vesicle shedding was stimulated, albeit to a lesser extent, by Shigella, another Gram-negative enteric pathogen (Fig. S3B) [28]. However, vesicle shedding was not stimulated by EPEC-conditioned media [29], heat-killed EPEC, or 10 μg/ml E. coli O55:B5 LPS (Fig. 4C). These findings indicate that microvillar vesicle shedding is upregulated in response to EPEC exposure, and that this response requires live, intact bacteria.
Figure 4. EPEC stimulates LV release from IECs.
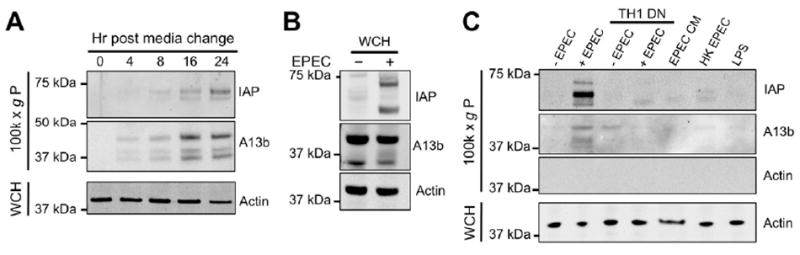
(A) LVs accumulate in CACO-2BBE cell culture media over time. Media was subject to ultra-speed centrifugation and 100,000 × g pellets were processed for western blotting with probes for IAP and annexin A13b; whole cell homogenates (WCH) from each time point were processed in parallel and probed for actin. (B) Whole cell IAP expression levels increase upon exposure to EPEC. (C) EPEC stimulates increased LV production from CACO-2BBE cells, indicated by the presence of IAP in the LV containing 100,000 × g pellet fraction. EPEC-stimulated LV production is attenuated by expression of a Myo1a dominant negative construct (TH1 DN). Shedding requires intact, live bacteria, as EPEC-conditioned media (CM), heat-killed (HK) EPEC, and purified LPS fail to stimulate LV production.
Conclusions
Previous work investigating the physiological function of IAP has focused on the activity of this enzyme as a soluble or cell-associated host defense factor in the gut lumen [2-5, 30]. The findings reported here suggest that microvillar vesicle shedding is a mechanism for distributing IAP activity into the mucous layer or gut lumen. Deploying IAP in this manner would enable chemical modification of bacterial toxins (i.e. dephosphorylation of LPS; Figs. 1A, B; S1) at sites distal to the apical surface of IECs. However, LVs also limit the attachment of pathogenic bacteria to IECs and inhibit bacterial population growth (Figs. 2, 3; S2). Moreover, IAP expression and LV production are upregulated by pathogenic Gram-negative bacteria (Figs. 4, S3). Based on these data and the fact that trillions of microvilli extend from the surface of IECs, we propose that microvillar vesicle shedding represents a powerful mechanism for limiting the potentially harmful impact of microbes and associated pro-inflammatory compounds that accumulate in the intestinal lumen. Future studies must focus on defining mechanisms that regulate LV production in response to microbial signals, mechanisms responsible for the impact of LVs on bacterial viability, and the role of LV production in animal models of gut disease.
Supplementary Material
HIGHLIGHTS.
Enterocyte microvillus-derived lumenal vesicles (LVs) detoxify bacterial toxins
LVs bind directly to bacteria and prevent pathogenic attachment to epithelial cells
LVs limit population growth of both commensal and pathogenic bacteria
LV production is a constitutive process, but is upregulated by bacterial stimulation
Acknowledgments
The authors thank all members of the Tyska laboratory for advice and support, Dr. Joseph Roland of the VUMC Epithelial Biology Center for assistance with GelCount analysis, Amy MacKenzie for assistance with rat tissue harvest, and Rebecca McRae for assistance with bacterial isolation and reagent preparation. EPEC strain E2348/69 was a generous gift of Dr. Gail Hecht, University of Illinois. Shigella strains were a generous gift of Dr. Benjamin Spiller, VUMC. Additionally, we thank the laboratory of Dr. Melanie Ohi for assistance with negative-stain TEM. This work was supported by National Institutes of Health R01-DK-075555 (MJT), American Heart Association predoctoral (DAS, REM) and postdoctoral (RN) fellowships and grant-in-aid 09GRNT2310188 (MJT), the VUMC Digestive Diseases Research Center (DK058404 to Dr. R. M. Peek), NCI CA46413 and GI Special Program of Research Excellence P50 95103 (RJC), and a Vanderbilt University Innovation and Discovery in Engineering And Science award (MJT). FAVS was performed in the Vanderbilt University Flow Cytometry Shared Resource, supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Louvard D, Kedinger M, Hauri HP. The differentiating intestinal epithelial cell: establishment and maintenance of functions through interactions between cellular structures. Annu Rev Cell Biol. 1992;8:157–195. doi: 10.1146/annurev.cb.08.110192.001105. [DOI] [PubMed] [Google Scholar]
- 2.Koyama I, Matsunaga T, Harada T, Hokari S, Komoda T. Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin Biochem. 2002;35:455–461. doi: 10.1016/s0009-9120(02)00330-2. [DOI] [PubMed] [Google Scholar]
- 3.Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007;2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldberg RF, Austen WG, Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, et al. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci U S A. 2008;105:3551–3556. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramasamy S, Nguyen DD, Eston MA, Nasrin Alam S, Moss AK, Ebrahimi F, Biswas B, Mostafa G, Chen KT, Kaliannan K, et al. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis. 2010;17:532–542. doi: 10.1002/ibd.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell RE, Tyska MJ. Myosin-1a powers the sliding of apical membrane along microvillar actin bundles. J Cell Biol. 2007;177:671–681. doi: 10.1083/jcb.200701144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McConnell RE, Higginbotham JN, Shifrin DA, Jr, Tabb DL, Coffey RJ, Tyska MJ. The enterocyte microvillus is a vesicle-generating organelle. J Cell Biol. 2009;185:1285–1298. doi: 10.1083/jcb.200902147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mooseker MS, Tilney LG. Organization of an actin filament-membrane complex. Filament polarity and membrane attachment in the microvilli of intestinal epithelial cells. J Cell Biol. 1975;67:725–743. doi: 10.1083/jcb.67.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins JH, Borysenko CW. The 110,000-dalton actin- and calmodulin-binding protein from intestinal brush border is a myosin-like ATPase. J Biol Chem. 1984;259:14128–14135. [PubMed] [Google Scholar]
- 10.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 11.Malo MS, Alam SN, Mostafa G, Zeller SJ, Johnson PV, Mohammad N, Chen KT, Moss AK, Ramasamy S, Faruqui A, et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut. 2010;59:1476–1484. doi: 10.1136/gut.2010.211706. [DOI] [PubMed] [Google Scholar]
- 12.Fishman WH, Green S, Inglis NI. L-phenylalanine: an organ specific, stereospecific inhibitor of human intestinal alkaline phosphatase. Nature. 1963;198:685–686. doi: 10.1038/198685b0. [DOI] [PubMed] [Google Scholar]
- 13.Cao Z, Li C, Higginbotham JN, Franklin JL, Tabb DL, Graves-Deal R, Hill S, Cheek K, Jerome WG, Lapierre LA, et al. Use of fluorescence-activated vesicle sorting for isolation of naked2-associated, basolaterally-targeted exocytic vesicles for proteomic analysis. Mol Cell Proteomics. 2008 doi: 10.1074/mcp.M700155-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vallance BA, Finlay BB. Exploitation of host cells by enteropathogenic Escherichia coli. Proc Natl Acad Sci U S A. 2000;97:8799–8806. doi: 10.1073/pnas.97.16.8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1876–1881. doi: 10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scaletsky IC, Silva ML, Trabulsi LR. Distinctive patterns of adherence of enteropathogenic Escherichia coli to HeLa cells. Infection and immunity. 1984;45:534–536. doi: 10.1128/iai.45.2.534-536.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wullaert A, Bonnet MC, Pasparakis M. NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146–158. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–1657. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 19.Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M. Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. Journal of immunology. 2001;167:1609–1616. doi: 10.4049/jimmunol.167.3.1609. [DOI] [PubMed] [Google Scholar]
- 20.Macconkey A. Lactose-Fermenting Bacteria in Faeces. J Hyg (Lond) 1905;5:333–379. doi: 10.1017/s002217240000259x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma R, Young C, Neu J. Molecular modulation of intestinal epithelial barrier: contribution of microbiota. J Biomed Biotechnol. 2010;2010:305879. doi: 10.1155/2010/305879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci. 1992;102:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- 24.Peterson MD, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. I. Ultrastructural analysis of cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:445–460. doi: 10.1242/jcs.105.2.445. [DOI] [PubMed] [Google Scholar]
- 25.Peterson MD, Bement WM, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. II. Changes in expression and localization of brush border proteins during cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:461–472. doi: 10.1242/jcs.105.2.461. [DOI] [PubMed] [Google Scholar]
- 26.Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol. 2006;297:374–386. doi: 10.1016/j.ydbio.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Tyska MJ, Mooseker MS. A role for myosin-1A in the localization of a brush border disaccharidase. J Cell Biol. 2004;165:395–405. doi: 10.1083/jcb.200310031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005;19:2645–2655. doi: 10.1101/gad.1299905. [DOI] [PubMed] [Google Scholar]
- 30.Chen KT, Malo MS, Beasley-Topliffe LK, Poelstra K, Millan JL, Mostafa G, Alam SN, Ramasamy S, Warren HS, Hohmann EL, et al. A Role for Intestinal Alkaline Phosphatase in the Maintenance of Local Gut Immunity. Dig Dis Sci. 2010 doi: 10.1007/s10620-010-1396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.