

Abstract
Mdm2 is the major negative regulator of p53 tumor suppressor activity. This oncoprotein is overexpressed in many human tumors that retain the wild type p53 allele. As such, targeted inhibition of Mdm2 is being considered as a therapeutic anticancer strategy. The N-terminal hydrophobic pocket of Mdm2 binds to p53 and thereby inhibits the transcription of p53 target genes. Additionally, the C-terminus of Mdm2 contains a RING domain with intrinsic ubiquitin E3 ligase activity. By recruiting E2 ubiquitin conjugating enzyme(s), Mdm2 acts as a molecular scaffold to facilitate p53 ubiquitination and proteasome-dependent degradation. Mdmx (Mdm4), an Mdm2 homolog, also has a RING domain and hetero-oligomerizes with Mdm2 to stimulate its E3 ligase activity. Recent studies have shown that C-terminal residues adjacent to the RING domain of both Mdm2 and Mdmx contribute to Mdm2 E3 ligase activity. However, the molecular mechanisms mediating this process remain unclear, and the biological consequences of inhibiting Mdm2/Mdmx co-operation or blocking Mdm2 ligase function are relatively unexplored. This study presents biochemical and cell biological data that further elucidate the mechanisms by which Mdm2 and Mdmx co-operate to regulate p53 level and activity. We use chemical and genetic approaches to demonstrate that functional inhibition of Mdm2 ubiquitin ligase activity is insufficient for p53 activation. This unexpected result suggests that concomitant treatment with Mdm2/Mdmx antagonists may be needed to achieve therapeutic benefit.
Keywords: Mdm2, ligase, p53, RING, ubiquitin, Nutlin
Introduction
The p53 protein is most often considered a stress-activated tumor suppressor but its functions likely evolved for other purposes (1). Recent studies indicate that p53 preserves genome integrity in the male germline, limits teratogen-induced developmental abnormalities, contributes to implantation and modulates metabolism (2). Its tumor suppressive role emerges when it is activated by genotoxic conditions or expression of oncogenes, whereupon it eliminates potentially tumorigenic cells via induction of cell cycle arrest, apoptosis, senescence or autophagy. Given the importance of p53 function in normal growth processes, as well as in response to tumor-initiating conditions, sophisticated mechanisms have evolved to limit p53 activity until it is absolutely required, and to then titrate its output in a stress and cell-appropriate fashion.
P53 activity is controlled by two broad mechanisms. First is a complex array of post-translational modifications that affect its ability to engage with relevant co-activators or negative regulators. This in turn determines p53 stability and abundance, and its ability to productively engage chromatin to regulate downstream target genes and micro-RNAs (3, 4). Second are mechanisms that control the stability and/or activity of its negative regulators, most significant of which are the related RING domain proteins, Mdm2 and Mdmx (also known as Mdm4) (5). Embryonic deletion of either protein engenders early lethality that is completely p53-dependent, underscoring their critical role during development. Conditional deletion of either Mdm2 or Mdmx indicates that while both restrict p53 activation in almost all tissues tested, Mdm2 loss often has a more profound effect (6). These data suggest a modulatory, role for Mdmx in many somatic tissues, with possibly more important roles in actively dividing tissues (7, 8).
Both Mdm2 and Mdmx contain a RING domain, variants of which are found in several hundred human proteins. In the case of Mdm2, the RING domain possesses E3 ubiquitin ligase activity: it recruits one or more E2 ubiquitin conjugating enzymes (hereafter E2, (9)) and facilitates transfer of ubiquitin from E2 to p53 and other substrates (10), leading to their proteasome-dependent degradation (11). As p53 is a transcription factor, Mdm2-dependent degradation is therefore an effective mechanism to modulate activation of p53 target genes. The binding of both Mdm2 and Mdmx to p53 also directly inhibits p53 transactivation function by preventing recruitment of transcriptional co-activators (8). Although Mdmx also has a RING domain, it does not possess intrinsic ubiquitin ligase activity (12). RING-mediated oligomerization is a conserved function of this domain, and Mdm2 is able to homo-oligomerize, as well as hetero-oligomerize with Mdmx. By analogy with other RING/RING heterodimeric complexes (such as Brca1/Bard1, where only Brca1 has ligase activity (13)), a model in which Mdmx enhances Mdm2-dependent p53 ubiquitylation has been proposed (12). However, the precise molecular mechanism(s) by which Mdmx modulates Mdm2 function remain to be defined.
Although Mdmx co-operates with Mdm2 to inhibit p53 function, Mdmx can also be targeted for Mdm2-dependent degradation (14). Following genotoxic stress, Mdmx downregulation contributes to p53 activation. Indeed, attenuating damage-induced Mdmx degradation in vivo reduces both basal and stress-induced p53 activities. This engenders both remarkable radioresistance, and dramatically increases sensitivity to Myc-induced lymphomagenesis (15). In addition to the Mdm2 and Mdmx RING domains, residues at the extreme C terminus of each protein are also important for regulation of Mdm2 ubiquitin ligase function (16, 17). Structural and functional analyses predict that C-terminal aromatic residues in both Mdm2 and Mdmx play a critical role in the context of Mdm2/Mdmx hetero-oligomers (16-19). Mdm2 point mutants in this region prevent p53 degradation, yet allow Mdmx degradation. Furthermore, Mdmx can restore Mdm2-directed ligase activity to these mutants, seemingly by providing the C-terminal residues in trans. These data suggest that the extreme C-terminus provides subtle structural elements that are critical for controlling p53 ubiquitylation; however, the mechanistic basis for these effects remains to be determined.
As both Mdm2 and Mdmx are potential therapeutic targets for cancer treatment (5), insight into their molecular interplay may inform new drug discovery and development strategies. Here, we investigate the effects of Mdm2 ligase inhibition on the control of p53 stability and activity. We show that the Mdmx extreme C-terminus comprises a key regulatory element affecting the degradation of endogenous p53 and Mdm2; it is also required for degradation of Mdmx in response to DNA damage. Using a genetic approach, we show that the inhibition of Mdm2 ligase function leads to stabilization of transcriptionally inactive p53. Furthermore, the stabilized p53 can be reactivated by attenuation of the interaction of p53 with either Mdm2 or Mdmx. These findings indicate that drugs designed to selectively inhibit Mdm2 ligase activity may, if used alone, not activate p53 sufficiently to elicit adequate anti-tumor effects. Rather, as they do engender significant increases in p53 abundance, they may achieve therapeutic benefits if used in combination with Mdm2 and/or Mdmx antagonists.
Results
Functional inhibition of Mdm2 stabilizes endogenous p53
By analogy with other heterodimeric E3 ligases, residues in the Mdm2 and Mdmx C- terminal tails may contribute to the correct structure for recruitment or processivity of the E2 conjugating enzyme(s) required for p53 degradation. While a previous study found that Mdm2 and Mdmx C-terminal point mutants (Mdm2Y489A and MdmxF488A, respectively) prevented Mdm2-dependent degradation of p53, the consequences for p53 activation were not explored (17). We therefore initiated a genetic approach to evaluate the functional consequences of Mdm2 ligase inhibition by generating U2OS cell lines expressing doxycycline (Dox)-inducible wild type (WT) and Mdm2Y489A and MdmxF488A. U2OS was chosen as the host cell since it retains a wild type p53 allele, and expresses a molecular excess of Mdm2 over Mdmx (20). This provides a situation in which the excess Mdm2 is a relevant physiological target for evaluating the effects of exogenously expressed Mdm2 or Mdmx mutants. A relatively high dose (100ng/ml) of doxycycline was used for comparisons between Mdm2 and Mdmx, since at lower doses we either failed to see robust increases in the levels of Dox-inducible Mdm2 or observed cell-to-cell heterogeneity in Mdm2 levels (data not shown). This is consistent with previous reports of differential expression of Mdm2 and Mdmx from the same promoter (21). Importantly, MdmxWT was downregulated by DNA damage at both low and high dose doxycycline (see Supplementary Figure 1C and D), indicating that the levels of induction achieved at the maximum Dox dose used for these studies is not saturating the capacity of the damage response system to induce Mdmx degradation.
Figure 1 shows the effects of Mdm2Y489A and MdmxF488A overexpression on levels of p53 and its downstream target, p21. As expected, induction of Mdm2WT led to a decrease in p53, and corresponding decrease in p21. In contrast, Mdm2Y489A expression led to an increase in p53, likely due to inhibition of endogenous Mdm2 E3 ligase activity (17). Note that despite the increase in p53 abundance, the levels of p21 were reduced by Mdm2Y489A expression (Figure 1 and Supplementary Figures 1E and F). Similarly, overexpression of MdmxF488A increased p53 steady state levels, yet p21 did not increase. The ability of MdmxF488A to stabilize p53, yet suppress p53-dependent transactivation was also observed at low dose (15ng/ml) doxycycline (Supplementary Figure 1A, B, E, F). At this dose of doxycycline, exogenous Mdmx levels were approximately four-fold those observed in MCF7 cells, a breast cancer cell line with Mdmx gene amplification (Danovi et al 2004). Together, these data suggest that while the extreme C-terminal aromatic residues of Mdm2 and Mdmx are required for p53 degradation, they are dispensable for suppression of p53 transcriptional activity (see below).
Figure 1. Expression of Mdm2Y489A or MdmxF488A leads to stabilization of p53.

Cell lines containing doxycycline-inducible Mdm proteins were treated with Doxycycline (100 ng/ml) for 24h and analyzed by western blot for the indicated proteins.
We next investigated the mechanism by which MdmxF488A causes stabilization of p53. Figure 2 shows that MdmxF488A expression increased the half-life of both endogenous Mdm2 and p53, and this was not accompanied by a change in Mdm2 mRNA levels (Figure 2A and 2B). This suggests that MdmxF488A causes post-transcriptional stabilization of Mdm2 by inhibiting Mdm2 ubiquitin ligase activity, and this leads to concomitant p53 stabilization. We tested the hypothesis that MdmxF488A inhibits ubiquitin ligase activity by determining the extent of p53 ubiquitylation in cell lines expressing equivalent levels of MdmxWT or MdmxF488A. Consistent with this proposal, the total levels of p53 are higher in cells expressing MdmxF488A compared to MdmxWT, and the amount of ubiquitylated p53 is significantly lower in cells expressing this mutant (Figure 2C). These data are consistent with a previous study in which p53 ubiquitylation was reduced by Mdm2Y489A (17).
Figure 2. Stabilization of p53 upon MdmxF488A expression is post-translational.

(A) Following induction of MdmxWT or MdmxF488A, cycloheximide was added for the indicated times prior to blotting for p53 and Mdm2. Band intensities were calculated using the LiCor/Odyssey image analysis system, and plotted as log2 values. (B) Mdm2 mRNA was analyzed by qPCR following addition of Dox, or the damaging agent NCS (300 ng/ml) as a positive control. (C) Cells were transfected with Histidine-tagged ubiquitin and doxycycline was added to induce MdmxWT or MdmxF488A. After 24h, cells were lyzed and ubiquitylated proteins were pulled down using Ni2+-agarose beads. Following SDS-PAGE, p53 was detected using DO-1. Numbers above each lane represent the ratio of ubiquitylated p53 species to the total amount of p53 in the lysate.
MdmxF488A binds Mdm2 and p53, but reduces p53 ubiquitylation
Like some other RING E3 ubiquitin ligase complexes, such as Brca1/Bard1, Mdm2 and Mdmx can hetero-oligomerize. As only Mdm2 possesses E3 ubiquitin ligase activity, a reasonable explanation for the above results is that MdmxF488A hetero-oligomerizes with Mdm2 and reduces its ability to degrade p53. However, MdmxF488A could also stabilize p53 by more indirect mechanisms. For example, MdmxF488A might antagonize p53-dependent transactivation of Mdm2, leading to a reduction in Mdm2 levels. This is unlikely, since Mdm2 mRNA levels are similar in the presence and absence of MdmxF488A (Figure 2B).
The inability of MdmxF488A to activate p53 while increasing p53 levels was surprising, and could be explained by its ability to suppress p53-dependent transactivation concomitant with binding to, and inactivation of, Mdm2 ligase function. To begin to explore this possibility, we first determined whether MdmxF488A could bind both Mdm2 and p53 using the proximity ligation in situ assay (PLISA, see Supplementary Figure 2 for a description of the method, and (22)).
PLISA confirmed that MdmxF488A binds endogenous Mdm2 and p53 in intact cells (Figures 3A and B). Induction of MdmxWT led to a small but detectable increase in the interaction between Mdmx and Mdm2, as shown by an increase in the number of PLISA foci (Figure 3A, panel ii). The PLISA signal for Mdmx/Mdm2 interaction was much more robust after MdmxF488A induction (Figure 3A, panel vi). This is in part due to the elevated Mdm2 levels in MdmxF488A expressing cells (see Supplementary Figure 3A and western blots in Figures 4 and 6). Interaction of Mdm2 with MdmxWT was detected in the nucleus, but appeared slightly enriched in the cytoplasm (Figure 3A, panel ii). This did not appear to be due to a preferential enrichment of either protein in the cytoplasm (Supplementary Figure 3C). In the case of MdmxF488A, the intensity of PLISA foci was greatest in the nuclei, although the Mdm2/Mdmx interaction was also detected in the cytoplasm (data not shown). These data confirm that the MdmxF488A mutation does not significantly interfere with Mdm2 binding, and that Mdm2 and MdmxF488A interact in both nuclear and cytoplasmic compartments in situ.
Figure 3. Proximity ligation assay (PLISA) reveals MdmxF488A binds to p53 and Mdm2.
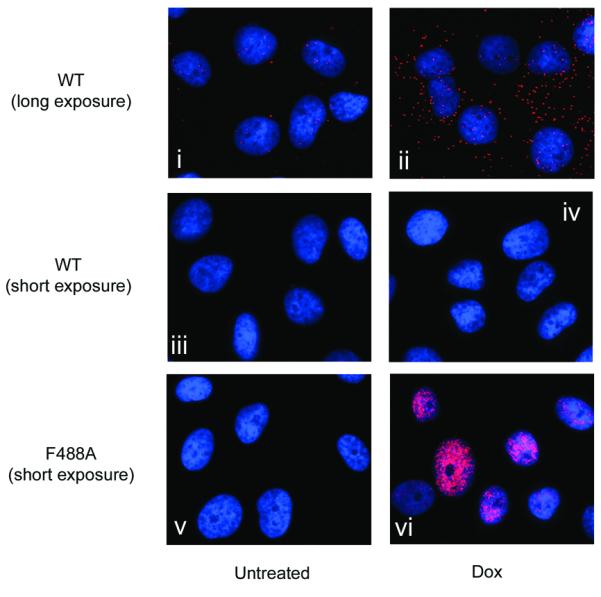
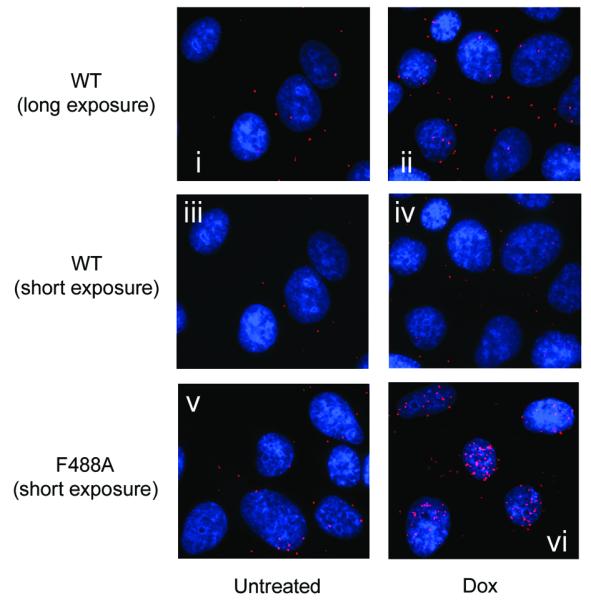
(A) Both MdmxWT and MdmxF488A interact with endogenous Mdm2. Cells were treated for 24h with doxycycline and then incubated with primary antibodies against Mdm2 and Mdmx. PLISA was then performed, and visualized under a fluorescence microscope. For MdmxWT, a longer exposure time was required to detect Mdmx/Mdm2 PLISA signals (panels i and ii). Panels iii and iv, (MdmxWT) and v and vi (MdmxF488A) were taken with the same exposure time (B) MdmxWT and MdmxF488A Mdmx interact with p53. Cells were treated as in (A) and then processed for PLISA after incubation with primary antibodies to detect Mdmx and p53. Exposures were as in (A).
Figure 4. MdmxF488A expression inhibits p53 ubiquitylation, but suppresses p53-dependent transactivation.

(A and B) Cells expressing Mdm2WT and Mdm2Y489A (A) or MdmxWT or MdmxF488A (B) were treated with 100 ng/ml doxycycline for 24h prior to addition of NCS (300 ng/ml, 5h) and analyzed by western blot for the indicated proteins and qPCR for p21 mRNA expression.
Figure 6. MdmxF488A antagonizes p53-dependent transactivation and promotes cell survival.

(A) Following induction of MdmxWT or MdmxF488A for 19h, cells were treated with 10 μM Nutlin for 24h and analyzed for the indicated proteins. (B) Cells treated as in (A) were analyzed for Mdm2 mRNA by qPCR. Results are from 3 replicate experiments. (C) MdmxWT or MdmxF488A cells were seeded into 6cm plates and colonies were allowed to form. Cells were then treated as in Figures 5A and B. (D) MdmxF488A and MdmxG57A/F488A cells were treated and processed as in (A). (E) MdmxF488A and MdmxG57A/F488A cells were treated and processed as in Figure 5B.
PLISA analyses revealed that MdmxWT and MdmxF488A also interact with p53 (Figure 3B). Again, the signal in the cells expressing MdmxF488A cell line was higher than in those expressing MdmxWT, in part because induction of the F488A mutant leads to increased p53 levels. Standard immunofluorescence showed that both MdmxWT and MdmxF488A were distributed throughout the nucleus and cytoplasm, but p53 was predominantly nuclear following induction of MdmxF488A (Supplementary Figures 3B and C). This likely contributes to the preferential localization of p53/Mdmx PLISA signal in the nucleus. Cytoplasmic PLISA signals were also observed at a higher intensity in MdmxF488A cells compared to MdmxWT, presumably due to the increased total p53 levels (data not shown). The PLISA signal faithfully reflects p53-Mdmx interaction since many fewer PLISA signals were observed in Dox-inducible cells expressing equivalent levels of the p53 binding-deficient Mdmx mutant, G57A (Supplementary Figure 3D). Conventional immunoprecipitation also confirmed that MdmxF488A is found in complex with Mdm2 and with p53 (Supplementary Figures 4A and B). Finally, we found that mutation of the Mdmx p53 binding site does not affect the ability of MdmxF488A to stabilize p53 (Supplementary Figure 4C and Figure 5C). Together, these data show that MdmxF488A binds both p53 and Mdm2, and suggest that the dominant negative effect of MdmxF488A is due to direct perturbation of Mdm2 ligase function, rather than sequestration of p53.
Both Mdm2Y489A and MdmxF488A suppresses p53 transactivation function
Since an increase in p53 level is often associated with increased p53 activity, we next determined whether the p53 stabilized in cells expressing Mdm2Y489A or MdmxF488A was transcriptionally active. As expected, induction of Mdm2WT reduced p53 level and transcriptional activity (Figure 4A, lane 2 and Figure 4C). By contrast, induction of Mdm2Y489A stabilized p53, yet was still able to suppress p53 activity (Figure 4A, lane 6 and Figure 4C). In fact, although Mdm2Y489A expression and treatment with the DNA damaging agent neocarzinostatin (NCS) led to similar increases in p53 level, p53 transcriptional activity was only increased by damage (compare lanes 6 and 7). These data demonstrate that ligase-deficient Mdm2 retains the capacity to suppress p53-dependent transactivation, presumably by binding to and occluding the p53 transactivation domain. Similar results were obtained by comparing the effects of MdmxWT and MdmxF488A (Figure 4B). Specifically, while MdmxF488A expression led to an increase in p53 level, there was no corresponding increase in p53 transcriptional activity (Figure 4B, compare lanes 5 and 6 and Figure 4D). Again, although p53 levels following MdmxF488A expression or DNA damage were similar, p53 was only active in the latter case (Figure 4B, compare lanes 6 and 7 and Figure 4D). By normalizing the amount of p21 mRNA to p53 protein, we found that both Mdm2Y489A and MdmxF488A reduce p53 specific activity (Supplementary Figure 5A and B).
Following DNA damage, we observed that MdmxWT but not MdmxF488A was downregulated (Figure 4B, compare lanes 6 and 8). This is consistent with a dominant negative function of MdmxF488A, since downregulation of Mdmx after DNA damage is an Mdm2-dependent process (14, 23). Similarly, Mdm2Y489A appeared significantly more stable than Mdm2WT following NCS treatment (Figure 4A, compare lanes 2 and 4, 6 and 8). Intriguingly, neither Mdm2Y489A nor MdmxF488A expression was sufficient to block damage-induced p53 activation (compare lanes 3 and 4, 7 and 8 in Figures 4C and D). Taken together, these data suggest that DNA damage signaling can counteract the suppressive effects of Mdm2 ligase inhibition on p53 activation. Additionally, the finding that DNA damage leads to downregulation of MdmxWT (Figure 4B) and destabilization of Mdm2WT (Figure 4A and Supplementary Figure 6) indicates that, although these proteins are overexpressed, they are still subject to regulation by endogenous damage signaling pathways.
Antagonizing the interaction between p53 and Mdm2Y489A or MdmxF488A derepresses p53 activity and elicits a cytotoxic response
Inhibition of Mdm2 ligase activity is suggested as a potential anticancer strategy in tumors with wild type p53 (24, 25). However, whether the ensuing p53 stabilization would be sufficient to elicit a cytotoxic response remains unclear. This is since inhibition of Mdm2 ligase activity would also stabilize Mdm2 itself, which could then interact with and antagonize p53. In this case, combinations of Mdm2 ligase inhibitors and Mdm2 antagonists, such as Nutlin-3a (hereafter Nutlin) may be required. We utilized the Mdm2Y489A and MdmxF488A inducible cell lines to investigate the potential for such an approach.
Although Mdm2Y489A stabilizes p53, expression of the protein did not affect the growth of U2OS cells with wild type p53 (Figure 5A and B). This is consistent with the ability of Mdm2Y489A to block p53-dependent transactivation. By contrast, Nutlin suppressed colony outgrowth in the presence of both Mdm2WT and Mdm2Y489A, even though the increase in p53 abundance was similar (data not shown). Thus, while inhibition of Mdm2 ligase activity stabilizes p53, concurrent treatment with an Mdm2 antagonist is required for a maximal cytotoxic response.
Figure 5. Mdm2Y489A antagonizes p53-dependent transactivation and promotes cell survival.

(A) Mdm2WT or Mdm2Y489A cells were seeded into 6cm plates and colonies were allowed to form. Cells were then treated with 100 ng/ml Doxycycline for 16h prior to addition of Nutlin for a further 48h. Drug was then replaced with regular medium. Following a further 6 days, cells were fixed and stained with crystal violet. (B) Following extraction with Sorenson’s buffer (50mM sodium citrate, 50mM citric acid in 50% (v/v) ethanol solution), total crystal violet stain was quantified by measuring absorbance at 590nm.
A prediction from these results is that cells in which Mdm2 ligase activity is blocked by the MdmxF488A mutant would not be sensitive to Nutlin, as numerous reports show that this drug interacts poorly with the Mdmx hydrophobic p53 binding pocket (26, 27). To address this, we repeated the colony outgrowth experiments with MdmxWT and MdmxF488A. Consistent with previous studies, induction of MdmxWT reduced basal levels of Mdm2 protein and mRNA by 50% (Figure 6A and B), whereas treatment with Nutlin alone lead to the expected increase in Mdm2 mRNA and protein (Figure 6A, lane 3). Induction of MdmxF488A or treatment with Nutlin both lead to similar increases in p53 and Mdm2 protein levels (Figure 6A, lanes 6 and 7), yet p53-dependent transactivation increased only with the Mdm2 antagonist (Figure 6B, bars 6 and 7 and Supplementary Figure 5D). This underscores the notion that MdmxF488A retains the ability to antagonize p53-dependent transcription while concomitantly stabilizing both Mdm2 and p53 proteins.
Since Nutlin is a less potent antagonist of Mdmx, Mdmx overexpression should attenuate Nutlin-induced p53 activation. Following Nutlin treatment, MdmxWT marginally inhibited p53 activation, while MdmxF488A expression had a more robust effect (compare Figure 6B lanes 3 and 4, and lanes 7 and 8). Consistent with this observation, only MdmxF488A appeared to attenuate Nutlin-induced p53 specific activity (Supplementary Figure 5C).
The above results show that while MdmxF488A blocks Mdm2 E3 ligase activity and stabilizes p53, its ability to inhibit p53-dependent transactivation remains intact. A prediction from the preceding results is that while p53 levels are elevated, cells expressing MdmxF488A should continue to proliferate due to inhibition of p53 transactivation. Consistent with this, expression of MdmxF488A alone did not inhibit growth, despite the high level of p53 under these conditions (Figure 6C). These findings suggest that inhibiting p53 degradation can be tolerated, providing the Mdmx/p53 interaction is preserved. Conversely, disrupting Mdmx/p53 binding under these conditions should lead to increased p53-dependent toxicity. In order to test this, we introduced a second mutation into MdmxF488A. This mutation (G57A) reduces the interaction of Mdmx with p53 ((28) and Supplementary Figures 3 and 7). Figure 6D shows that although MdmxF488A and MdmxG57A/F488A each lead to p53 accumulation, only MdmxG57A/F488A causes an increase in p53 activity (measured as an increase in the level of p21 protein). Importantly, the elevated p53 activity in MdmxG57A/F488A was not due to higher total p53 levels, or a low Mdmx:p53 ratio (Supplementary Figure 7). Interestingly, although MdmxG57A/F488A and Nutlin stabilized p53 to similar levels, induction of p21 was much higher after Nutlin treatment. Additionally, although prolonged expression of MdmxG57A/F488A Mdmx was associated with a marked decrease in cell viability compared with MdmxF488A, cell survival was significantly higher than following Nutlin treatment (Figure 6E, compare lanes 2 and 6). In theory, this could be due either to the increased Mdm2 levels resulting from MdmxG57A/F488A, inhibition of Mdm2 E3 ligase activity, or because MdmxG57A/F488A has some residual binding to p53 (Supplementary Figure 7). We used Nutlin, an Mdm2-selective antagonist (27), as a chemical probe to ascertain whether a putative Mdm2/MdmxG57A/F488A heterodimer might bind to and inhibit p53 function. Indeed, we found that the viability of cells expressing MdmxG57A/F488A was further decreased by Nutlin treatment (Figure 6E, compare lanes 6 and 8), indicating that the elevated level of stabilized Mdm2 is a significant determinant of p53 activity following induction of MdmxG57A/F488A. Together, these data suggest inhibition of Mdm2 ligase activity may be most effective when combined with either Mdm2 or Mdmx antagonists.
Discussion
The functional co-operation between Mdm2 and Mdmx may be a critical component for regulating p53 levels in normal cells. Therefore, understanding the mechanistic basis for this may aid rational design of Mdm2/Mdmx targeted therapeutics. Mdm2/Mdmx binding proceeds via RING/RING domain interaction, which is utilized by a large number of other RING domain-containing human proteins. Thus, findings from studies of Mdm2 and Mdmx may provide greater insight into molecular mechanisms in other RING E3 ligase-associated pathways. In this manuscript, we provide further insight into the regulation of p53 by Mdm2 and Mdmx, and show that the co-operation between the two proteins is critical for p53 abundance control. Additionally, our data suggest that correct Mdm2/Mdmx hetero-oligomerization is critical for degradation of Mdmx following DNA damage. These findings indicate that regulating Mdm2/Mdmx hetero-oligomerization is likely to be a key determinant of p53-dependent responses to genotoxic stress.
Previous studies indicated that MdmxF488A inhibits Mdm2-dependent degradation of p53 (17). We confirmed and extended these observations by showing that expression of MdmxF488A leads to stabilization of non-ubiquitylated, endogenous p53. RING E3 ubiquitin ligases (including Mdm2) bind to E2s, and facilitate the transfer of ubiquitin to their target substrates (in this case, p53). Since we show the interaction between Mdm2 and p53 is not perturbed in the presence of MdmxF488A, the most likely explanation is that transfer of ubiquitin to p53 is being inhibited. As structural studies suggest the MdmxF488A mutation perturbs the function of the Mdm2/Mdmx heterodimer (19), we infer this mutation may prevent the recruitment of E2 to the complex. Alternatively, E2 may be recruited, but unable to transfer its ubiquitin cargo to p53. This would be consistent with proposals that RING E3 ligases can allosterically activate E2 to facilitate ubiquitin transfer (29, 30). Since MdmxF488A can be detected in complexes containing both p53 and Mdm2, we suggest that MdmxF488A induction is effectively a “substrate trap”, and may facilitate detection of intermediates in the degradation of p53. We also show that optimal Mdmx degradation after DNA damage requires an intact Mdmx extreme C-terminus. As damage-induced phosphorylation of MdmxF488A and its binding to Mdm2 are unperturbed, the failure to degrade Mdmx is likely due to defects in Mdm2-dependent ubiquitylation. Recent studies have shown that following DNA damage, degradation of Mdmx is associated with an increase in p53 transactivation function (15, 31). In the present study, p53 activation was equivalent in cells expressing both MdmxWT and MdmxF488A, even though the latter protein was not degraded after damage. It is likely that additional damage-induced modifications to p53 itself, and possibly to the RING domains of both Mdm2 and Mdmx are responsible for this effect (32, 33). Together with the finding that phosphorylation of the Mdm2 and Mdmx N-termini also contribute to p53 activation (34, 35), these data highlight the redundancy built into damage signaling pathways to ensure a ‘failsafe’ p53 response to genotoxic stress.
Earlier studies provided compelling evidence for the functional co-operation between Mdm2 and Mdmx (16-19, 36, 37). Modeling this molecular interaction in vivo is beginning to provide further insight into its biological importance. For example, a point mutation of the Mdm2 RING domain that destroys RING structure (C462A) and blocks Mdm2 ligase activity triggers p53 stabilization and leads to p53-dependent embryonic lethality (38). Additionally, mutation or deletion of the Mdmx RING also prevents Mdm2/Mdmx interaction and triggers p53-dependent lethality in vivo (39, 40). In these systems, Mdm2 remains unstable, likely explaining the inability to control p53 activity under these conditions. Therefore, it is still unclear whether complete inhibition of Mdm2 ligase activity, in the context of structurally intact Mdm2/Mdmx RING domains, would be sufficient to activate p53. Our in vitro data suggest that a high level of structurally intact but ligase-deficient Mdm2 can still block p53 activity. Whether this is recapitulated at physiological levels in vivo remains an open question. An in vivo study of MdmxF488A and Mdm2Y489A (17) would also provides a direct test of whether ligase-independent RING domain activities of Mdm2 contribute to its biological function.
Our results also have implications for the design of drugs that block Mdm2 ligase activity by inhibiting the function of the Mdm2/Mdmx hetero-oligomer. We show here that perturbing the function of the hetero-oligomer increases p53 levels, suggesting ligase inhibition would be a viable anticancer strategy. However, in our system, p53 stabilization was not sufficient to trigger its activation, since it was antagonized by stabilized Mdm2 or high levels of exogenous Mdmx. Thus, we infer that increasing p53 level by blocking Mdm2 ligase activity may not be sufficient to trigger a robust p53 response. Previous studies lead to the discovery of the HLI series of compounds, which block Mdm2 ligase activity and lead to p53 stabilization (24, 41). In contrast to our results, treatment with HLI compounds also activated p53-dependent transcription. One explanation for this discrepancy is that the levels of exogenous Mdm2 or Mdmx used in our system exceed those present following treatment of tumor cells with HLI compounds. However, our results with MdmxG57A/F488A show the increase of endogenous Mdm2 following Mdm2 ligase inhibition can also block p53-dependent transactivation. We suggest, therefore that there are cases in which Mdm2 ligase inhibition alone may not be sufficient to trigger p53 activation. In such instances, either treatment with antagonists such as Nutlin as single agents, or in combination with ligase inhibitors may be more effective.
Materials and Methods
Cell culture and drug treatments
MCF7 cells were cultured in DMEM/10% FBS with Ciprofloxacin (CIP, 10 μg/ml). U2OS cells with doxycycline-inducible human Mdmx expression cassettes were generated as previously described (42) and cultured in DMEM/10% FBS/CIP/G418 (400 μg/ml). Doxycycline (SIGMA, St Louis, MO) was dissolved in water as stocks of 500 μg/ml and frozen at −20°C. Neocarzinostatin (stock 1 mg/ml in sodium acetate) was obtained from Robert Schultz at NCI, the proteasome inhibitor MG132 was from Calbiochem and Nutlin-3a was a kind gift of Lyubomir Vassilev (Hoffman-La Roche, Nutley, NJ). Unless otherwise stated, Nutlin-3a was used at a final concentration of 10 μM. Cycloheximide (US Biologicals) was dissolved in 0.1% ethanol and used at a final concentration of 100 μg/ml.
Western blotting, immunoprecipitation and antibodies
Cells were lyzed in Oren Buffer (50 mM Tris, pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40, 1 mM PMSF, 1 mM sodium vanadate, 10 mM NaF, and Complete Mini Protease Inhibitors (Roche)), at 4°C for 30 min. Following SDS-PAGE, proteins were transferred to PVDF membrane (Millipore, MA). The following antibodies were used for western blotting: p53, DO1 (Calbiochem, mouse, 1:1000) or FL-393 (Santa Cruz, rabbit, 1: 1500); Mdmx, BL1258 (Bethyl Laboratories, 1:10000, overnight); Mdm2, triple mouse monoclonal cocktail of IF-2 (Calbiochem), SMP-14 (Santa Cruz) and 4B2 (Calbiochem) (1:500 each, overnight); p21, C-19 (Santa Cruz, rabbit 1:1500 or Cip/Waf1 (Transduction Labs, mouse IgG2a, 1:500); actin, (rabbit, 1:20000, SIGMA, MO). Peroxidase-conjugated secondary antibody was used at 1:10000 (Jackson Immunochemicals). For LiCor far-red western analysis, lysis was as above, but protein was transferred to Immobilon-Fluor (Millipore, MA) prior to blocking with BioRad PBS/Casein Blocker per the manufacturer’s instructions. Species-specific secondary antibodies for LiCor analysis were conjugate to Alexa Fluor 680 (Molecular Probes) or IRD-800 (Rockland, PA). Analysis was performed using Odyssey v3.0 software. For immunoprecipitation analyses, cells were harvested on the plate in Oren Buffer. Typically, 500μg-1mg of protein was used for immunoprecipitation. The antibodies used were: DO1 or agarose bead-conjugated FL393 (p53), anti-HA (HA.11, Covance), BL-1258 (Human Mdmx, Bethyl Labs). 2μg of each antibody per milligram of protein was used. Immobilized recombinant protein A was used for antibody pulldown, and immunoprecipitates were washed and eluted as described (43).
Proximity ligation in situ assay
U2OS cells expressing a doxycycline-inducible HA-HDMX construct (20) were seeded onto coverslips and treated with doxycyline for 24 h. Cells were fixed in 3.7% para-formaldehyde, washed in PBS, and permeabilized in 0.2% Triton X-100 for 5 min. Coverslips were then blocked in 10% normal goat serum in PBS (NGS) for 2 h. For Mdmx/p53 PLISA, primary antibodies HA.11 (Covance, 1:500) and FL-393 (Santa Cruz, 1:1000) were diluted in PBS/EDTA/0.2% Triton X-100/2% NGS and incubated at 4°C overnight. For Mdmx/Mdm2 PLISA, primary antibodies BL-1258 (1:1000) and 5B10 (1:500) were used. Note that some background staining in the absence of doxycycline is observed using BL-1258, but the inducible nature of the system permitted clear distinction of Mdmx/Mdm2 complex formation. Following washes with TBS/0.05% Tween-20, a proximity ligation in situ assay (PLISA) was performed according to the manufacturer’s protocol (Detection Kit 613, OLink Bioscience, Sweden) with the following exception: goat anti-rabbit (minus) and anti-mouse (plus) PLISA probes were diluted in NGS at 1:10 instead of 1:5. Coverslips were mounted on microscope slides and images acquired using OpenLab software (Perkin-Elmer/Improvision, UK) and a Zeiss Axioplan 2 microscope with 63x magnification. Post-image capture, nuclear (Hoechst) images were contrast-enhanced and all PLISA images (red foci) from untreated and Dox-treated cells were contrast-enhanced simultaneously to reduce background (non-focal) signals. RGB planes of nuclei and PLISA foci were then overlaid.
Ubiquitylation assay
U2OS were transfected with His-tagged ubiquitin using FuGene HD (Roche) at a 2:5 ratio (DNA to FuGeneHD). After 6h, transfection reagent was replaced with regular medium in the presence or absence of doxycycline to induce MdmxWT or MdmxF488A. 24h later, cells were lysed, and pulldown of ubiquitylated proteins was performed using agarose-conjugated Ni2+ beads (Invitrogen, licensed from Qiagen). Lysis and wash conditions were as previously described (44, 45). Following SDS-PAGE and transfer to PVDF membrane, ubiquitylated p53 was visualized using DO-1 antibody.
Quantitative PCR
Total RNA was prepared using QiaShredder and RNeasy RNA isolation kits per the manufacturer’s instructions (Qiagen, CA). 2μg of total RNA per sample was used for cDNA synthesis with random hexamer primers, using SuperScriptIII Reverse Transcriptase system (Invitrogen, CA). Real time quantitative PCR was performed using an ABI PRISM 7700 Sequence Detection System, using Platinum SYBR SuperMix (Invitrogen) with ROX as an internal standard. Changes in gene expression were normalized to 18S mRNA. Primer sequences supplied on request.
Colony outgrowth
For colony formation assays, cells were plated at 3000/6cm plate and colonies were allowed to form (~2 days) before addition of Doxycycline for 24h prior to treatment with 10μM Nutlin (for 2 days). After a further 5-6 days, cells were fixed in 3.7% para-formaldehyde and stained with crystal violet. Images of colonies were taken on the plate, and then dye was extracted in Sorenson’s buffer (50mM sodium citrate, 50mM citric acid in 50% (v/v) ethanol solution) and quantified by absorbance at 590nm.
Supplementary Material
Supplementary Figure 3 Validation of PLISA assay (A) Cells expressing MdmxWT or MdmxF488A were treated with doxycycline for 24h prior to fixation and processing for Mdm2 and Mdmx by conventional immunofluorescence. The primary antibodies (5B10 and BL1258) were the same as those used in Figure 2B. (B) Cells expressing MdmxWT or MdmxF488A Mdmx were treated with doxycycline for 24h prior to fixation and processing for p53 and Mdmx by conventional immunofluorescence. The primary antibodies (FL-393 and anti-HA) were the same as those used in Figure 2B. (C) Subcellular localization of Mdm2, MdmxWT, MdmxF488A and p53. Following induction of exogenous Mdmx with 100 ng/ml Dox for 24h, cells were separated into nuclear and cytoplasmic fractions and probed by western blot. (D) Cells expressing MdmxWT or MdmxG57A were treated with doxycycline for 24h and analyzed by western blot for the indicated proteins. In parallel, cells on coverslips were analyzed by PLISA for Mdmx/p53 interaction. Note that despite similar levels of both Mdmx and p53, PLISA signals were significantly lower in cells expressing the p53-binding mutant MdmxG57A. Note that a combination of high doxycycline dose and addition of proteasome inhibitor was used in order to demonstrate that MdmxG57A interacts very weakly with p53 compared to MdmxWT, despite high levels of Mdmx, Mdm2 and p53 under these conditions. This likely contributes to the increased concentration of p53/MdmxWT complexes in the nucleus compared to Figure 3B.
Supplementary Figure 4 p53/Mdm2/MdmxF488A complexes (A) Following induction of MdmxF488A, cells were lyzed and either MdmxF488A or p53 was immunoprecipitated. The panel shows that both Mdm2 and p53 can be found in complex with MdmxF488A. Note that interaction between MdmxWT and p53 was not detected in these cells unless proteasome inhibitor was added (right panels). We infer this is since the complex containing Mdm2, p53 and wild type Mdmx is unstable under the conditions used. (B) Both MdmxWT and MdmxF488A interact with endogenous Mdm2. Following induction of HA-tagged exogenous Mdmx with Dox (100 ng/ml) for 24h, (in the presence or absence of MG132 for the final 6h) Mdmx was immunoprecipitated with HA antibody. As a negative control, lysates from MdmxF488A expressing cells treated with MG132 were incubated with mouse IgG. Although Mdm2 levels were not stabilized under these conditions, ImageJ analysis (data not shown) of p53 revealed a 2-fold increase in p53 in cells expressing MdmxWT but not in cells expressing MdmxF488A, consistent with the notion that MdmxF488A is already acting effectively as an inhibitor of p53 degradation. (C) The double point mutant MdmxG57A/F488A retains the ability to stabilize p53. U2OS cells harboring the rtTA cassette were transfected with MdmxF488A or Mdmx G57A/F488A and then treated with 20 ng/ml Dox for 24h prior to blotting for the indicated proteins.
Supplementary Figure 5 Mdm2Y489A and MdmxF488A suppresses basal but not damage-induced p53 activity. p53 ‘specific activity’ was determined following treatment with either NCS (A and B) or Nutlin (C ). Specific activity was calculated by normalizing the amount of p21 (NCS) or Mdm2 (Nutlin) mRNA from qPCR to the amount of p53 (determined by LiCor/Odyssey). Analysis was performed on data from Figures 4 and 6. (D) Cells were treated as in Figure 6B and analysed for p21 mRNA expression.
Supplementary Figure 6 Mdm2WT, but not Mdm2Y489A Mdm2 is destabilized upon DNA damage. Cells were treated with doxcycline for 24h prior to addition of 300ng/ml NCS for an additional 4h. Cycloheximide was then added (t=0) and cells were harvested at the indicated times. Band intensities were quantified by LiCor/Odyssey and plotted as log2 of the band intensity (where intensity at t=0 is 1).
Supplementary Figure 7 Binding of MdmxF488A to p53 is compromised by introduction of the G57A mutation. (A) Following addition of doxycycline for 24h, Mdmx was immunoprecipitated with HA.11 antibody (Covance) and western blot for Mdmx (BL-1258) and p53 (FL-393) was performed. Note that although significantly more Mdmx was immunoprecipitated in the MdmxG57A/F488A mutant, proportionally less p53 was co-immunoprecipitated compared to MdmxF488A. Graph in right panel is the LiCor quantification of ratio of p53 to Mdmx in the immunoprecipitates. (B) Despite the induction of similar amounts of p53 protein (left panel), the level of p21 was significantly lower in both MdmxF488A and MdmxG57A/F488A cells (right panel). However, (C) shows that MdmxG57A/F488A was a weaker suppressor of p53 dependent transactivation than MdmxF488A. Specifically, p21 levels were higher in the double mutant cells, despite a higher Mdmx/p53 ratio in following MdmxG57A/F488A expression (see also Figure 6).
Supplementary Figure 1 Quantitative analyses of doxycycline-inducible Mdmx and Mdm2 cell lines. Doxycycline was added at the indicated concentrations for 24h and cells were analyzed by western (A) the LiCor/Odyssey imaging system (B) to quantify levels of exogenous Mdmx and endogenous p53. MCF7 breast cancer epithelial cells were used for comparison. (C) Doxycycline was added at the indicated concentrations for 24h and cells were analyzed by western blot. (D) Doxycycline was added at the indicated concentrations for 24h; cells were treated with 300 ng/ml NCS for 5h where indicated. Lower panels in both (C) and (D) are based on quantifications of Mdmx band intensities from the upper panels. The level of p53 (E) and p21 (F) protein in the presence or absence of Mdm2, Mdmx or the indicated mutants is shown. Data were obtained by analysis of band intensities from Figure 1.
Supplementary Figure 2 Schematic of PLISA. Primary antibodies against two proteins of interest are incubated with fixed cells on coverslips. PLISA probes (yellow and green) are species-specific secondary antibodies conjugated to oligonucleotides. If the antibodies are in close proximity, they can hybridize to a DNA template for rolling circle amplification (center panel). Following rolling circle amplification, coverslips are incubated with a fluorescently labeled oligonucleotide that binds to target sequences of the rolling circle template (right panel). The rolling circle amplification can only occur when the PLA probes are in proximity; thus no signal is obtained with proteins that do not interact (for example if the binding site in protein B is mutated (lower panel).
Acknowledgments
Work in this manuscript was funded by a National Institutes of Health R01 Grant CA61449 awarded to GMW. We thank all members of the Wahl lab for input on various aspects of this project, Dimitris Xirodimas (Dundee University) for advice on the ubiquitylation assays and Masha Poyurovsky (Columbia University) for productive discussions. We also thank Rachel Klevit and Chris Pierini (University of Washington) for their comments and insight into ubiquitylation during this project.
Footnotes
Conflict of interest The authors declare no conflict of interests in this manuscript.
Supplementary information is available at Oncogene’s website.
References
- 1.Aranda-Anzaldo A, Dent MA. Reassessing the role of p53 in cancer and ageing from an evolutionary perspective. Mech Ageing Dev. 2007;128(4):293–302. doi: 10.1016/j.mad.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol. 2010;2(12):a000893. doi: 10.1101/cshperspect.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol. 2010;2(8):a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7(11):819–22. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol Cancer Res. 2009;7(1):1–11. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007;27(15):5479–85. doi: 10.1128/MCB.00555-06. Epub 2007 May 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maetens M, Doumont G, Clercq SD, Francoz S, Froment P, Bellefroid E, et al. Distinct roles of Mdm2 and Mdm4 in red cell production. Blood. 2007;109(6):2630–3. doi: 10.1182/blood-2006-03-013656. [DOI] [PubMed] [Google Scholar]
- 8.Marine JC, Dyer MA, Jochemsen AG. MDMX: from bench to bedside. J Cell Sci. 2007;120(Pt 3):371–8. doi: 10.1242/jcs.03362. [DOI] [PubMed] [Google Scholar]
- 9.Saville MK, Sparks A, Xirodimas DP, Wardrop J, Stevenson LF, Bourdon JC, et al. Regulation of p53 by the ubiquitin-conjugating enzymes UbcH5B/C in vivo. J Biol Chem. 2004;279(40):42169–81. doi: 10.1074/jbc.M403362200. Epub 2004 Jul 26. [DOI] [PubMed] [Google Scholar]
- 10.Marine JC, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010;17(1):93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- 11.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275(12):8945–51. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 12.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A. 2003;100(21):12009–14. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, et al. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem. 2001;276(18):14537–40. doi: 10.1074/jbc.C000881200. [DOI] [PubMed] [Google Scholar]
- 14.Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278(46):45946–53. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- 15.Wang YV, Leblanc M, Wade M, Jochemsen AG, Wahl GM. Increased radioresistance and accelerated B cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell. 2009;16(1):33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poyurovsky MV, Priest C, Kentsis A, Borden KL, Pan ZQ, Pavletich N, et al. The Mdm2 RING domain C-terminus is required for supramolecular assembly and ubiquitin ligase activity. Embo J. 2007;26(1):90–101. doi: 10.1038/sj.emboj.7601465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uldrijan S, Pannekoek WJ, Vousden KH. An essential function of the extreme C-terminus of MDM2 can be provided by MDMX. Embo J. 2007;26(1):102–12. doi: 10.1038/sj.emboj.7601469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kostic M, Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE. Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol. 2006;363(2):433–50. doi: 10.1016/j.jmb.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 19.Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15(5):841–8. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- 20.Wang YV, Wade M, Wong E, Li YC, Rodewald LW, Wahl GM. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc Natl Acad Sci U S A. 2007;104(30):12365–70. doi: 10.1073/pnas.0701497104. Epub 2007 Jul 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Clercq S, Gembarska A, Denecker G, Maetens M, Naessens M, Haigh K, et al. Widespread overexpression of epitope-tagged Mdm4 does not accelerate tumor formation in vivo. Mol Cell Biol. 2010;30(22):5394–405. doi: 10.1128/MCB.00330-10. Epub 2010 Sep 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 23.Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci U S A. 2005;102(14):5056–61. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell. 2005;7(6):547–59. doi: 10.1016/j.ccr.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 25.Weissman AM, Yang Y, Kitagaki J, Sasiela CA, Beutler JA, O’Keefe BR. Inhibiting Hdm2 and ubiquitin-activating enzyme: targeting the ubiquitin conjugating system in cancer. Ernst Schering Found Symp Proc. 2008;(1):171–90. doi: 10.1007/2789_2008_108. [DOI] [PubMed] [Google Scholar]
- 26.Joseph TL, Madhumalar A, Brown CJ, Lane DP, Verma C. Differential binding of p53 and nutlin to MDM2 and MDMX: Computational studies. Cell Cycle. 2010;9(6):1167–81. doi: 10.4161/cc.9.6.11067. [DOI] [PubMed] [Google Scholar]
- 27.Popowicz GM, Czarna A, Holak TA. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7(15):2441–3. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 28.Wade M, Rodewald LW, Espinosa JM, Wahl GM. BH3 activation blocks Hdmx suppression of apoptosis and co-operates with Nutlin to induce cell death. Cell Cycle. 2008;7(13):1973–82. doi: 10.4161/cc.7.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das R, Mariano J, Tsai YC, Kalathur RC, Kostova Z, Li J, et al. Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol Cell. 2009;34(6):674–85. doi: 10.1016/j.molcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ozkan E, Yu H, Deisenhofer J. Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc Natl Acad Sci U S A. 2005;102(52):18890–5. doi: 10.1073/pnas.0509418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. Embo J. 2005;24(19):3411–22. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1(6):a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. Embo J. 2009;28(24):3857–67. doi: 10.1038/emboj.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dastidar SG, Raghunathan D, Nicholson J, Hupp TR, Lane DP, Verma CS. Chemical states of the N-terminal “lid” of MDM2 regulate p53 binding: simulations reveal complexities of modulation. Cell Cycle. 2011;10(1):82–9. doi: 10.4161/cc.10.1.14345. [DOI] [PubMed] [Google Scholar]
- 35.Zuckerman V, Lenos K, Popowicz GM, Silberman I, Grossman T, Marine JC, et al. c-Abl phosphorylates Hdmx and regulates its interaction with p53. J Biol Chem. 2009;284(6):4031–9. doi: 10.1074/jbc.M809211200. [DOI] [PubMed] [Google Scholar]
- 36.Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, et al. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277(22):19251–4. doi: 10.1074/jbc.C200150200. [DOI] [PubMed] [Google Scholar]
- 37.Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447(1):5–9. doi: 10.1016/s0014-5793(99)00254-9. [DOI] [PubMed] [Google Scholar]
- 38.Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, et al. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12(4):355–66. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 39.Huang L, Yan Z, Liao X, Li Y, Yang J, Wang ZG, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc Natl Acad Sci U S A. 2011;108(29):12001–6. doi: 10.1073/pnas.1102309108. Epub 2011 Jul 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pant V, Xiong S, Iwakuma T, Quintas-Cardama A, Lozano G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci U S A. 2011;108(29):11995–2000. doi: 10.1073/pnas.1102241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitagaki J, Agama KK, Pommier Y, Yang Y, Weissman AM. Targeting tumor cells expressing p53 with a water-soluble inhibitor of Hdm2. Mol Cancer Ther. 2008;7(8):2445–54. doi: 10.1158/1535-7163.MCT-08-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong ET, Kolman JL, Li YC, Mesner LD, Hillen W, Berens C, et al. Reproducible doxycycline-inducible transgene expression at specific loci generated by Cre-recombinase mediated cassette exchange. Nucleic Acids Res. 2005;33(17):e147. doi: 10.1093/nar/gni145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stommel JM, Wahl GM. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. Embo J. 2004;23(7):1547–56. doi: 10.1038/sj.emboj.7600145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT. SUMO-1 modification activates the transcriptional response of p53. Embo J. 1999;18(22):6455–61. doi: 10.1093/emboj/18.22.6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xirodimas D, Saville MK, Edling C, Lane DP, Lain S. Different effects of p14ARF on the levels of ubiquitinated p53 and Mdm2 in vivo. Oncogene. 2001;20(36):4972–83. doi: 10.1038/sj.onc.1204656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 3 Validation of PLISA assay (A) Cells expressing MdmxWT or MdmxF488A were treated with doxycycline for 24h prior to fixation and processing for Mdm2 and Mdmx by conventional immunofluorescence. The primary antibodies (5B10 and BL1258) were the same as those used in Figure 2B. (B) Cells expressing MdmxWT or MdmxF488A Mdmx were treated with doxycycline for 24h prior to fixation and processing for p53 and Mdmx by conventional immunofluorescence. The primary antibodies (FL-393 and anti-HA) were the same as those used in Figure 2B. (C) Subcellular localization of Mdm2, MdmxWT, MdmxF488A and p53. Following induction of exogenous Mdmx with 100 ng/ml Dox for 24h, cells were separated into nuclear and cytoplasmic fractions and probed by western blot. (D) Cells expressing MdmxWT or MdmxG57A were treated with doxycycline for 24h and analyzed by western blot for the indicated proteins. In parallel, cells on coverslips were analyzed by PLISA for Mdmx/p53 interaction. Note that despite similar levels of both Mdmx and p53, PLISA signals were significantly lower in cells expressing the p53-binding mutant MdmxG57A. Note that a combination of high doxycycline dose and addition of proteasome inhibitor was used in order to demonstrate that MdmxG57A interacts very weakly with p53 compared to MdmxWT, despite high levels of Mdmx, Mdm2 and p53 under these conditions. This likely contributes to the increased concentration of p53/MdmxWT complexes in the nucleus compared to Figure 3B.
Supplementary Figure 4 p53/Mdm2/MdmxF488A complexes (A) Following induction of MdmxF488A, cells were lyzed and either MdmxF488A or p53 was immunoprecipitated. The panel shows that both Mdm2 and p53 can be found in complex with MdmxF488A. Note that interaction between MdmxWT and p53 was not detected in these cells unless proteasome inhibitor was added (right panels). We infer this is since the complex containing Mdm2, p53 and wild type Mdmx is unstable under the conditions used. (B) Both MdmxWT and MdmxF488A interact with endogenous Mdm2. Following induction of HA-tagged exogenous Mdmx with Dox (100 ng/ml) for 24h, (in the presence or absence of MG132 for the final 6h) Mdmx was immunoprecipitated with HA antibody. As a negative control, lysates from MdmxF488A expressing cells treated with MG132 were incubated with mouse IgG. Although Mdm2 levels were not stabilized under these conditions, ImageJ analysis (data not shown) of p53 revealed a 2-fold increase in p53 in cells expressing MdmxWT but not in cells expressing MdmxF488A, consistent with the notion that MdmxF488A is already acting effectively as an inhibitor of p53 degradation. (C) The double point mutant MdmxG57A/F488A retains the ability to stabilize p53. U2OS cells harboring the rtTA cassette were transfected with MdmxF488A or Mdmx G57A/F488A and then treated with 20 ng/ml Dox for 24h prior to blotting for the indicated proteins.
Supplementary Figure 5 Mdm2Y489A and MdmxF488A suppresses basal but not damage-induced p53 activity. p53 ‘specific activity’ was determined following treatment with either NCS (A and B) or Nutlin (C ). Specific activity was calculated by normalizing the amount of p21 (NCS) or Mdm2 (Nutlin) mRNA from qPCR to the amount of p53 (determined by LiCor/Odyssey). Analysis was performed on data from Figures 4 and 6. (D) Cells were treated as in Figure 6B and analysed for p21 mRNA expression.
Supplementary Figure 6 Mdm2WT, but not Mdm2Y489A Mdm2 is destabilized upon DNA damage. Cells were treated with doxcycline for 24h prior to addition of 300ng/ml NCS for an additional 4h. Cycloheximide was then added (t=0) and cells were harvested at the indicated times. Band intensities were quantified by LiCor/Odyssey and plotted as log2 of the band intensity (where intensity at t=0 is 1).
Supplementary Figure 7 Binding of MdmxF488A to p53 is compromised by introduction of the G57A mutation. (A) Following addition of doxycycline for 24h, Mdmx was immunoprecipitated with HA.11 antibody (Covance) and western blot for Mdmx (BL-1258) and p53 (FL-393) was performed. Note that although significantly more Mdmx was immunoprecipitated in the MdmxG57A/F488A mutant, proportionally less p53 was co-immunoprecipitated compared to MdmxF488A. Graph in right panel is the LiCor quantification of ratio of p53 to Mdmx in the immunoprecipitates. (B) Despite the induction of similar amounts of p53 protein (left panel), the level of p21 was significantly lower in both MdmxF488A and MdmxG57A/F488A cells (right panel). However, (C) shows that MdmxG57A/F488A was a weaker suppressor of p53 dependent transactivation than MdmxF488A. Specifically, p21 levels were higher in the double mutant cells, despite a higher Mdmx/p53 ratio in following MdmxG57A/F488A expression (see also Figure 6).
Supplementary Figure 1 Quantitative analyses of doxycycline-inducible Mdmx and Mdm2 cell lines. Doxycycline was added at the indicated concentrations for 24h and cells were analyzed by western (A) the LiCor/Odyssey imaging system (B) to quantify levels of exogenous Mdmx and endogenous p53. MCF7 breast cancer epithelial cells were used for comparison. (C) Doxycycline was added at the indicated concentrations for 24h and cells were analyzed by western blot. (D) Doxycycline was added at the indicated concentrations for 24h; cells were treated with 300 ng/ml NCS for 5h where indicated. Lower panels in both (C) and (D) are based on quantifications of Mdmx band intensities from the upper panels. The level of p53 (E) and p21 (F) protein in the presence or absence of Mdm2, Mdmx or the indicated mutants is shown. Data were obtained by analysis of band intensities from Figure 1.
Supplementary Figure 2 Schematic of PLISA. Primary antibodies against two proteins of interest are incubated with fixed cells on coverslips. PLISA probes (yellow and green) are species-specific secondary antibodies conjugated to oligonucleotides. If the antibodies are in close proximity, they can hybridize to a DNA template for rolling circle amplification (center panel). Following rolling circle amplification, coverslips are incubated with a fluorescently labeled oligonucleotide that binds to target sequences of the rolling circle template (right panel). The rolling circle amplification can only occur when the PLA probes are in proximity; thus no signal is obtained with proteins that do not interact (for example if the binding site in protein B is mutated (lower panel).