

Abstract
Multiciliate cells function prominently in the respiratory system, brain ependyma, and female reproductive tract to produce vigorous fluid flow along epithelial surfaces. These specialized cells form during development when epithelial progenitors undergo an unusual form of ciliogenesis, in which they assemble and project hundreds of motile cilia. Notch inhibits multiciliate cell formation in diverse epithelia, but how progenitors overcome lateral inhibition and initiate multiciliate cell differentiation is unknown. Here we identify a coiled-coil protein, termed multicilin, which is Notch regulated and highly expressed in developing epithelia where multiciliate cells form. Inhibiting multicilin function specifically blocks multiciliate cell formation in the Xenopus skin and kidney, while ectopic expression induces the differentiation of multiciliate cells in ectopic locations. Multicilin localizes to the nucleus, where it directly activates the expression of genes required for multiciliate cell formation, including FoxJ1 and genes mediating centriole assembly. Multicilin is also necessary and sufficient to promote multiciliate cell differentiation in mouse airway epithelial cultures. These findings suggest that multicilin initiates multiciliate cell differentiation in diverse tissues, by coordinately promoting the transcriptional changes required for motile ciliogenesis and centriole assembly.
Cilia are microtubule-based organelles that project from the cell surface, and in vertebrates have diversified into structurally different subtypes1. Primary cilia are generally short, immotile, and extended by a variety of cell types as a means to sense mechanical and chemical stimuli. Motile cilia are extended by specialized epithelial cells and used to generate fluid flow along their surfaces. Cilium diversity enables vertebrate cells to carry out specialized functions and is one factor underlying the spectrum of phenotypes observed in the human ciliopathies1. How cilium diversity is generated is an important problem in organelle biogenesis, and a poorly understood aspect of cell type differentiation.
Cilium diversity arises in part via the differential expression of components required for cilium subtype differentiation. FoxJ1, a winged-helix transcription factor, exemplifies this mechanism by activating gene expression required for motile cilia formation2. Motile cilia first appear in the embryo when node cells extend a single cilium to generate a flow that establishes left-right asymmetry3. Motile cilia also form during organogenesis by multiciliate cells (MCCs) that differentiate within the respiratory airways, ependyma, and oviduct4. In FoxJ1 mutants, primary cilia are unaffected, but cilia formation is disrupted in both MCCs and the cells mediating left-right patterning5–8. In gain-of-function experiments, FoxJ1 is sufficient to induce the formation of an ectopic motile monocilium7,8. Thus, FoxJ1 activates gene expression required for motile ciliogenesis but acts downstream of the mechanism that determines cilium number.
Cilium number is determined by the centriole, a microtubule-based structure that anchors the cilium, and comprises the core of the centrosome. In G0/1, the mother centriole docks at the plasma membrane and serves as a basal body to nucleate the outgrowth of the ciliary axoneme9. Typically, cells form a monocilium using a centriole-pair that is only duplicated during S-phase. During MCC differentiation, however, hundreds of centrioles are generated in postmitotic progenitors, to serve as basal bodies required for multiple cilia. This novel form of centriole assembly still occurs in MCC precursors in FoxJ1 mutant mice, suggesting that FoxJ1 is not required for this process5,6. The hallmark of MCC differentiation is the activation of a novel centriole assembly pathway along with motile cilia outgrowth, but the underlying mechanism that triggers these events is unknown.
Results
Identification and expression of MCI in Xenopus
The formation of MCCs in various organ systems, including the embryonic Xenopus skin, is known to be restricted by Notch signaling10–13. We therefore compared RNA expression in isolated Xenopus skin after manipulating Notch activity (see Methods), identifying genes that are repressed by Notch, and thus likely to promote MCC differentiation. The top gene in this comparison encodes a small coiled-coil protein that we have termed multicilin (MCI) for the reasons discussed below.
MCI is the Xenopus ortholog of human Idas, a protein related to the cell cycle regulators Geminin and GEMC1 (Supplementary Fig. 1a)14–16. MCI is encoded in a region of the X. tropicalis genome that is syntenic to mammalian genomes containing MCI orthologs and flanked by CCNO and CDC20B. CCNO is functionally uncharacterized, but also strongly upregulated in our microarray analysis. Three members of the MIR449 family embedded in the CDC20B gene are expressed during, and required for, MCC differentiation17. Thus, MCI lies in a genomic region that appears to have evolved to play a major role in MCC formation.
MCI RNA expression in Xenopus embryos presages other markers of MCC differentiation, including FoxJ1 and α-tubulin7 (Supplementary Fig. 1c–e, data not shown), occurring in a spotty pattern that correlates best with MCCs rather than other differentiated cell types18–20. MCI expression is lost by stage 26, when MCCs in the skin are fully differentiated, but is then detected in the developing nephrostomes of the kidneys where MCCs form at later stages (Supplementary Fig. 1f,g). Consistent with the microarray results, MCI RNA expression in the skin is inversely affected by the levels of Notch signaling (Supplementary Fig. 1n,o). Thus, expression of MCI RNA is transiently detected in epithelia with differentiating MCCs in a Notch regulated manner.
Knock down of MCI activity inhibits the formation of MCCs in the skin
To examine MCI function in skin development, we injected embryos with a morpholino (MCI-MOSPL) designed to target and inhibit splicing of the MCI preRNA21. Splicing of endogenous MCI RNA was inhibited in morphants, as predicted, resulting in MCI transcripts that should encode a truncated MCI protein (Supplementary Fig. 2a). Specificity of the morphant phenotype was verified by a rescue experiment, (Supplementary Fig. 2d–f), and by identical phenotypes obtained with dominant-negative MCI mutants (see below). A second MCI morpholino targeting the MCI start site proved less effective, suggesting that it only partially blocked MCI function (Supplementary Fig. 2b–c,g).
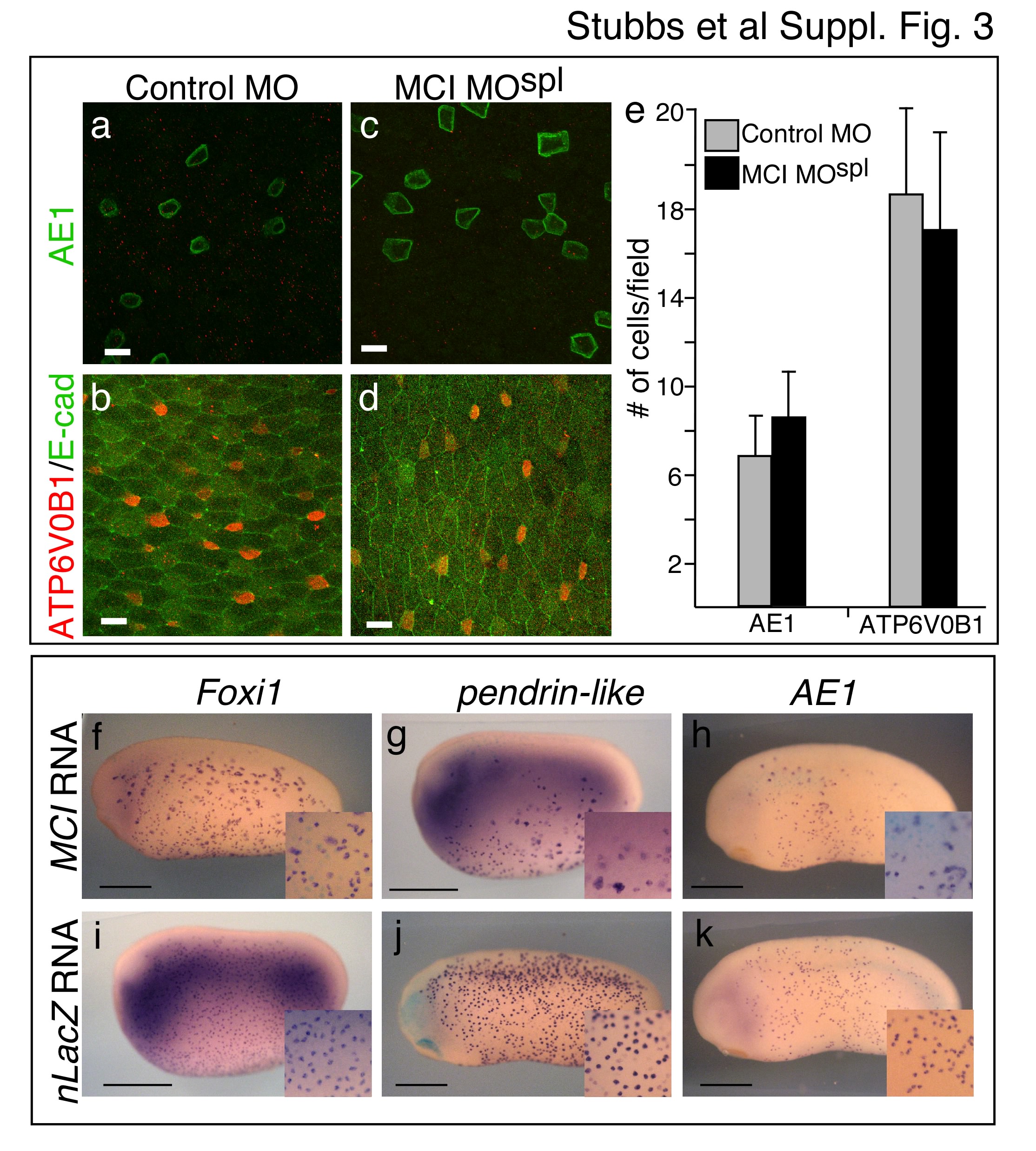
MCI-MOSPL morphants developed normally based on external morphology, but completely lacked MCCs in the skin based on cilia staining (Fig. 1b versus Fig. 1a). MCI morphants also lacked other features of MCCs, such as multiple centrioles, suggesting an early block in MCC differentiation, not just a block in basal body docking as seen in FoxJ1 morphants7 (Fig. 1c–f). MCCs first arise as precursors within the basal layer of the ectoderm, and differentiate by intercalating into the outer layer, along with an equal number of precursors for proton secreting cells (PSCs)19,22. In MCI-MOSPL morphants, the total number of intercalating cells is cut in half, with only intercalating PSCs evident, consistent with a selective loss of MCC precursors (Fig. 1i,j). In addition, PSCs numbers and subtypes were unchanged in MCI morphants compared to controls (Supplementary Fig. 3a–e). Thus, in the absence of MCI, epithelial progenitors fail selectively to give rise to MCCs, most likely remaining as undifferentiated basal cells.
Figure 1. MCI morphants lack MCCs.

(a–b) Confocal images taken of the skin from embryos that were injected with a control morpholino (a) or a MCI splicing morpholino (MCI-MOspl, b), fixed at stage 28, and stained with ZO-1 (red) and acetylated-tubulin (green) antibodies to label cell boundaries and cilia, respectively. (c–f) Confocal images of the skin from embryos injected with a control morpholino (c,e) or with MCI-MOspl (d,f), along with Hyls-GFP RNA to label centrioles (green, e,f). At stage 28, embryos were fixed, stained with rhodamine-phallodin (red) to label actin, and imaged. (g) Diagram of a transplant assay where the outer epithelium (OL) was isolated at stage 10 from a donor embryo injected with mRFP RNA and transplanted onto the inner layer (IL) of a host embryo injected with mGFP RNA. (h–i) Host embryos were also injected with a control morpholino (h) or with MCI-MOspl (i), fixed at stage 28 when intercalation and differentiation is complete, and imaged by confocal microscopy after staining for cilia (blue). (j) Quantification of outer cells (OC, red), ciliated cells (CCs, green/blue) or proton secreting cells (PSCs, green) for transplants onto control and MCI-MOspl injected embryos, showing the average (±s.d.) for 15 fields from 5 embryos. CC formation is dramatically reduced (* p=6×10−9) while PSC and OC numbers are statistically the same. Scale bars=10 microns. Arrowheads (a,c,e,h) and arrows (h) denote MCCs and PSCs, respectively.
MCI induces ectopic MCC differentiation
To determine if MCI can promote MCC differentiation, a myc-tagged form of MCI (MT-MCI) was misexpressed in embryos by RNA injection (Fig. 2b). At high levels, injection of MT-MCI RNA was toxic, while at lower concentrations, injected embryos survived through gastrulation and showed two phenotypes (Fig. 2b versus 2a): (1) outer epithelial cells were in some cases 2–4 times larger than normal and when extreme in size, nonciliated, and (2) the number of MCCs increased dramatically. Since apical size of the epithelial cells decreases in the early embryo through stage 10 without cell growth (Supplementary Fig. 4a), the former result suggests that ectopic MCI induces very early cell cycle exit (see below). The latter result suggests that MCI is sufficient to promote MCC differentiation.
Figure 2. MCI induces MCC differentiation.

(a,b) Confocal images of the skin in embryos injected at the two-cell stage with MT-MCI and mRFP RNA (b) or mRFP RNA alone (a), fixed at stage 28, stained with an acetylated tubulin antibody to label cilia (green). Asterisks in b denote unusually large cells. (c,d) Confocal images of the skin in embryos injected with MCI-HGR RNA along with mRFP RNA as a tracer treated with DEX at stage 11.5 (d), and then fixed at stage 28 and stained for cilia (green). (e–g) The outer epithelium from donor embryos injected with mRFP RNA (red) was transplanted onto host embryos injected with mGFP RNA (green) as illustrated in Fig. 1g. Host (f) and donor (g) embryos were also injected with MCI-HGR RNA. Embryos were treated with DEX at stage 11.5 and fixed at stage 28. Shown are confocal images of a control transplant (e), a transplant onto a host injected with MCI-HGR RNA (f), or a transplant from a donor injected with MCI-HGR RNA (g), after staining for cilia (blue). (h) Cell types in the skin were scored as MCCs (green with blue cilia staining), PSCs (green with no cilia staining), outer cells (red), or outer cells with cilia (red with blue cilia). Data is presented as an average of 10–15 fields (±s.d.) obtained from at least three transplants where the asterisks denote experimental values significantly different from control values (p<.005). Scale bars =20microns
MCI toxicity at early developmental stages led us to generate a glucocorticoid-inducible form (MCI-HGR)23. Embryos injected with MCI-HGR RNA developed normally in the absence of Dexamethasone (DEX), (Fig. 2c). However, when MCI-HGR expressing embryos were treated with DEX at the beginning of gastrulation, MCC differentiation was broadly induced across the skin (Fig. 2d), resulting in areas where all of the cells were multiciliated. The skin cell types that respond to MCI were determined by restricting MCI-HGR to different layers of the ectoderm (Fig. 1g). When confined to the outer layer, MCI-HGR efficiently induced MCCs but had little or no effect on the number of MCCs and PSCs derived from the underlying basal layer (Fig. 2g,h). Conversely, when MCI-HGR was expressed in just the basal layer, the number of intercalating MCCs increased at least three-fold, the number of PSCs dropped, and the outer cells in the transplant remained unciliated (Fig. 2f,h). Thus, MCI-HGR induces MCC differentiation in a layer autonomous fashion, including ectopically in the outer epithelium. In addition, MCI can inhibit PSC intercalation (Fig. 2h) and the number of cells expressing PSC markers (Supplementary Fig. 3f–k), suggesting that it can redirect cell fate. However PSCs did not increase in MCI morphants (Fig. 1j), indicating that MCC and PSC fates are normally established independently.
MCI promotes MCC differentiation downstream of Notch by coordinately activating several pathways
The results above suggest that MCI acts as a cell fate switch that allows progenitor cells to overcome lateral inhibition and form a MCC. We examined this possibility further by expressing different concentrations of MCI-HGR RNA along with RNA encoding an activated form of Notch, the intracellular domain (ICD). Under conditions where ICD efficiently represses endogenous MCC differentiation (Fig. 3c,f), ectopic MCC differentiation was still induced by high concentration of MCI-HGR RNA (Fig. 3b,f). At a lower concentration of MCI-HGR, however, ICD regained inhibitory activity, reducing the number of ectopic MCCs 2-fold (Fig. 3e,f). Significantly, under these conditions, a fraction of MCCs induced by MCI-HGR failed to complete differentiation, resulting in cells that extend a single cilium but do not assemble centrioles, while in others, centriole assembly occurred but axoneme elongation failed (Fig. 3e). Thus, MCI acts downstream of Notch to promote MCC formation, but Notch inhibits MCI expression and its ability to promote different aspects of the differentiation process, including centriole assembly and motile cilia extension. MCI promotes both processes cell autonomously, based on the analysis of MCI expressing clones (Supplementary Fig. 4b–e). In addition when MCI-HGR is expressed along with a FoxJ1 morpholino7, MCI still induces centriole formation, but ciliogenesis is severely disrupted, to the same extent as that seen in the MCCs of FoxJ1 morphants (Fig. 3g–i), suggesting that MCI induces motile cilia outgrowth via FoxJ1.
Figure 3. MCI acts downstream of Notch to promote centriole assembly and motile cilia extension via FoxJ1.

(a–e) Shown are confocal images of the skin in embryos injected with varying amounts of MCI-HGR RNA (a,d), ICD RNA (c), or both RNAs (b,e), along with mRFP (red) and Centrin4-GFP RNA (green) as a tracer and to mark centrioles, respectively. Embryos were treated with DEX at stage 11.5, fixed at stage 28 and stained with acetylated tubulin to visualize cilia (blue). In embryos injected with ICD and 37pgs of MCI-HGR RNA (e), MCC differentiation was sometimes incomplete, resulting in cells extending cilia but failing to undergo centriole assembly (arrowhead) or vice versa (arrow). Average number of centrioles (lower right, ±s.d.) is statistically the same with or without ICD at 150pg of MCI-HGR but different at 37pg of MCI-HGR RNA (p=6×10−6). Scale bars=20 microns. (f) The average percentage of MCCs (multiple centrioles and cilia) per field (±s.d) is plotted based on data obtained from three fields from four embryos (n=12). Values marked with an asterisk are highly significant (p<.005). (g–h) Shown are confocal images of the skin of embryos injected with MCI-HGR RNA, Centrin4-RFP and mRFP RNA, followed by a FoxJ1 (h) or a control morpholino (g)7. Embryos were treated with DEX at stage 11.5, fixed at stage 28 and stained with an acetylated tubulin antibody to label cilia (green). Scale bars=20microns. (i) Cilia number per cell was scored from two fields from five embryos (n=10) for each condition, and plotted as the average fraction of cells (±s.d.) with different cilia number as indicated per field.
MCI induces cell cycle exit and centriole assembly
The molecular pathway that induces centriole assembly in dividing cells is activated during the G1/S transition. By contrast, centriole assembly in MCCs occurs postmitotically. We therefore asked whether MCI activity drives cell cycle exit, using EdU-labeling to score the proportion of cells in the outer layer that still cycle after inducing MCI-HGR activity with DEX. The results show that MCI-HGR caused a marked decrease in EdU labeling within 1–3hrs after DEX induction, suggesting that MCI activity rapidly induces cell cycle exit (Supplementary Fig. 4f–h).
Centriole assembly in MCCs is poorly characterized molecularly but is known to occur at novel sites, called deuterosomes24. To determine whether MCI induces centriole assembly via similar structures, we scored foci of Sas6-GFP, a protein that seeds new centrioles when they multiply in MCCs25. In control embryos, a Sas6-GFP tracer lightly labels centrioles in outer layer cells as well as basal bodies in MCCs (Fig. 4a–e). By contrast, in MCI-HGR RNA injected embryos, numerous, large, bright foci of Sas6-GFP form in outer cells, starting approximately 4–6 hours after the addition of DEX, or slightly after cell cycle exit as measured above (Fig. 4f–j,k). The formation of these foci peaks at 8hrs post DEX, before dispersing into a large number of centrioles/basal bodies at 10hrs (Fig. 4j). When MCI-HGR injected embryos are fixed and imaged by TEM at 8hrs post-DEX, centriole assembly sites with the same morphology as deuterosomes are detected in outer cells (Fig. 4l). Centriole assembly induced by MCI is efficient, ultimately resulting in approximately the same number as that found in endogenous MCCs (Fig. 4m–o), with a similar distribution around the mean, and with proper polarity along the A-P axis (Supplementary Fig. 4i–l). These findings strongly suggest that MCI induces centriole assembly and basal body formation by the same mechanism operating in MCCs.
Figure 4. MCI induced centriole assembly.

(a–j) Shown are confocal images of the skin of control (a–e) or MCI-HGR RNA injected embryos (f–j), also expressing SAS6-GFP (green) and mRFP(red). Embryos were treated with DEX at stage 11.5, followed by fixation at the time intervals indicated. In control embryos, Sas6-GFP lightly labels centrioles (a,c arrowhead) in outer cells and basal bodies/centrioles in MCCs (d,e, arrow). By contrast, large bright Sas6-GFP foci are induced in outer cells by MCI-HGR (g,i arrowhead). Scale bar (a)=20microns (k) Plot showing the average fraction of cells (±s.d.) per field with different SAS-GFP foci number at the indicated time point, based on twelve fields taken from four embryos injected with MCI-HGR RNA. In control embryos, every outer cell scored contained 0–2 centrioles labeled with Sas6-GFP (data not shown). (l) Shown is a TEM image taken of an embryo injected with MCI-HGR RNA and treated with DEX for 8hrs. Structures similar to deuterosomes (arrows) in a pigmented (arrowhead) outer cell are located apical to the nucleus. Scale bar=500nm. (m–n) Shown are confocal images of the skin in embryos injected with mRFP and Hyls1-GFP RNA to mark cell boundaries (red) and centrioles (green), respectively, either alone (m) or with MCI-HGR RNA (n). At stage 11.5, embryos were treated with Dex, and then fixed at stage 28. Scale bars= 20microns (o) The average centriole number per cell (±s.d.) is plotted for control and MCI-HGR expressing embryos, using data obtained by scoring cells in at least three fields from five embryos. The slight increase in basal body number obtained with 150pg is significantly different (p<0.05) from control MCCs.
MCI is a direct regulator of MCC gene expression
MCI could mediate a cell fate switch by activating gene expression required for MCC differentiation, a possibility consistent with the response of MCI-HGR to DEX treatment, and the nuclear localization of ectopically expressed, tagged forms of MCI (Supplementary Fig. 5a–b), similar to human Idas14. Indeed, the expression of FoxJ1 and α-tubulin was lost in embryos injected with MCI-MOSPL (Fig. 5b,f) and conversely, dramatically upregulated in embryos injected with MCI-HGR RNA (Fig. 5d,h). The response to MCI is likely to be mediated through the proximal promoters of these genes, since MCI-HGR also induced the expression of transgenes containing GFP driven by 1.5kb of genomic DNA lying upstream of translation start site in the FoxJ1 or α-tubulin genes (Supplementary Fig. 5c–j)22.
Figure 5. MCI activates MCC gene expression.

(a–h) Shown is the expression of α-tubulin (a–d) or for FoxJ1 (e–h) in embryos injected with a control morpholino (a,e), with MCI-MOspl (b, f) or with MCI-HGR RNA along with nlacz RNA as a tracer (d,h) or alone as a control (c,g). RNA injected embryos were treated with DEX at stage 11.5, and all embryos were fixed at stage 26, and stained with X-gal (light blue). Scale bar=0.5mm. Insert shows a higher power (i) Animal caps were isolated at stage 10 from embryos injected with ICD RNA, MCI-HGR RNA (MCI), or the two RNAs together, treated with DEX at st11, and extracted for total RNA at stage 12, approximately two hours later. Shown is the log level of expression (±s.d.) relative to ICD values set at zero, as measured in triplicate using QT-PCR and after normalization against a ubiquitously expressed RNA, ODC. (j–m) Single animal blastomeres were injected with MCI-HGR RNA at the two-cell stage. At stage 11, half of the injected and uninjected control embryos were treated with cyclohexemide (CHX) for one hour, and then additionally treated with DEX for another hour to induce MCI-HGR activity. CHX is sufficient to block the onset of α-tubulin expression in the controls (compare k to j), indicating that the CHX treatment is effective, but does not block the activation of α-tubulin by MCI-HGR (m). Scale bar (j)=0.2mm.
We next used quantitative PCR to analyze MCI transcriptional activity in animal cap assays. MCI-HGR strongly induced FoxJ1 and α-tubulin expression in animal caps even in the presence of ICD, as described above (Fig. 5i). We then used microarrays to compare RNA expression in animal caps injected with ICD RNA alone, to that in animal caps injected with both ICD and MCI-HGR RNAs (Supplementary Table 2). As expected, genes upregulated by MCI-HGR included FoxJ1 and α-tubulin, as well as those previously shown in microarrays to be induced in animal caps by FoxJ1 (e.g. Tektins, ccdc78)7. MCI also induced genes already known to be expressed in MCCs but poorly characterized functionally (e.g Sox-7)26, genes potentially involved in MCC differentiation based on encoded structural motifs (e.g. ccdc45, ccdc52), and genes encoding centriole components (e.g. CEP76). Genes identified in this microarray analysis need to be validated (as shown above for FoxJ1 and α-tubulin), but are consistent with the model that MCI selectively activates gene expression required for MCC differentiation, but not for other cell types, including PSCs (Supplementary Fig. 3f–h).
We next asked whether MCI activates MCC gene expression directly. When embryos injected with MCI-HGR RNA were treated with DEX at stage 10.5, induction of α-tubulin expression occurs rapidly, within 1 hour (Fig. 5l, data not shown). Cycloheximide (CHX) treatment of control embryos between stage 11 and 12, blocks the onset of endogenous α-tubulin expression (Fig. 5k). In embryos injected with MCI-HGR, however, α-tubulin expression was still strongly activated in the presence of CHX, when DEX was added after the first hour of treatment (Fig. 5m). FoxJ1 expression was also induced by MCI-HGR following 1 hour of DEX treatment (data not shown), indicating the MCI may directly regulate the genes required for MCC differentiation in parallel.
Because MCI localizes to the nucleus and rapidly activates gene expression, we tested whether MCI behaves as a transcriptional activator or repressor when recruited to DNA. MCI was fused to the DNA binding domain of Gal4 and co-transfected into HEK293 cells along with a plasmid containing UAS binding sites upstream of a luciferase reporter. In this assay, MCI strongly activated transcription of the reporter gene at least 10–100 fold over basal transcription (Supplementary Fig. 5k), while Geminin, as a control, had no significant activity. Deletion mutants of MCI tested in this assay indicate that the transcriptional activation domain is not discrete, but distributed over several regions (Supplementary Fig. 5k).
Structural features of MCI required for MCC differentiation
To gain further insight into how MCI functions molecularly, we mapped the domains in MCI required for MCC differentiation using deletion constructs (Supplementary Fig. 6a). An MCI mutant lacking the coiled-coil domain (MCIΔ180–213) not only failed to induce MCC differentiation, but instead blocked both the appearance of MCCs in the skin and the expression of FoxJ1 and α-tubulin (Fig. 6c,g, Supplementary Fig. 6b,m). As in MCI morphants, MCIΔ180–213 blocked the formation of MCCs specifically, since the number of intercalating PSCs were unchanged (Supplementary Fig. 6c–e,r). Further deletion analysis (Fig. 6d,h, Supplementary Fig. 6b,n,s, data not shown) showed that the 40 amino acids at the carboxy terminus are sufficient to generate this strong dominant-negative phenotype, suggesting that it contains a critical domain required for MCI function.
Figure 6. Domains required for MCI function.

(a) Diagram of MCI. (b–e) Shown are embryos injected with RNA encoding different MCI mutants as indicated, fixed at stage 13/14, and stained for α-tubulin RNA expression (f–i) Confocal images of the skin of embryos injected with RNA encoding different MCI mutants, fixed at stage 28, and stained with ZO-1 (red) and acetylated tubulin (green) antibodies to label cell boundaries and cilia, respectively. Scale bars=0.5mm (b–e), 10 microns (f–h), or 20 microns (i).
Consistent with this interpretation, a form of MCI lacking CT (MCIΔ334–374) failed to induce ectopic MCC differentiation (Fig. 6i, Supplementary Fig. 6f) but did induce the large cell phenotype and reduced EdU incorporation consistent with premature cell cycle arrest (Supplementary Fig. 6g–j), as did a form containing just the coiled-coil domain (data not shown). MCIΔ334–374 did not qualitatively change the expression of the PSC marker, Foxi1, or the MCC markers, FoxJ1 and α-tubulin (Fig. 6e, Supplementary Fig. 6o,t) but did cause weak ciliogenesis defects (Fig. 6i). In sum, both the coiled-coil and CT domain are required for MCI to function as a transcriptional regulator during MCC differentiation. The former domain can induce cell cycle exit but not MCC gene expression while the latter is a potent specific inhibitor of MCC differentiation.
MCI is required for MCC differentiation in diverse epithelia
We next asked whether MCI function is required for other cells to form motile cilia. In Xenopus, motile monociliated cells that mediate left-right patterning arise on the gastrocoel roof plate (GRP) during gastrulation and extend cilia prior to the MCCs in the skin27. MCI RNA was not detectable in the superficial mesoderm that gives rise to the GRP (data not shown). Moreover, when MCI-MOSPL or the potent MCI dominant negative mutant was targeted to the mesoderm, motile cilia still appeared on the GRP (Supplementary Fig. 7a–e), suggesting that MCI is not required for motile cilium formation per se, but only when multiple motile cilia form per cell.
We next determined whether MCI is required when MCCs form within epithelia derived from other germ layers. MCI is expressed in Xenopus embryos in the developing nephrostomes, a derivative of the lateral mesoderm (Supplementary Fig. 1). In MCI morphants, the formation of MCCs within the kidney appears to be blocked based on α-tubulin expression, while MCI-HGR induced expanded regions of α-tubulin expression both in the kidney as well as ectopically in neighboring tissue (Supplementary Fig. 7f–i).
We also examined the expression and function of MCI in primary cultures of mouse tracheal epithelial cells (MTEC), in which MCC differentiation is promoted by exposure to an air-liquid interface (ALI)28. Based on RT-PCR, expression of mouse MCI (Pubmed Gene ID 622408) RNA markedly increases in MTEC cultures closely following the time course of MCC differentiation (Supplementary Fig. 8a)25. We next used lentiviral gene transfer to introduce either a myc-tagged form of full-length mouse MCI (MT-MCI) or a dominant-negative form that lacks the coiled-coil domain (MT-MCIΔCC), then analyzed for MCC differentiation. Under these conditions, approximately 20% of the control infected cells (GFP) differentiate into MCCs as marked either with pericentrin (basal body marker) or FoxJ1 staining, when assayed at 4 days or at both 4 and 8 days post-ALI, respectively (Fig. 7d). Strikingly, a vast majority (95–97%) of the cells in the MTEC cultures infected with MT-MCI form MCCs, while a vast majority (95–97%) of the cells infected with MT-MCIΔCCdo not. In cultures treated with DAPT, a Notch inhibitor, the number of cells expressing FoxJ1 increases approximately two-fold, along with the levels of MCI expression (Supplementary Fig. 8f–h). In these cultures, MT-MCIΔCCalso blocked FoxJ1 expression (Supplementary Fig. 8f), suggesting the MCI is required downstream of Notch regulation to promote MCC differentiation, as shown above in Xenopus skin.
Figure 7. Mouse MCI governs MCC differentiation in MTEC cultures.

(a–c) MTEC cultures were established, infected with lentivirus expressing MT-MCI, MT-MCIΔCC or GFP as a control, and switched to ALI culture conditions. At 4 days post-ALI, cultures were fixed, stained for infected cells (green) using an antibody to the myc-tag or GFP, to pericentrin (red) to identify MCCs and to E-cadherin (blue) to label cell boundaries. Shown are examples of cultures infected with a virus expressing GFP (a), MT-MCI (b) or MT-MCI-ΔCC (c). Arrows and asterisks denote infected cells that are pericentrin positive or negative, respectively. The insert in panel b shows a higher power image of pericentrin staining that marks clusters of nascent basal bodies. Scale bars = 50 microns (d) Percentage of infected cells that were ciliated versus nonciliated based on FoxJ1 or pericentrin staining under each condition, where n=total # of infected cells scored under each condition (±s.d.). Values marked with an asterisk are statistically significant (p<1×10 −10) from the values obtained for the GFP controls.
Discussion
Our results identify MCI as a key regulator of MCC differentiation based on its ability to coordinately promote cell cycle exit, deuterosome-mediated centriole assembly, and FoxJ1 expression (Supplementary Fig. 9). MCI functions in developing epithelia derived from all three germ layers, ranging from Xenopus skin and kidney to mouse trachea, suggesting that MCC differentiation is driven in epithelial progenitors by a MCI-mediated program regardless of embryonic lineage. Since MCI is positioned at the top of a hierarchy that directs epithelial progenitors into the MCC fate, mechanisms that promote or inhibit MCC precursor formation are likely to target MCI. An example of the latter is the Notch pathway, which restricts MCC differentiation in a variety of tissues and species11–13,29,30, and which we show here represses MCI expression and activity. Identification of MCI, therefore, is an important step towards determining how MCCs are selected out by Notch, programmed to differentiate, and for exploring how these cells are lost in disease states and restored during tissue repair.
One implication of our findings is that MCI determines cell fate not as a sequence specific DNA binding protein as in most cases, but rather as scaffolding protein that is cell type specific in expression and action. MCI presumably acts in a multi-protein complex that could conceivably function on or off DNA but is necessary and sufficient to activate gene expression associated with MCC differentiation. MCI may also function in part via its ability to heterodimerize with Geminin14, a dual-function protein that acts broadly to inhibit embryonic gene expression associated with differentiation31. Indeed, overexpressing Geminin in embryos does not induce MCC formation but inhibits their differentiation (data not shown)31. However, MCI is unlikely to act solely as a Geminin inhibitor, since the coiled-coil domain of MCI, which is sufficient to bind Geminin14 (data not shown), does not promote MCC formation. Moreover, MCI activates only MCC gene expression, a small subset of the embryonic gene expression inhibited by Geminin31. Thus, identification of additional binding partners for MCI is likely to be an essential step in revealing how MCI activates the genes required to acquire the MCC fate.
Centriole assembly in MCCs occurs through two pathways: approximately 10% of the centrioles form orthogonally to an existing centriole, much like in dividing cells, however the vast majority assembles around deuterosomes24. The centriolar pathway could be driven by increased expression of factors that initiate centriole assembly at the G1/S transition, which can modestly amplify centriole number when overexpressed32–36. Our findings suggest that the acentriolar pathway may be driven by MCI, as evidenced by its ability to induce the appearance of ectopic deuterosomes (Fig. 4l), resulting in the appropriate centriole number (Fig. 4o). Since this response correlates with the ability of MCI to activate transcription, we also suggest that MCI transcriptional targets are directly involved in initiating centriole assembly via the deuterosome-mediated, acentriolar pathway. Further characterization of these targets, therefore, will likely identify an alternative centriole assembly pathway that underlies this striking example of organelle biogenesis.
Materials and Methods
RNA Isolation and Microarray
Animal caps were isolated from embryos injected at the 4-cell stages with RNA encoding ICD to activate Notch signaling or dominant-negative forms of Suppressor of Hairless (Su(H)) or human mastermind (HMM) to block Notch signaling37,38. Total RNA was isolated using the proteinase K method from ectoderm explanted at stage 10, and harvested at the equivalent of stage 12. Total RNA was used to generate labeled complimentary RNA (cRNA) that was hybridized to Xenopus laevis v2 Genome Array chips (Affymetrix # 901214). Microarray data were obtained from two independent experiments in which embryos were injected with ICD RNA alone, and two independent experiments in which embryos were injected with either dnSu(H) or dnHMM RNA. These data sets were analyzed using Bullfrog analysis software using a pairwise comparison, with the minimum fold change set at 3 39. The top gene upregulated (168-fold) when Notch was blocked versus activated corresponds to Unigene Xl.55698. Microarray data has been submitted to GEO (GSE32331). Analysis of MCI-HGR regulated genes was carried out as described above, except that embryos were injected with both MCI-HGR and ICD RNAs, or with just ICD RNA alone. Isolated caps were treated with DEX at stage 11 and then harvested four hours later for analysis.
Constructs
A Xenopus clone for MCI was isolated using PCR from a stage 17 cDNA library, verified by sequencing and cloned downstream of the 6X myc-tags in CS240 to generate a RNA template for myc-tagged MCI (MT-MCI). RNA templates for the deletion mutations of MCI were generated in the CS2-MT vector using restriction sites or PCR, followed by sequencing. A RNA template for MCI-HGR was constructed by fusing sequences encoding the ligand binding domain of the human glucocorticoid receptor (HGR) to the MT-MCI carboxy terminus23. Templates for generating synthetic RNA encoding Hyls1-GFP, MT-ICD, Centrin4-RFP, Clamp-GFP, mGFP, or mRFP in vitro have been described previously7,22,41,42.
A cDNA encoding mouse MCI was obtained from the Riken mouse FANTOMTM clone library (clone ID 5830438C23). Both the full-length mouse MCI, and a form lacking the coiled-coil domain (MT-MCIΔCC, Δ175–238) were generated by PCR, sequenced, and inserted downstream of the six myc tags in CS2. Sequences encoding MT-MCI and MT-MCIΔCC were transferred into the pRRL.sin-18.PPT.PGK.GFP.pre vector25,43, and used to generate lentiviruses using protocols previously described25.
Xenopus laevis fertilizations and microinjections
Xenopus embryos were obtained by in vitro fertilization using standard protocols44. Embryos were injected at the two-cell stage with capped, synthetic mRNAs, typically using 1–5 ng/embryos. Morpholinos (Genetools) directed against MCI RNA targeted the initiation codon (Supplementary Table 1), or the splice donor site at the junction between exon5 and intron 5 (Supplementary Table 1). The FoxJ1 morpholino has been described previously7. Efficacy of the splicing morpholino was assayed using RT-PCR (Supplementary Fig. 2). Unless indicated otherwise, embryos were injected with 40–50ng of morpholino.
In-situ Hybridization and Immunofluorescence
Whole mount, in situ hybridizations were performed in embryos injected with RNAs or morpholinos where the injected side was marked by a LacZ RNA tracer and stained with X-Gal as described previously7. RNA probes to detect Xenopus α-tubulin, FoxJ1, Foxi1, AE1, and pendrin-like were previously described7,19 and the MCI probe consisted of just the coding region.
Embryos were fixed quickly for visualization of GFP fusion proteins and actin staining with phallodin using a formaldehyde and gluteraldehyde-based quick fix7. For antibody staining, embryos were fixed in 4% paraformaldehyde in Phosphate Buffered Saline (PBS) for 1 hour on ice followed by dehydration in 100% ethanol. In the case of the gastrocoel roof plate, dorsal explants were placed underneath a glass coverslip just prior to fixation, in order to flatten out the curvature of the GRP and to press the cilia against the cell surface for measurement7. Fixed tissues were rehydrated, washed with PBS/0.1% TritonX-100 (PBT), and blocked with PBT containing 10% heat-inactivated normal goat serum (PBT/HIGS) for at least one hour. Embryos were incubated with primary antibody in blocking solution overnight as follows: Rabbit anti-ZO-1 (Zymed 1:200), mouse monoclonal anti-acetylated α-tubulin (Sigma, 1:200–1:1000), mouse anti-myc (9E10, 1:10), rabbit anti-GFP (Molecular Probes, 1:1000), or rabbit anti-dsRed (Clontech 1:1000). After washing, embryos were incubated overnight in Cy2, Cy3, or Cy5 labeled Goat anti-IgG of the appropriate species (all used at 1:500, Jackson ImmunoResearch), washed in PBT and then mounted in PVA/DABCO. Mounted embryos were imaged on a BioRad Radiance 2100 confocal mounted to a Zeiss inverted microscope using a 40X or 63X objective. Typically, data was collected from three randomly chosen fields from five embryos using the tracer to detect injected regions. Statistical significance was gauged in all experiments using a two-tailed t-test.
Xenopus transgenics
A 1.5 kb genomic fragment lying upstream of the translational start in the X. tropicalis FoxJ1 gene was isolated by PCR, and cloned upstream of GFP using CS2 as the backbone. FoxJ1-GFP transgenics were generated employing the Kroll and Amaya method as modified by Sparrow et al, using sperm nuclei transfer45,46. The 1.6k α-tubulin-GFP F1 transgenics were generated using sperm nuclei derived from an F0 male as described previously22. Transgenic embryos were injected at the two-cell stage with MCI-HGR or FoxJ1 RNA, along with mRFP as a tracer or as a control, treated with DEX at stage 11, fixed at stage 26, and imaged, after staining for cilia using an acetylated antibody.
MTEC cultures, lentiviral manipulation, immunofluorescence and RT-PCR
MTEC were cultured from wildtype C57BL/6J (The Jackson Laboratory) mice of six weeks of age or older, as previously described 25. All procedures involving animals were approved by the Institutional Animal Care and Use Committee in accordance with established guidelines for animal care. In short, airway epithelial cells were isolated from trachea by protease digestion, contaminating fibroblasts were removed by preplating the suspension, and the remaining, unadhered epithelial cells were seeded onto collagen coated Transwell-Clear permeable membranes (Corning). Cells were grown in MTEC complete medium 28 until air-liquid interface (ALI) was created approximately two days after confluence by adding MTEC basal medium + 2% NuSerum28. Beating cilia were observed by phase microscopy 3 days after ALI creation. To block Notch activity, MTEC were treated with 1 μM DAPT in ALI media for 72 h from ALI + day 1 to day 4. Lentivirus production and infection of MTEC were carried out as previously described 25. In short, lentivirus expressing GFP or myc-tagged MCI cDNA was produced by transient cotransfection of 293T/17 cells (ATCC) with the lentiviral transfer vector pRRL.sin-18.PPT.PGK.GFP.pre 43 and helper vectors (pCMVDR8.74 packaging vector and pMD2.VSVG-envelope vector) 47 using the calcium phosphate coprecipitation method. The lentiviral supernatant was concentrated by centrifugation to achieve 107–108 infectious units/ml. To infect MTEC, tight junctions were first disrupted by EGTA treatment, then cells were spin infected with lentivirus. For indirect immunofluorescence, MTEC were fixed in 4% paraformaldehyde at room temperature for 10 min, then filters were excised from plastic supports. Samples were blocked in 10% normal horse serum (Invitrogen) and 0.1% Triton X-100 in PBS for 1 h at room temperature. Primary antibodies were applied to filters at 37 oC for 1 h: Foxj1 (1:500, eBioscience), pericentrin (1:250, Abcam), myc (1:200, A14, Santa Cruz) and E-cadherin (1:250, ECCD-2, Invitrogen). Alexa dye-conjugated secondary antibodies (1:250, Invitrogen) were applied to filters at room temperature for 30 min. Filters were mounted with 12 mm #1.5 coverslips (Erie Scientific) using Mowiol mounting medium containing N-propyl gallate (Sigma). For RT-PCR, RNA was extracted from MTEC using Qiagen RNeasy Mini kit (Qiagen) after cells were treated with a 1:1 mix of Nonenzymatic Cell Dissociation Solution (Sigma) and 0.5% Trypsin (Invitrogen) for 20 min. cDNA was generated using the SuperScript III First Strand Synthesis kit (Invitrogen) and used to amplify GAPDH (control) and MCI.
Quantitative RT-PCR
Embryos were injected at the two-cell stage with ICD and/or MCI-HGR RNA. Animal caps were dissected at stage 10, treated with DEX at stage 11, and than harvested for total RNA at stage 13. cDNA templates were generated from 3μg of RNA using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative RT-PCR reactions were performed using the ABI Prism 7900HT Thermal Cycler, using primers for FoxJ1 and α-tubulin or for ornithine decarboxylase (ODC) as a normalization control (Supplementary Table 1). Data was analyzed using Applied Biosystems Sequence Detection System (SDS) software.
EdU Assays
Embryos were injected at the two-cell stage with MCI-HGR and mRFP RNA or mRFP RNA alone. Dexamethasone (Dex) was added to embryos at mid-gastrula stages. 1 hour, 3 hour or 7 hours after addition of Dex embryos were injected four times (10nL/drop) into the gastrocoel with 10mM EdU (Invitrogen). Embryos were allowed to develop until stage 28, fixed for 1hr in 3.7% Formaldehyde in 1X PBS, and then dehydrated into ethanol. Embryos were stained using the Invitrogen Click-iT Alexa 488 kit (Invitrogen C10337). Briefly embryos were rehydrated into PBT (1X PBS with 0.5% Triton X-100). Embryos were permeabilized by washing 1hr in PBT at room temp then incubated for 1hr in EdU reaction buffer made up per kit instructions. Embryos were washed several times in PBT then mounted in PVA/DABCO and imaged as for other staining.
Reporter assays
HEK293 cells were cotransfected with140ng of firefly luciferase reporter construct (Promega pGL4.23 #E4811) and 150ng of expression constructs where MCI fragments were cloned in frame with the Gal4 DNA binding domain in the pCMV-BD vector (Agilent #211342). Cells were transfected in 48-well plates using FuGENE 6 (Roche #1181443001) together with 10ng of the renilla luciferase pGL4.74 plasmid (Promega #E6921) as a transfection efficiency control. Firefly and renilla luciferase activity was measured 40–48 hours after transfection, using the Dual-Glo Luciferase Assay System (Promega #E2920). Values are mean ± standard deviation for three individual experiments.
Transmission Electron Microscopy
Embryos were fixed in 2% glutaraldehyde in 0.1M sodium cacodylate buffer, rinsed, postfixed in 1% osmium tetroxide and 1% potassium ferrocyanide, rinsed, en bloc stained in 1% uranyl acetate, dehydrated with glycol methacrylate and embedded in Epon. Thin sections (~60nm) were cut on an ultramicrotome, collected onto formvar-coated slot grids and stained with 2% uranyl acetate and 0.2% lead citrate. The sections were examined at 80kV in a Zeiss Libra 120 PLUS EF-TEM Transmission Electron Microscope.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The work described here was supported by NIH grants RO1 GM096021 (C.K) and R01 GM059823 (J.A.) and an AP Giannini Postdoctoral Fellowship (E.K.V). The authors thank Evan Campbell for technical assistance and members of the lab for comments on the manuscript.
Footnotes
Author Contribution:
J.S., E.V. and C.K. carried out the experimental analysis. All authors participated in aspects of project planning, data analysis, and manuscript preparation.
Competing Financial Interest Statement
The authors have no competing financial interests.
References
- 1.Sharma N, Berbari NF, Yoder BK. Ciliary dysfunction in developmental abnormalities and diseases. Curr Top Dev Biol. 2008;85:371–427. doi: 10.1016/S0070-2153(08)00813-2. [DOI] [PubMed] [Google Scholar]
- 2.Thomas J, et al. Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell. 2010;102:499–513. doi: 10.1042/BC20100035. [DOI] [PubMed] [Google Scholar]
- 3.Basu B, Brueckner M. Cilia multifunctional organelles at the center of vertebrate left-right asymmetry. Curr Top Dev Biol. 2008;85:151–74. doi: 10.1016/S0070-2153(08)00806-5. [DOI] [PubMed] [Google Scholar]
- 4.Satir P, Christensen ST. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 5.Brody SL, Yan XH, Wuerffel MK, Song SK, Shapiro SD. Ciliogenesis and left-right axis defects in forkhead factor HFH-4-null mice. Am J Respir Cell Mol Biol. 2000;23:45–51. doi: 10.1165/ajrcmb.23.1.4070. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Knowles HJ, Hebert JL, Hackett BP. Mutation of the mouse hepatocyte nuclear factor/forkhead homologue 4 gene results in an absence of cilia and random left-right asymmetry. J Clin Invest. 1998;102:1077–82. doi: 10.1172/JCI4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stubbs JL, Oishi I, Izpisua Belmonte JC, Kintner C. The forkhead protein Foxj1 specifies node-like cilia in Xenopus and zebrafish embryos. Nat Genet. 2008;40:1454–60. doi: 10.1038/ng.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu X, Ng CP, Habacher H, Roy S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat Genet. 2008;40:1445–53. doi: 10.1038/ng.263. [DOI] [PubMed] [Google Scholar]
- 9.Marshall WF. Basal bodies platforms for building cilia. Curr Top Dev Biol. 2008;85:1–22. doi: 10.1016/S0070-2153(08)00801-6. [DOI] [PubMed] [Google Scholar]
- 10.Deblandre GA, Wettstein DA, Koyano-Nakagawa N, Kintner C. A two-step mechanism generates the spacing pattern of the ciliated cells in the skin of Xenopus embryos. Development. 1999;126:4715–28. doi: 10.1242/dev.126.21.4715. [DOI] [PubMed] [Google Scholar]
- 11.Guseh JS, et al. Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development. 2009;136:1751–9. doi: 10.1242/dev.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morimoto M, et al. Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. J Cell Sci. 2010;123:213–24. doi: 10.1242/jcs.058669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsao PN, et al. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development. 2009;136:2297–307. doi: 10.1242/dev.034884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pefani DE, et al. Idas, a novel phylogenetically conserved geminin-related protein, binds to geminin and is required for cell cycle progression. The Journal of biological chemistry. 2011;286:23234–46. doi: 10.1074/jbc.M110.207688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balestrini A, Cosentino C, Errico A, Garner E, Costanzo V. GEMC1 is a TopBP1-interacting protein required for chromosomal DNA replication. Nat Cell Biol. 2010;12:484–91. doi: 10.1038/ncb2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo S, Kroll KL. Geminin’s double life: chromatin connections that regulate transcription at the transition from proliferation to differentiation. Cell Cycle. 2006;5:374–9. doi: 10.4161/cc.5.4.2438. [DOI] [PubMed] [Google Scholar]
- 17.Marcet B, et al. Control of vertebrate multiciliogenesis by miR-449 through direct repression of the Delta/Notch pathway. Nat Cell Biol. 2011 doi: 10.1038/ncb2241. [DOI] [PubMed] [Google Scholar]
- 18.Hayes JM, et al. Identification of novel ciliogenesis factors using a new in vivo model for mucociliary epithelial development. Dev Biol. 2007;312:115–30. doi: 10.1016/j.ydbio.2007.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quigley IK, Stubbs JL, Kintner C. Specification of ion transport cells in the Xenopus larval skin. Development. 2011;138:705–14. doi: 10.1242/dev.055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dubaissi E, Papalopulu N. Embryonic frog epidermis: a model for the study of cell-cell interactions in the development of mucociliary disease. Dis Model Mech. 2010;4:179–92. doi: 10.1242/dmm.006494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heasman J. Morpholino oligos: making sense of antisense? Dev Biol. 2002;243:209–14. doi: 10.1006/dbio.2001.0565. [DOI] [PubMed] [Google Scholar]
- 22.Stubbs JL, Davidson L, Keller R, Kintner C. Radial intercalation of ciliated cells during Xenopus skin development. Development. 2006;133:2507–15. doi: 10.1242/dev.02417. [DOI] [PubMed] [Google Scholar]
- 23.Kolm PJ, Sive HL. Efficient hormone-inducible protein function in Xenopus laevis. Dev Biol. 1995;171:267–72. doi: 10.1006/dbio.1995.1279. [DOI] [PubMed] [Google Scholar]
- 24.Dirksen ER. Centriole and basal body formation during ciliogenesis revisited. Biol Cell. 1991;72:31–8. doi: 10.1016/0248-4900(91)90075-x. [DOI] [PubMed] [Google Scholar]
- 25.Vladar EK, Stearns T. Molecular characterization of centriole assembly in ciliated epithelial cells. J Cell Biol. 2007;178:31–42. doi: 10.1083/jcb.200703064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fawcett SR, Klymkowsky MW. Embryonic expression of Xenopus laevis SOX7. Gene expression patterns: GEP. 2004;4:29–33. doi: 10.1016/j.modgep.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Schweickert A, et al. Cilia-driven leftward flow determines laterality in Xenopus. Curr Biol. 2007;17:60–6. doi: 10.1016/j.cub.2006.10.067. [DOI] [PubMed] [Google Scholar]
- 28.You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1315–21. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Pathak N, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134:1111–22. doi: 10.1242/dev.02806. [DOI] [PubMed] [Google Scholar]
- 30.Ma M, Jiang YJ. Jagged2a-notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genet. 2007;3:e18. doi: 10.1371/journal.pgen.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim JW, Hummert P, Mills JC, Kroll KL. Geminin cooperates with Polycomb to restrain multi-lineage commitment in the early embryo. Development. 2011;138:33–44. doi: 10.1242/dev.059824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dzhindzhev NS, et al. Asterless is a scaffold for the onset of centriole assembly. Nature. 2010;467:714–8. doi: 10.1038/nature09445. [DOI] [PubMed] [Google Scholar]
- 33.Bettencourt-Dias M, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol. 2005;15:2199–207. doi: 10.1016/j.cub.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 34.Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T. Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol. 2010;191:721–9. doi: 10.1083/jcb.201006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cizmecioglu O, et al. Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to the centrosome. J Cell Biol. 2010;191:731–9. doi: 10.1083/jcb.201007107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol. 2005;7:1140–6. doi: 10.1038/ncb1320. [DOI] [PubMed] [Google Scholar]
- 37.Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397–411. doi: 10.1101/gad.991602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wettstein DA, Turner DL, Kintner C. The Xenopus homolog of Drosophila Suppressor of Hairless mediates Notch signaling during primary neurogenesis. Development. 1997;124:693–702. doi: 10.1242/dev.124.3.693. [DOI] [PubMed] [Google Scholar]
- 39.Zapala MA, Lockhart DJ, Pankratz DG, Garcia AJ, Barlow C. Software and methods for oligonucleotide and cDNA array data analysis. Genome Biol. 2002;3:SOFTWARE0001. doi: 10.1186/gb-2002-3-6-software0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner DL, Weintraub H. Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 1994;8:1434–47. doi: 10.1101/gad.8.12.1434. [DOI] [PubMed] [Google Scholar]
- 41.Dammermann A, et al. The hydrolethalus syndrome protein HYLS-1 links core centriole structure to cilia formation. Genes Dev. 2009;23:2046–59. doi: 10.1101/gad.1810409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitchell B, et al. The PCP pathway instructs the planar orientation of ciliated cells in the Xenopus larval skin. Curr Biol. 2009;19:924–9. doi: 10.1016/j.cub.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Follenzi A, Ailles LE, Bakovic S, Geuna M, Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet. 2000;25:217–22. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- 44.Sive H, Grainger RM, Harland RM. The early development of Xenopus laevis: a laboratory manual. Cold Spring Harbor Press; Plainview, NY: 1998. [Google Scholar]
- 45.Amaya E, Kroll KL. A method for generating transgenic frog embryos. Methods Mol Biol. 1999;97:393–414. doi: 10.1385/1-59259-270-8:393. [DOI] [PubMed] [Google Scholar]
- 46.Sparrow DB, Latinkic B, Mohun TJ. A simplified method of generating transgenic Xenopus. Nucleic Acids Res. 2000;28:E12. doi: 10.1093/nar/28.4.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dull T, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.