

The search for new biofuels has generated increased interest in biochemical pathways that produce hydrocarbons.[1] Although hydrocarbons are simple molecules, the biosynthesis of molecules that lack any chemical functional groups is surprisingly challenging.[2] Biochemical reactions that remove functionality, such as decarboxylations, dehydrations and reduction of double bonds, invariably rely on the presence of adjacent functional groups to stabilize unfavorable transition states. Enzymes involved in hydrocarbon biosynthesis are therefore of interest both for applications in biofuels production and because of the unusual and chemically difficult reactions they catalyze.[3] One enzyme that has attracted particular interest, is aldehyde decarbonylase (AD), which catalyzes the decarbonylation of long-chain fatty aldehydes, to the corresponding alkanes.[4]
AD was originally identified in studies on the biosynthesis of hydrocarbon waxes by higher plants and algae.[5] These enzymes are integral membrane proteins of Mr ~ 70,000 Da that convert long-chain aldehydes, derived from fatty acids, to alkanes and carbon monoxide. The enzymes require divalent metal ions for activity;[4b, 6] however, the identity of the metal remains unclear. It was reported that the pea enzyme is active with Co, Cu or Zn[6] whereas the Botryococcus braunii enzyme uses Co.[4b] Sequence analysis of cer1, a gene encoding a putative AD from Arabidopsis thaliana, indicates that it is member of the sterol oxidase class of non-heme iron-dependent oxygenases.[7]
More recently, a soluble version of AD was discovered in cyanobacteria (cAD).[4c] Serendipitously, a crystal structure for cAD from Prochlorococcus marinus MIT9313, had previously been solved as part of a structural proteomics project, although no function had been assigned.[8] The structure revealed that cAD is member of the non-heme dinuclear iron oxygenase family of enzymes exemplified by methane monoxygenase, type I ribonucleotide reductase, and ferritin.[9] In all these enzymes, the di-iron centre is contained within an anti-parallel four-α-helix bundle in which two histidines and four carboxylates (either from aspartate or glutamate) supply the protein ligands to the metal ions. The dinuclear iron centre of cAD superimposes very closely on those of the other enzymes; however in the structure the carboxylate of an adventitiously bound long-chain fatty acid bridges the two iron atoms, displacing one of the glutamate ligands (Figure 1).
Figure 1.
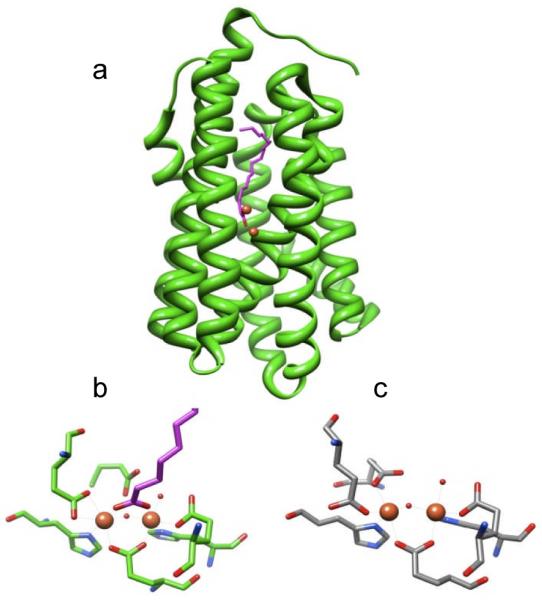
Structure of cAD from P. Marinus (PDB ID 2OC5A). a) ribbon diagram showing position of iron atoms and co-crystallized fatty acid. b) structure of the di-iron cluster showing protein ligands and co-crystallized fatty acid which displaces one Glu ligand. c) comparison with the di-iron cluster of methane monooxygenase from Methylococcus capsulatus (Bath) (PDB ID 1XVC)
The reactions catalyzed by this structural class of di-iron enzymes invariably involve molecular oxygen[9] and typically require an external reducing system in the form of ferredoxin, ferredoxin oxidoreductase and NADPH, to reduce the ‘resting’di-ferric form of the enzyme to the active di-ferrous form at the start of each turn-over. It was established that cAD also requires this external reducing system to catalyze the conversion of aldehydes to alkanes.[4c]
Most recently it was shown that, rather than producing CO as a co-product, cAD converts aldehydes to alkanes and formate.[10] Formally this appears to be a hydrolysis reaction, albeit a very unusual one. However, it was found that, like other members of di-iron oxygenase family, cAD utilizes molecular oxygen and that one atom of oxygen is incorporated into formate.[11] This led the authors to propose that cAD catalyzes a “cryptic” oxidation reaction (Scheme 1) in which an iron-peroxo species attacks the carbonyl group to facilitate the chemically difficult scission of the C1-C2 bond; finally, further reduction of oxygen was proposed to occur so that overall O2 is reduced to water.
Scheme 1.
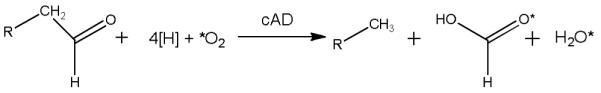
“Cryptic” oxidation of aldehydes to alkanes and formate proposed for the oxygen-dependent reaction of cAD
Here we report that cAD catalyzes the decarbonylation of aldehydes under anaerobic conditions to give the same products: i.e. an alkane and formate. Moreover, this reaction occurs at rates that are 100 – 1000 times faster than those reported for the oxygen-dependent reaction. We also present evidence that the aldehyde substrate interacts with metal site and demonstrate, for the first time, that iron is the active metal in cAD. Intriguingly, even under anaerobic conditions a reducing system is still required. Overall, our results point to a new and fundamentally different mode of reactivity for non-heme di-iron enzymes.
The experiments we report were conducted on cAD from Prochlorococcus. marinus MIT9313. The enzyme was over-expressed in E. coli from a synthetic gene, codon-optimized for expression in E. Coli, and purified by standard methods (see supporting information).
Given the very unusual nature of the reaction, and that several different metal ions have been reported as necessary for AD activity, we considered it essential to establish the metal requirements for cAD. In particular, it has been speculated that cAD may be Mn-requiring enzyme, analogous to the Mn/Fe-dependent ribonucleotide reductases.[4c, 10-12] The enzyme, as isolated, was pale brown and the u.v. – visible spectrum exhibited a shoulder at 350 nm characteristic of ferric iron (see Figure S3 in supporting information). Metal analysis by ICP-MS established that in a typical preparation only 30 - 35 % of the purified enzyme contained metal ions (assuming 2 metal ions per protein). The following metal composition represents that found in one enzyme preparation: Fe = 17.8 %; Zn = 9.6 %; Ni = 5.1 %; Mn = 3.0 %; Cu < 0.2 % and Co < 0.1 %.
Endogenous metals could be removed from cAD by first incubating the enzyme with 10 mM ferrozine and 5 mM sodium dithionite overnight,[13] followed by dialysis against buffers containing 10 mM EDTA and 10 mM NTA. This allowed the apo-enzyme to be reconstituted with biological relevant divalent metal ions, including Mn, Fe, Co, Ni, Cu and Zn, and their activity tested. Only Fe(II) supported alkane formation; the activity of other metals was indistinguishable from background activity.
We also investigated whether a hetero-metal cluster may support activity, or promote enhanced activity. Assays were conducted in which the molar ratio of Fe(II) to the second metal ion was varied between 0 and 100 % and cAD activity plotted as function of iron percentage (see figure S4 in supporting information). Mn, Co, Cu and Ni had no effect on activity whereas and Zn appeared to be slightly inhibitory. Thus cAD appears to function as a di-nuclear iron enzyme.
Having established that cAD is iron-dependent, we investigated the kinetics and cofactor requirements for the production of alkanes by the enzyme. Although the conversion of n-octadecanal to n-heptadecane by cAD was established in the initial identification of the enzyme,[4c] no kinetic information was reported. We confirmed that heptadecane formation was absolutely dependent upon the presence of ferredoxin, ferredoxin oxidoreductase and NADPH. However, based on assay conditions reported by Schirmer et al,[4c] i.e. 5μM cAD, 10 μM ferrous ammonium sulfate, 300 μM octadecanal; 30 μg/mL spinach ferredoxin, 0.04 U/mL ferredoxin reductase and 800 μM NADPH, the reaction proceeded extremely slowly and typically only ~ 1 turn-over could be achieved in 1 hour.
During the course of these initial investigations, we rapidly established that cAD is fully active under strictly anaerobic conditions (see Figure S5 in supporting information) and thus does not require oxygen for activity. Indeed, activity was ~ 40 % higher than in the presence of oxygen. Therefore, subsequent studies were conducted anaerobically. This observation is particularly surprising given that all other di-iron enzymes in this structural family catalyze reactions involving O2, and the recent observation that cAD incorporates oxygen into formate under aerobic conditions.[11] Clearly a “cryptic” oxidation mechanism cannot operate anaerobically and so the reaction must occur by a fundamentally different mechanism.
Because of the poor activity observed with the ferredoxin system, we investigated other biochemical reducing systems. We discovered that the commonly used 5-methylphenazinium methylsulfate (PMS):NADH reducing system[14] could effectively substitute for the ferredoxin system, and resulted in greatly improved activity. Replacing the ferredoxin system with 75 μM PMS and 750 μM NADH in the above assay allowed reaction rates to be determined from the initial time points, with turn-over numbers approaching 0.4 min−1 at 37 °C. The reaction rate increased linearly with increasing octadecanal concentration and showed no sign of saturation at concentrations up to 500μM (Figure 2). An apparent kcat/Km of ~ 1 min−1mM−1 may be calculated from these data, but as discussed below this probably significantly underestimates the true value of kcat/Km.
Figure 2.
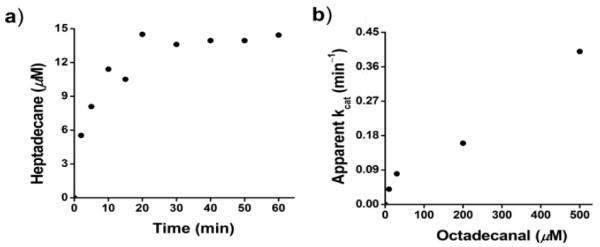
Kinetics of heptadecane formation by cAD. a) time course of heptadecane formation (octadecanal concentration = 200μM); b) rate of reaction as a function of substrate concentration.
Scant kinetic data has been reported for the oxygen-dependent reaction of cAD; Warui et al estimated that only ~ 3 turnovers had occurred in 20 h.[10] This probably underestimates the rate of the oxygen-dependent reaction, but based on this number cAD is 1000 to 100-fold more active in the anaerobic reaction with the PMS/NADH reducing system.
The slow rates of catalysis exhibited by cAD undoubtedly derive in part from the very poor solubility of the substrate. Addition of detergents such Triton did not improve activity noticeably. Assays had to be shaken vigorously to obtain reproducible results. Even at low concentrations the substrate appears to be present as micelles in solution; and it is very likely that reaction kinetics are dominated by phase transfer of substrate molecules from micelles. Furthermore, the ferredoxin system seemed to be inactivated by continuous shaking, which may explain the poor activity observed in using this reducing system.
We note that Li et al[11] reported that cAD was inactive in the absence of O2. The reason for the discrepancy with our results is unclear. However, they used cAD from a different cyanobacterium, Nostoc punctiforme, which has ~ 60 % sequence identity with the P. marinus enzyme. Sequence differences could conceivably alter substrate binding in the active site. Their experiments were also conducted under different assay conditions, with a reducing system that supports much lower activity to start with.
We established that the anaerobic reaction of cAD with octadecanal also produces formate. Formate was confirmed as the co-product by derivatizating the products of reaction with 2-nitrophenyl hydrazine (2-NPH)[15] and subsequent analysis by reverse phase HPLC and mass spectroscopy (Figure 3c). GC analysis of heptadecane from the same reaction confirmed that formate and heptadecane were produced stoichiometrically during the reaction, making it unlikely that formate results from a side-reaction. Attempts to detect CO formation by cAD, either by using myoglobin to bind CO[16] (see Figure S6 in supporting information) or by MS analysis of the reaction head space, were all negative.
Figure 3.

Identification of the products of the cAD reaction. a) GC-MS chromatograph demonstrating the conversion of octadecanal to heptadecane; b) mass spectra of heptadecane demonstrating that hydrogen derives from the solvent and not from the aldehyde proton; c) mass spectra of recovered 2-NPH-derivatized formate demonstrating that the aldehyde proton or deuteron is retained in formate.
When the reaction was performed in D2O, the product alkane contained deuterium, the labeling pattern being independent of whether the substrate was deuterated (Figure 3b). (Some additional deuterium derives from exchange of the α-protons of the aldehyde with solvent during the reaction; however control experiments established that this accounts for less than half of the deuterium incorporation.) Whereas when the reaction was performed with deuterated octadecanal the aldehyde proton was retained in formate, as determined by MS analysis of the 2-NPH derivative (Figure 3c).
Thus it appears that under anaerobic conditions the conversion of aldehydes to alkanes and formate catalyzed by cAD is a true hydrolysis reaction, as shown in Scheme 2.
Scheme 2.
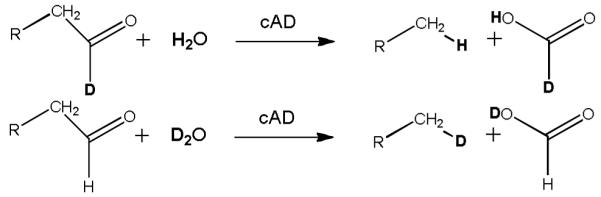
Oxygen-independent hydrolytic conversion of aldehydes to alkanes and formate catalyzed by cAD
Given that the cAD reaction does not require O2, and is redox neutral, we re-considered the role of the external reducing system in the reaction. For most iron-dependent oxygenases the reducing system functions to reduce Fe(III) to Fe(II), which then reacts with oxygen.[9e, 17] In aerobic conditions the reducing system may function to reduce the ferric form of cAD to the ferrous form. However, if this were the only function of the reducing system it should not be required under anaerobic conditions. To test this, we examined the activity of apo-cAD reconstituted with Fe(II) under rigorously anaerobic conditions in the absence of external reductant. Reactions containing 100 μM apo-cAD reconstituted with 200 μM Fe(II) were incubated with 200 μM octadecanal for periods of up to 24 h. However no heptadecane formation could be detected. (The limit of detection in the experiment was < 1 μM heptadecane). Thus an external reducing system appears to be absolutely required for the formation of alkanes. This strongly suggests that it performs a catalytic role in the reaction rather than simply being required to maintain the enzyme in the ferrous state.
By analogy with the reaction of di-iron enzymes with O2, we hypothesized that the first step in the cAD reaction may involve electron transfer from Fe(II) to the carbonyl group of the aldehyde. To test this, we investigated the interaction of the substrate with the di-iron centre by EPR spectroscopy (Figure 4). The apo-enzyme was reconstituted with Fe(II) under anaerobic conditions, (holo-enzyme concentration 425 μM) and spectra recorded at 77 K and at 6 K. Experiments were conducted in the absence of the reducing system so that turn-over could not occur. At 77 K the enzyme is EPR-silent, as expected for a di-ferrous iron center,[18] however at 6 K the presence of the di-ferrous center is confirmed by a broad, weak resonance centred at g = 13. Addition of heptanal, 600 μM, to holo-cAD resulted in the appearance of a signal at g = 4.3 characteristic of a high-spin ferric ion. (Heptanal, rather than octadecanal, was used in this experiment due to its higher solubility. Heptanal is a substrate for cAD, see supporting information)
Figure 4.
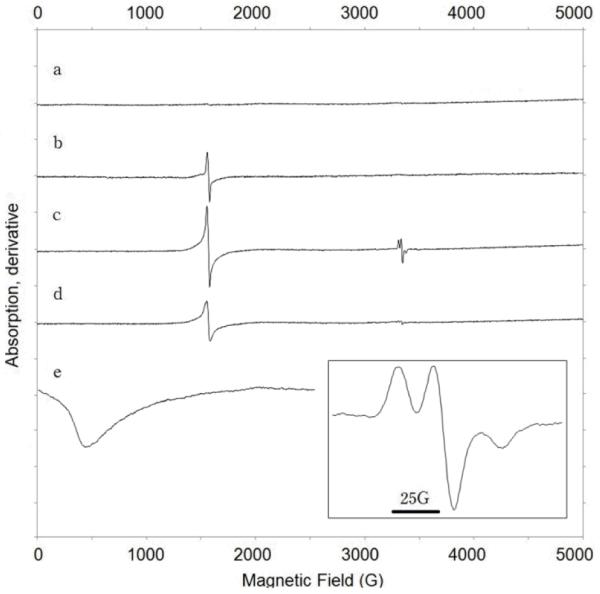
EPR spectra of cAD prepared under anaerobic conditions: a) apo-cAD reconstituted with Fe(II) to form di-ferrous enzyme; b) addition of heptanal to di-ferrous cAD; c) addition of heptanal and spin trapping reagent PBN to di-ferrous cAD. inset: expansion of g = 2 region of this spectrum showing PBN radical-adduct; d) addition of PBN only to di-ferrous cAD. e) spectrum of di-ferrous enzyme ion at 6 K (20 mW microwave power). All spectra except e) were recorded at 9.395 GHz; microwave power, 2 mW; modulation amplitude, 16 G; temperature, 77 K, and are plotted on the same scale.
This observation is consistent with Fe(II) transferring an electron to the substrate to generate Fe(III) and, presumably, a ketyl radical that is too unstable to observe directly. Further support for the generation of an organic radical came from experiments in which the spin-trapping agent N-tert-Butyl-α-phenylnitrone (phenyl-N-t-butylnitrone, PBN), which has been used to trap radical intermediates in other enzyme reactions[19], was included in the reaction. The ferric signal is now accompanied by a characteristic signal for the PBN nitroxide radical-adduct at g = 2. The hyperfine structure of the nitroxide radical is broadened, indicating that it results from PBN intercalated within the protein[19], rather than free in solution. This is consistent with PBN reacting with the substrate radical, or possibly a protein-based radical, as it is formed at the active site of the enzyme. Addition of PBN in the absence of heptanal (Figure 4, spectrum d) appears to result in some oxidation of Fe(II), but very little radical-adduct is observed.
Although many details of this remarkable reaction remain to be elucidated, we wish to propose a tentative mechanism that accounts for the experimental observations above (Scheme 3). Our results provide evidence that in the first step aldehyde binding to the di-ferrous iron centre initiates electron transfer from iron to the substrate. This would result in an iron-bound ketyl radical, 2, analogous to the superoxo species formed by other di-iron enzymes in their reaction with O2. At this point, injection of an additional electron from the reducing system would serve both to reduce the iron cluster, and to generate the more reactive ketyl radical anion, 3. We then propose that 3 undergoes homolytic cleavage of the C1-C2 bond to form a methylene radical and that concomitantly the formyl group becomes covalently bound to iron (4). Proton-coupled electron transfer (with water or a protein side-chain supplying the proton) would result in alkane formation (5) and a ferric-hydroxyl species that could serve to hydrolyze iron-formyl complex. Lastly, return of an electron to the reducing system and hydrolysis of the iron-formyl complex to give formate and regenerate the ferrous cluster, 6, must occur. The precise mechanism and timing of these latter steps must remains more speculative.
Scheme 3.
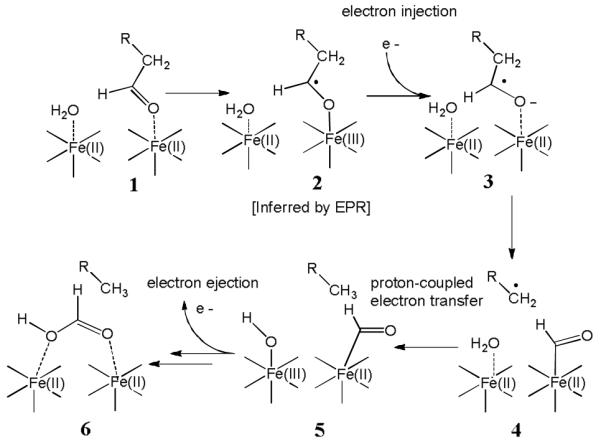
A mechanistic proposal for cAD that accounts for the reported experimental observations.
Although we are not aware of a direct mechanistic precedent for C-C scission by a ketyl radical, similar species have been shown to facilitate C-O cleavage in hydroxyglutaryl-CoA dehydratase.[20] The methylene radical, 4, would be highly unstable and short-lived, but similar species are established features of the mechanisms of adenosylcobalamin and radical-SAM enzymes.[21] This aspect of the mechanism could be probed with substrates that incorporate “radical clocks” such as cyclopropyl rings.[22] Iron-formyl complexes have been characterized in di-iron compounds that model the hydrogenase active site, although these are stabilized by CO ligands.[23] We note that transiently reducing the iron centre during the reaction would stabilize the formation of the proposed iron formyl complex (intermediates 4 and 5), and in thereby facilitate decarbonylation. It might also allow these intermediates to be trapped at low temperatures by rapid freeze-quench methods to permit their spectroscopic characterization.
In conclusion, it appears that cAD can catalyze the same reaction by two quite different mechanisms. The oxygen-independent decarbonylation of aldehydes catalyzed by cAD represents a radical departure from the “conventional” oxygen activation chemistry catalyzed by di-iron oxygenase enzymes. The enzyme appears to exploit the reduced di-iron center to facilitate novel radical chemistry that requires electron transfer from an external reductant. The hydrolysis of aldehydes in this manner presents a formidable challenge because breaking the C1-C2 bond either heterolytically, to produce a carbanion at C2, or homolytically to produce a radical would result in highly unstable intermediates. Indeed, we are unaware of a similar reaction in either the biological realm or in conventional chemistry. Further studies are planned to uncover the mechanism of this unique reaction.
The extremely low activity so far observed for the enzyme presents a major barrier to its use in biofuels applications. Our studies have identified conditions where the enzyme is much more active, although substrate solubility appears to be an important factor limiting catalytic rates. Moreover, an enzyme that is capable of working in anoxic environments may have practical advantages in technological applications.
Supplementary Material
Footnotes
This work was supported in part by grants from the American Chemical Society Petroleum Research Fund 48781 ND4, European Union FP-7 256808 and the National Institutes of Health GM 093088 to E.N.G.M.
Contributor Information
Debasis Das, Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, (U.S.A.).
Dr. Bekir E. Eser, Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, (U.S.A.)
Aaron Sciore, Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, (U.S.A.).
E. Neil G. Marsh, Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, (U.S.A.).
Jaehong Han, School of Biological Sciences, Chung-Ang University, Anseong 456-756, (Korea)
References
- [1].a) Connor MR, Atsumi S, Biomed J. Biotechnol. 2010 doi: 10.1155/2010/541698. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keasling JD. Science. 2010;330:1355. doi: 10.1126/science.1193990. [DOI] [PubMed] [Google Scholar]; c) Steen EJ, Kang YS, Bokinsky G, Hu ZH, Schirmer A, McClure A, del Cardayre SB, Keasling JD. Nature. 2010;463:559. doi: 10.1038/nature08721. [DOI] [PubMed] [Google Scholar]; d) Clarke ND. Curr. Opin. Struct. Biol. 2010;20:527. doi: 10.1016/j.sbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- [2].Buist PH. Nat. Prod. Rep. 2007;24:1110. doi: 10.1039/b508584p. [DOI] [PubMed] [Google Scholar]
- [3].Ragsdale SW. Chem. Rev. 2006;106:2217. doi: 10.1021/cr0503153. [DOI] [PubMed] [Google Scholar]
- [4].a) Cheesbrough TM, Kolattukudy PE. Proc. Natl. Acad. Sci. (USA) 1984;81:6613. doi: 10.1073/pnas.81.21.6613. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dennis M, Kolattukudy PE. Proc. Natl. Acad. Sci. (USA) 1992;89:5306. doi: 10.1073/pnas.89.12.5306. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB. Science. 2010;329:559. doi: 10.1126/science.1187936. [DOI] [PubMed] [Google Scholar]
- [5].Kunst L, Samuels AL. Prog. Lipid Res. 2003;42:51. doi: 10.1016/s0163-7827(02)00045-0. [DOI] [PubMed] [Google Scholar]
- [6].Schneider-Belhaddad F, Kolattukudy P. Arch. Biochem. Biophys. 2000;377:341. doi: 10.1006/abbi.2000.1798. [DOI] [PubMed] [Google Scholar]
- [7].Aarts MGM, Keijzer CJ, Stiekema WJ, Pereira A. Plant Cell. 1995;7:2115. doi: 10.1105/tpc.7.12.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Unpublished, structure solved by Joint Center of Structural Genomics (protein database entry PDB|2OC5|A).
- [9].a) Baik MH, Newcomb M, Friesner RA, Lippard SJ. Chem. Rev. 2003;103:2385. doi: 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]; b) Sjoberg BM. Metal Sites in Proteins and Models. 1997;88:139. [Google Scholar]; c) Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem. Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]; d) Theil EC, Goss DJ. Chem. Rev. 2009;109:4568. doi: 10.1021/cr900052g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wallar BJ, Lipscomb JD. Chem. Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]; f) Feig AL, Lippard SJ. Chem. Rev. 1994;94:759. [Google Scholar]; g) Lange SJ, Que L. Curr. Opin. Chem. Biol. 1998;2:159. doi: 10.1016/s1367-5931(98)80057-4. [DOI] [PubMed] [Google Scholar]
- [10].Warui DM, Li N, Norgaard H, Krebs C, Bollinger JM, Booker SJ. J. Am. Chem. Soc. 2011;133:3316. doi: 10.1021/ja111607x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li N, Norgaard H, Warui DM, Booker SJ, Krebs C, Bollinger JM. J. Am. Chem. Soc. 2011 doi: 10.1021/ja2013517. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Jiang W, Yun D, Saleh L, Barr EW, Xing G, Hoffart LM, Maslak MA, Krebs C, Bollinger JM. Science. 2007;316:1188. doi: 10.1126/science.1141179. [DOI] [PubMed] [Google Scholar]; b) Jiang W, Yun D, Saleh L, Bollinger JM, Krebs C. Biochemistry. 2008;47:13736. doi: 10.1021/bi8017625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stookey LL. Anal. Chem. 1970;42:779. [Google Scholar]
- [14].Halaka FG, Babcock GT, Dye JL. J. Biol. Chem. 1982;257:1458. [PubMed] [Google Scholar]
- [15].Peters R, Hellenbrand J, Mengerink Y, Van der Wal S. J Chromatogr A. 2004;1031:35. doi: 10.1016/j.chroma.2003.10.100. [DOI] [PubMed] [Google Scholar]
- [16].Wilks A, Ortiz de Montellano PR. J Biol Chem. 1992;267:8827. [PubMed] [Google Scholar]
- [17].Que L. Acc. Chem. Res. 2007;40:493. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- [18].Hendrick MP, Munck E, Fox BG, Lipscomb JD. J. Am. Chem. Soc. 1990;112:5861. [Google Scholar]
- [19].Kolberg M, Bleifuss G, Sjoberg BM, Graslund A, Lubitz W, Lendzian F, Lassmann G. Arch. Biochem. Biophys. 2002;397:57. doi: 10.1006/abbi.2001.2658. [DOI] [PubMed] [Google Scholar]
- [20].Kim J, Darley DJ, Buckel W, Pierik AJ. Nature. 2008;452:239. doi: 10.1038/nature06637. [DOI] [PubMed] [Google Scholar]
- [21].Marsh ENG, Patterson DP, Li L. ChemBioChem. 2010;11:604. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Newcomb M, Hollenberg PF, Coon MJ. Arch. Biochem. Biophys. 2003;409:72. doi: 10.1016/s0003-9861(02)00445-9. [DOI] [PubMed] [Google Scholar]
- [23].Borg SJ, Behrsing T, Best SP, Razavet M, Liu XM, Pickett CJ. J. Am. Chem. Soc. 2004;126:16988. doi: 10.1021/ja045281f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.