

Abstract
A deeper understanding of the mechanism of complement activation may help to elucidate the pathogenesis of IgA nephropathy (IgAN). Traditionally, the activation of an alternative pathway (AP) has been recognized as an enhancer mechanism of glomerular damage. This paper documents contemporary information concerning the possible pathological mechanisms of the lectin pathway (LP) in the circulation and in the glomerulus. The circulating initiator of LP activation is not fully understood. However, ligands for mannose-binding lectin (MBL) which are among the starter molecules of the LP are aberrant glycosylated molecules-containing immune complex. Recent reports have focused on N-glycans on secretory IgA as a candidate ligand. Mesangial deposits of MBL are seen in 25% of patients with IgAN. Mesangial deposits of MBL and C4 and/or C4 breakdown products are implicated as markers for disease progression of IgAN. On the other hand, patients with MBL deficiency tend to show better clinical presentation and lower levels of urinary protein and serum creatinine than MBL-sufficient patients. It is now recognized that involvement of AP and LP constitutes an additional mechanism for explaining the progression of IgAN.
1. Introduction
IgA nephropathy (IgAN) is the most common form of primary glomerulonephritis. It is generally agreed that up to 30% of patients with IgAN eventually progress to end-stage kidney disease [1]. The diagnosis of IgAN can be confirmed only by renal biopsy that shows mesangial cell proliferation and mesangial IgA deposits. In the circulation, serum levels of IgA are increased in approximately 50% of patients with IgAN. Recently, the key feature has been considered to be the deficiency of galactose in the hinge region of the IgA1 heavy chains and it is thought that these abnormal IgA1 glycosylations reduce clearance of immune complexes which may then be sufficient to trigger glomerular mesangial deposition [2].
With respect to complement deposition, C3 is highly detected in glomeruli and the detection rate is found in more than 90% of patients of IgAN [3, 4]. Since other complement molecules, such as properdin and C5, are colocated in the mesangial areas with IgA, but C1q is absent and there is an increase of C3 breakdown products in the serum of patients with IgAN, we had recognized that alternative pathway (AP) activation is involved in the pathogenesis of IgAN [5]. After discovering new molecules which belong to the lectin pathway (LP), we came to believe there is accumulating evidence to support a hypothesis of the occurrence of LP activation in the pathogenesis of IgAN. The issues surrounding these scenarios with LP activation are reviewed to summarize their potential significance in the pathogenesis of IgAN. In order to explain the updated knowledge in plain terms, the main body was separated into three parts, “In circulation,” “ In kidney,” and “Infection and clinical course.”
2. In Circulation
2.1. Serum Levels of MBL in Patients with IgA Nephropathy
We previously measured the serum concentrations of MBL in 20 healthy controls and in 80 patients with primary glomerular disease (Figure 1). The serum concentrations of MBL from the patients presented a wider distribution than in healthy controls, but a comparison of the mean levels from each group showed no significant difference. Since MBL levels are altered by more than 3-4 times baseline levels during severe infection [6], production of MBL might be enhanced under some context, such as inflammation. Our colleague, Onda, had reported that the patients with IgAN showed hypercomplementemia; the mean of CH50 titer and serum levels of C4, factor B and properdin in patients with IgAN were significantly higher than those in normal controls (Table 1) [7]. Although hypercomplementemia is derived from chronic inflammation which accelerates the production of complement components in IgAN, it is interesting to note that serum concentrations of C3 and MBL were relatively lower than in normal controls. Lhotta et al. [8] and Roos et al. [9] also previously described no significant difference of mean levels of serum MBL between healthy controls and IgAN patients. This implies that consumption of C3 may increase by LP activation, thereby adding to AP activation.
Figure 1.
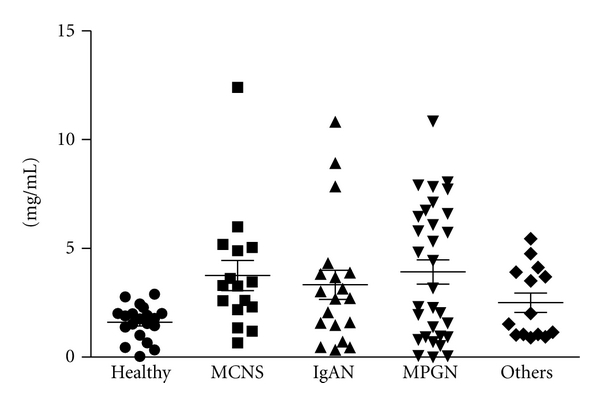
The serum concentrations of MBL were measured in 20 healthy controls and in 80 patients of primary glomerular diseases. The mean levels of MBL showed no statistical difference between each disease. However, some cases of primary glomerulonephritis presented higher concentrations of MBL compared with healthy controls. Abbreviations. MCNS, minimal change nephrotic syndrome (n = 16); IgAN, IgA nephropathy (n = 19); MPGN, membranoproliferative glomerulonephritis (n = 31); others (n = 14).
Table 1.
Serum levels of complement components in patients with IgA nephropathy [7].
| n | CH50 | C1q | C4 | C3 | C5 | B | P | MBL | |
|---|---|---|---|---|---|---|---|---|---|
| (U/mL) | (mg/dL) | (mg/dL) | (mg/dL) | (%) | (%) | (mg/mL) | (mg/mL) | ||
| IgA nephropathy | 50 | 44.0 ± 8.1* | 13.4 ± 2.8 | 28 ± 11* | 101 ± 26 | 122 ± 28 | 114 ± 32* | 32.6 ± 27.0* | 1.8 ± 1.8 |
| Healthy controls | 50 | 33.5 ± 5.4 | 12.6 ± 1.7 | 21 ± 5 | 106 ± 17 | 112 ± 17 | 95 ± 18 | 21.0 ± 24.0 | 2.1 ± 1.8 |
% expresses a percentage of pooled normal human serum, *: P < 0.01 IgA nephropathy versus healthy controls.
2.2. Initiators of Lectin Pathway Activation
From the 1980s, researchers have explored the essential molecules in the pathogenesis of IgAN. Tomino et al. deduced by an immunohistochemistry technique that the mesangial deposition of IgA1 is critical for glomerular damage, and a subsequent study showed that the polymeric form of IgA1 is predominantly eluted from renal tissues of IgAN patients [10, 11]. Conley et al. also confirmed a more predominantly glomerular deposition of IgA1 than that of IgA2 [12]. Recently, unique O-linked glycosylation sites at the hinge region of IgA1 have received much attention. From the analysis of the circulating immune complex, the galactose deficiency (under-O-glycosylated IgA) has been pointed out in the IgA1 molecules of patients with IgAN [13–15], although a high circulating overload alone does not induce the glomerular damage. As demonstrated in the current reviews, synthesis and binding of autoantibodies directed against galactose-deficient IgA1 (GalNAc residues on the hinge region) are required for the formation of pathogenic IgA1-containing immune complexes [2, 16]. Excesses of these relatively large complexes might lodge into the paramesangial area and might activate LP.
Since under-O-glycosylated IgA leads to polymerization of IgA [17] and purified polymeric IgA from patients with IgAN can activate LP [18], an important question is whether aberrant IgA can activate LP. Prominent ligands for MBL are mannose and N-acetyl-glucosamine (GlcNAc), and ligands for ficolins are GlcNAc. Because galactose and sialic acid are usually present in mammalian glycoproteins, they cannot be captured by MBL and ficolins [19]. Although such an under-O-glycosylated IgA1 from patients with IgAN could potentially interact with plant lectins [20], MBL and ficolins have no lectin activity for GalNAc residues. To the best of our knowledge, galactose-deficient IgA1 itself is able to activate C3 directly [9] but no direct clinical evidence from human studies is available which has demonstrated that MBL and ficolins can bind to aberrant IgA1 in human studies.
2.3. Other Candidate Ligands of MBL
Secretory IgA (SIgA) is the first line of defense in protecting the respiratory and intestinal tract, and the serum concentration of SIgA is very low (2-3 μg/mL). Since the N-glycans on the heavy chains of both SIgA1 and SIgA2 present terminal GlcNAc and mannose residues, they could be recognized by MBL and ficolins [21]. Oortwijn et al. clearly demonstrated glomerular staining of SIgA with MBL and C4d in the patients with IgAN [22]. Further extended study is needed to clarify the pathological role of SIgA and complement activation.
2.4. MBL Deficiency
Functional MBL deficiency is probably the most common immunodeficiency in humans, involving 10 to 20% of the population. It is characterized by low levels of circulating functional multimers due to a number of genetic polymorphisms within the coding (codon 54 of exon 1 is most common in humans and it determines serum concentration and carbohydrate recognition ability) and promoter regions of the MBL2 gene [23]. Our preliminary comparisons of the clinical backgrounds of MBL-sufficient patients of IgAN and MBL deficient patients of IgAN were determined by the analysis of polymorphisms of codon 54 (Table 2). The means of urinary protein and glomerular filtration rate (GFR) in MBL deficient subjects were better than in MBL-sufficient subjects, but lacked statistical significance. Pirulli et al. showed that polymorphism of codon 54 did not appear to have a primary involvement in the susceptibility and severity of IgAN in Italian patients [24]. They also reported a similar distribution of polymorphism frequency between healthy volunteers and IgAN patients. Nevertheless, the Pirulli's data and ours could have been influenced by the low number of IgAN patients and this might be due to a racial difference. Of great interest is whether the difference can be found between MBL deficiency and MBL sufficiency at the onset and progression of IgAN.
Table 2.
Clinical background compared with MBL sufficient and MBL deficient of IgA nephropathy.
| MBL sufficient (n = 55) | MBL deficient (n = 6) | |
|---|---|---|
| Gender (M : F) | 22 : 33 | 3 : 3 |
| Age (y) | 30.6 ± 8.7 | 26.5 ± 6.7 |
| Serum creatinine (mg/dL) | 0.83 ± 0.30 | 0.71 ± 0.14 |
| Estimated GFR (mL/min/1.73 m2) | 85.3 ± 30.9 | 98.4 ± 13.8 |
| Urinary protein (g/g·creatinine) | 1.45 ± 1.58 | 0.69 ± 0.97 |
| History of macrohematuria (%) | 27.3 | 33.3 |
| IgA (mg/dL) | 316.9 ± 112.9 | 265.2 ± 69.6 |
| C3 (mg/dL) | 97.8 ± 15.8 | 93.2 ± 2.6 |
| C4 (mg/dL) | 21.9 ± 6.1 | 19.8 ± 6.6 |
| CH50 (Unit/mL) | 40.1 ± 6.7 | 34.4 ± 3.4 |
3. In Kidney
3.1. Evidence of Glomerular Deposition of Mannose-Binding Lectin
Endo et al. first demonstrated that glomerular deposition of MBL/MASP-1 occurred in in the 25% of the cases of IgAN and that this was not observed in normal kidney and that the frequency was higher than that found in the other forms of glomerulonephritis [25]. In a subsequent paper, Lhotta et al. also confirmed that 27% of patients with IgAN had mesangial deposits of MBL [8]. Furthermore, the recent study by Roos et al. reported that the most MBL-positive patients presented mesangial deposits of L-ficolin and IgA1 but not IgA2 [9]. These reports suggested that MBL binds to immune complexes which contain aberrant IgA and/or IgG. Contrary to the above reports, Hisano et al. showed that patients with mesangial deposits of IgA1 and C3c showed no deposits of C4, MBL, and MASP-1. Moreover, patients with mesangial deposits of IgA1, IgA2, and C3c showed deposition of C4, MBL, and MASP-1. Furthermore, C1q deposition was not found in any patient. They concluded that AP activation occurs as a result of mesangial deposition of IgA1 and that LP activation is associated with mesangial deposition of IgA2 [26]. It is clear, however, that a determination of the initiator of LP activation in the mesangium has not yet been carried out to complete satisfaction.
In IgAN, the protease-damaged surface of glomerular resident cells or apoptotic cells can be recognized by MBL. In regard to patients of lupus nephritis, we recently proposed that positive glomerular staining for annexin V would be seen in the majority of patients who had confirmed glomerular deposits of MBL, L-ficolin, and properdin [27]. Further examinations will also be necessary to elucidate the initial activator of LP in situ.
3.2. Clinical Significance of Lectin Pathway Activation in Glomeruli
Endo et al. reported that the patients with glomerular deposits of MBL/MASP-1 deposition were young and that the duration of the disease prior to renal biopsy was short compared with that in patients without MBL/MASP-1 deposition [25]. In considering histological alterations, patients with glomerular deposits of MBL/MASP-1 have a more distinct activity index than chronicity index. Roos et al. compared IgAN patients who had negative glomerular staining for MBL with those who had positive staining for MBL [9]. They concluded that the activation of the LP was associated with more severe renal damage, as demonstrated by proteinuria, decreased renal function, and more severe histological findings, such as mesangial proliferation, crescent formation, glomerular sclerosis, and interstitial fibrosis. Accordingly, it is possible to envisage a scenario in which glomerular activation of LP might play an enhancer of progression of glomerular damage. Thus, we should perform immunofluorescent analysis of C4 and/or C4 breakdown products (C4c, C4d), and this should be a routine procedure in histological work and the presence of mesangial deposits of C4 may help us to identify patients with worse prognosis [28].
Contrary to the studies described above, Hisano et al. showed that there were no differences in clinical and pathological severity parameters for the patients with mesangial deposits of MBL [26]. In their paper, patients with mesangial deposits of MBL and IgA2 constituted more than half of the patients examined, which was a higher proportion than those described in the reports of Endo et al. and Roos et al. Because IgAN is chronic disease of long duration and shows a broad spectrum of clinical presentations, it is difficult to plan to compare their clinical course. At any rate, LP activation in the glomerulus can be identified in the context of histological examinations.
4. Infection and Clinical Course of IgA Nephropathy
Previous reports had indicated that glomerular deposits of MBL were present in the patients of postinfectious diseases and that causative pathogens were variable, such as Streptococcus [29, 30], Propionibacterium [31], Mucor [32], and hepatitis C virus [33]. In IgAN, several pathogenic viral and bacterial antigens have been proposed as being responsible for the formation of mesangial deposits of IgA and infection might trigger the onset and progression of the diseases [34, 35]. Up to now, there has benn only one report which has explored the association between MBL polymorphism and clinical course of IgAN. Gong et al. described the clinical significance of MBL polymorphism of codon 54 in patients with IgAN [36]. In this report, the patients carrying the variant allele (GAC) had episodes of upper respiratory or gastrointestinal infections prior to the onset, or exacerbation of IgAN which are absent in wild homozygotes (GGC/GGC). The association between infection and progression of glomerular damage via LP activation is a promising area of research for obtaining a better understanding of the pathogenesis of IgAN.
5. Conclusion
It appears that the activations of AP and LP are very much involved in the pathogenesis of IgAN. The traces of LP activation in the fluid phase and local glomerular tissues have been shown to accumulate in a subpopulation of IgAN. However, further studies will be essential to confirm the ligands for the MBL in the pathogenesis of IgAN.
References
- 1.Donadio JV, Grande JP. IgA nephropathy. New England Journal of Medicine. 2002;347(10):738–748. doi: 10.1056/NEJMra020109. [DOI] [PubMed] [Google Scholar]
- 2.Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. Journal of the American Society of Nephrology. 2011;22(10):1795–1803. doi: 10.1681/ASN.2011050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vangelista A, Frascà GM, Mondini S, Bonomini V. Idiopathic IgA mesangial nephropathy: immunohistological features. Contributions to Nephrology. 1984;40:167–173. doi: 10.1159/000409745. [DOI] [PubMed] [Google Scholar]
- 4.Haas M. Histologic subclassification of IgA nephropathy: a clinicopathologic study of 244 cases. American Journal of Kidney Diseases. 1997;29(6):829–842. doi: 10.1016/s0272-6386(97)90456-x. [DOI] [PubMed] [Google Scholar]
- 5.Wyatt RJ, Julian BA. Activation of complement in IgA nephropathy. American Journal of Kidney Diseases. 1988;12(5):437–442. doi: 10.1016/s0272-6386(88)80042-8. [DOI] [PubMed] [Google Scholar]
- 6.Dean MM, Minchinton RM, Heatley S, Eisen DP. Mannose binding lectin acute phase activity in patients with severe infection. Journal of Clinical Immunology. 2005;25(4):346–352. doi: 10.1007/s10875-005-4702-1. [DOI] [PubMed] [Google Scholar]
- 7.Onda K, Ohi H, Tamano M, et al. Hypercomplementemia in adult patients with IgA nephropathy. Journal of Clinical Laboratory Analysis. 2007;21(2):77–84. doi: 10.1002/jcla.20154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lhotta K, Würzner R, König P. Glomerular deposition of mannose-binding lectin in human glomerulonephritis. Nephrology Dialysis Transplantation. 1999;14(4):881–886. doi: 10.1093/ndt/14.4.881. [DOI] [PubMed] [Google Scholar]
- 9.Roos A, Rastaldi MP, Calvaresi N, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. Journal of the American Society of Nephrology. 2006;17(6):1724–1734. doi: 10.1681/ASN.2005090923. [DOI] [PubMed] [Google Scholar]
- 10.Tomino Y, Endoh M, Nomoto Y, Sakai H. Immunoglobulin A1 and IgA nephropathy. New England Journal of Medicine. 1981;305(19):1159–1160. doi: 10.1056/NEJM198111053051922. [DOI] [PubMed] [Google Scholar]
- 11.Tomino Y, Sakai H, Miura M. Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clinical and Experimental Immunology. 1982;49(2):419–425. [PMC free article] [PubMed] [Google Scholar]
- 12.Conley ME, Cooper MD, Michael AF. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. Journal of Clinical Investigation. 1980;66(6):1432–1436. doi: 10.1172/JCI109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hiki Y, Odani H, Takahashi M, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney International. 2001;59(3):1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- 14.Allen AC, Harper SJ, Feehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clinical and Experimental Immunology. 1995;100(3):470–474. doi: 10.1111/j.1365-2249.1995.tb03724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney International. 1997;52(2):509–516. doi: 10.1038/ki.1997.361. [DOI] [PubMed] [Google Scholar]
- 16.Glassock RJ. The pathogenesis of IgA nephropathy. Current Opinion in Nephrology and Hypertension. 2011;20(2):153–160. doi: 10.1097/MNH.0b013e3283436f5c. [DOI] [PubMed] [Google Scholar]
- 17.Kokubo T, Hiki Y, Iwase H, et al. Protective role of IgA1 glycans against IgA1 self- aggregation and adhesion to extracellular matrix proteins. Journal of the American Society of Nephrology. 1998;9(11):2048–2054. doi: 10.1681/ASN.V9112048. [DOI] [PubMed] [Google Scholar]
- 18.Roos A, Bouwman LH, Van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the Mannan-Binding lectin pathway. Journal of Immunology. 2001;167(5):2861–2868. doi: 10.4049/jimmunol.167.5.2861. [DOI] [PubMed] [Google Scholar]
- 19.Drickamer K. Engineering galactose-binding activity into a C-type mannose-binding protein. Nature. 1992;360(6400):183–186. doi: 10.1038/360183a0. [DOI] [PubMed] [Google Scholar]
- 20.Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN) Kidney International. 2004;65(5):1544–1547. doi: 10.1111/j.1523-1755.2004.05407.x. [DOI] [PubMed] [Google Scholar]
- 21.Royle L, Roos A, Harvey DJ, et al. Secretory IgA N- and O-glycans provide a link between the innate and adaptive immune systems. Journal of Biological Chemistry. 2003;278(22):20140–20153. doi: 10.1074/jbc.M301436200. [DOI] [PubMed] [Google Scholar]
- 22.Oortwijn BD, Rastaldi MP, Roos A, Mattinzoli D, Daha MR, Van Kooten C. Demonstration of secretory IgA in kidneys of patients with IgA nephropathy. Nephrology Dialysis Transplantation. 2007;22(11):3191–3195. doi: 10.1093/ndt/gfm346. [DOI] [PubMed] [Google Scholar]
- 23.Garred P, Larsen F, Seyfarth J, Fujita R, Madsen HO. Mannose-binding lectin and its genetic variants. Genes and Immunity. 2006;7(2):85–94. doi: 10.1038/sj.gene.6364283. [DOI] [PubMed] [Google Scholar]
- 24.Pirulli D, Boniotto M, Vatta L, et al. Polymorphisms in the promoter region and at codon 54 of the MBL2 gene are not associated with IgA nephropathy. Nephrology Dialysis Transplantation. 2001;16(4):759–764. doi: 10.1093/ndt/16.4.759. [DOI] [PubMed] [Google Scholar]
- 25.Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M, Fujita T. Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrology Dialysis Transplantation. 1998;13(8):1984–1990. doi: 10.1093/ndt/13.8.1984. [DOI] [PubMed] [Google Scholar]
- 26.Hisano S, Matsushita M, Fujita T, Endo Y, Takebayashi S. Mesangial IgA2 deposits and lectin pathway—mediated complement activation in IgA glomerulonephritis. American Journal of Kidney Diseases. 2001;38(5):1082–1088. doi: 10.1053/ajkd.2001.28611. [DOI] [PubMed] [Google Scholar]
- 27.Sato N, Ohsawa I, Nagamachi S, et al. Significance of glomerular activation of the alternative pathway and lectin pathway in lupus nephritis. Lupus. 2011;20(13):1378–1386. doi: 10.1177/0961203311415561. [DOI] [PubMed] [Google Scholar]
- 28.Espinosa M, Ortega R, Gómez-Carrasco JM, et al. Mesangial C4d deposition: a new prognostic factor in IgA nephropathy. Nephrology Dialysis Transplantation. 2009;24(3):886–891. doi: 10.1093/ndt/gfn563. [DOI] [PubMed] [Google Scholar]
- 29.Hisano S, Matsushita M, Fujita T, Takeshita M, Iwasaki H. Activation of the lectin complement pathway in post-streptococcal acute glomerulonephritis. Pathology International. 2007;57(6):351–357. doi: 10.1111/j.1440-1827.2007.02107.x. [DOI] [PubMed] [Google Scholar]
- 30.Ohsawa I, Ohi H, Endo M, Fujita T, Matsushita M, Fujita T. Evidence of lectin complement pathway activation in poststreptococcal glomerulonephritis. Kidney International. 1999;56(3):1158–1160. doi: 10.1046/j.1523-1755.1999.00642.x. [DOI] [PubMed] [Google Scholar]
- 31.Hagiwara S, Ohi H, Eishi Y, et al. A case of renal sarcoidosis with complement activation via the lectin pathway. American Journal of Kidney Diseases. 2005;45(3):580–587. doi: 10.1053/j.ajkd.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 32.Kusaba G, Ohsawa I, Ishii M, et al. Evidence of immunopathological traces in mucormycosis: an autopsy case. Clinical and Experimental Nephrology. 2010;14(4):396–400. doi: 10.1007/s10157-010-0289-9. [DOI] [PubMed] [Google Scholar]
- 33.Ohsawa I, Ohi H, Tamano M, et al. Cryoprecipitate of patients with cryoglobulinemic glomerulonephritis contains molecules of the lectin complement pathway. Clinical Immunology. 2001;101(1):59–66. doi: 10.1006/clim.2001.5098. [DOI] [PubMed] [Google Scholar]
- 34.Van Es LA. Pathogenesis of IgA nephropathy. Kidney International. 1992;41(6):1720–1729. doi: 10.1038/ki.1992.246. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki S, Nakatomi Y, Sato H, Tsukada H, Arakawa M. Haemophilus parainfluenzae antigen and antibody in renal biopsy samples and serum of patients with IgA neuropathy. Lancet. 1994;343(8888):12–16. doi: 10.1016/s0140-6736(94)90875-3. [DOI] [PubMed] [Google Scholar]
- 36.Gong R, Liu Z, Li L. Mannose-binding lectin gene polymorphism associated with the patterns of glomerular immune deposition in IgA nephropathy. Scandinavian Journal of Urology and Nephrology. 2001;35(3):228–232. doi: 10.1080/003655901750292015. [DOI] [PubMed] [Google Scholar]