

Abstract
The early proinflammatory cytokine tumor necrosis factor (TNF) is released in significant quantities by the activated immune system in response to infection, leukemia, autoimmune disorders, and radiation sickness. Nausea, emesis, and anorexia are common features of these disorders. TNF action on vagal afferent terminals in the brainstem is a likely cause of the malaise associated with these disorders. Our previous work has shown that TNF action to excite vagal afferents occurs as a result of sensitization of ryanodine channels in afferent nerve terminals. For millennia, cannabinoids (CB) have been used to combat the visceral malaise associated with chronic disease, although the mechanism of action has not been clear. Previous work in culture systems suggests that CB1 agonists can suppress neurotransmission by downregulating ryanodine channels through a protein kinase A (PKA)-dependent mechanism. Laser confocal calcium imaging methods were used to directly examine effects of CB1 cannabinoid agonists and TNF on visceral afferent signaling in the rat hindbrain. CB1 agonists blocked the effects of TNF to amplify vagal afferent responsiveness; blockade of PKA with H89 also eliminated the TNF amplification effect. These results help to explain the effectiveness of cannabinoids in blocking the malaise generated by TNF-releasing disease processes by opposing effects on ryanodine channels.
Introduction
Immune activation in response to infection, injury, or chronic disease causes high levels of cytokine release. Tumor necrosis factor-α (TNF) is one of the early, proinflammatory cytokines released under these conditions. TNF is produced by a variety of cell types including macrophages, lymphoid cells, mast cells, endothelial cells, cardiac myocytes, adipose tissue, and fibroblasts, as well as neurons, microglia, and astrocytes (Barrientos et al., 2008; Bashir et al., 2009; Hedayat et al., 2010; Park and Bowers, 2010; Maenhaut and Van de Voorde, 2011). Thus, infection, cancer, heart failure, or exposure to radiation can evoke production of TNF by both immune and nonimmune cell types.
Disease states causing release of large quantities of TNF are associated with “signs” of illness such as fever, gastric hypomotility, suppressed appetite, nausea, and vomiting. Systemic injections of TNF elicit the same signs of illness observed in chronic disease (see review, Hermann and Rogers, 2008).
TNF causes gastrointestinal symptoms (gastric stasis) by acting on vagal afferent fibers synapsing on medullary nuclei of the solitary tract (NST) that form vagal gastroinhibitory reflexes (Rogers et al., 2006b; Hermann and Rogers, 2008, 2009). Our neurophysiological studies suggested that TNF amplifies the responsiveness of vagal afferent pathways to otherwise innocuous sensory signals (Emch et al., 2000). Calcium imaging of vagal afferent terminals in NST slice preparations show that TNF effects are due to sensitization of the ryanodine-based calcium-induced-calcium-release (CICR) mechanism (Rogers et al., 2006b).
Cannabinoid (CB) agonists have been used to ameliorate the malaise of chronic disease for thousands of years (Ben Amar, 2006). More recently, cannabinoids have been used in response to antineoplastic therapies that are associated with the generation of large amounts of TNF (Pogrebniak et al., 1991; Gorter, 1999; Machado Rocha et al., 2008). CB1 receptors in the brain have been associated with the anti-illness effects of cannabinoids (Davis et al., 2007; Slatkin, 2007).
One possible consequence of CB1 receptor activation is the suppression of the presynaptic CICR mechanism mediated by ryanodine channels (Zhuang et al., 2005). Specifically, activation of the CB1 receptor in cultured neurons activates the G-protein subunit Gi (i.e., decreases activity of cAMP-dependent protein kinase), ultimately, deactivating protein kinase A (PKA).
Ryanodine CICR channels are upregulated by PKA phosphorylation (Lanner et al., 2010). PKA may also upregulate CD38, the enzyme responsible for generating cyclic ADP ribose (cADPR), a potent activator of CICR ryanodine channels (Deshpande et al., 2003; Rogers et al., 2006b). Activation of PKA is one of many mechanisms the TNF transduction apparatus can engage (Pan et al., 1997).
The following live cell imaging study of vagal afferent terminals in the NST tests the hypothesis that TNF effects to amplify visceral afferent synaptic transmission will be blocked by CB1 agonists and agents that block PKA.
Materials and Methods
Long–Evans rats of either sex (200–300 g) were obtained from the breeding colony located at Pennington Biomedical Research Center. Animals were maintained in a room with a 12:12 h light-dark cycle with constant temperature and humidity, and given food and water ad libitum. All experimental protocols were performed according to the guidelines set forth by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committees at the Pennington Biomedical Research Center.
Vagal afferent prelabeling for in vitro calcium imaging.
Techniques for performing calcium imaging studies of identified vagal afferent fibers and varicosities have been described in detail (Rogers et al., 2006a,b; Rogers and Hermann, 2008). Before surgery, glass microinjection pipettes are pulled from 1.8 mm OD starbore capillary tubing (Radnoti Glass Technologies) using Narishige Model 1D puller. Tips are beveled at ∼20° angle on a rotating diamond-coated glass disk (Sutter BV-10); final tip diameter ∼50 μm. Micropipettes were filled with 20% CalciumGreen 1-dextran 3000MW conjugate (CGD; Invitrogen) in a solution of 1% Triton X-100 and distilled water.
Rats are anesthetized with pentobarbital (Nembutal, 50 mg/kg, i.p.; Abbott Laboratories). Using aseptic technique, the nodose ganglion is exposed at the jugular foramen. The nodose appears as a pink translucent swelling of the vagus at the posterior lacerated foramen (Green, 1935). The micropipette containing CGD is connected to a source of pulsed air pressure (Picospritzer, General Valve) and the tip of the pipette is guided by hand through the sheath of the nodose ganglion. Pressure pulses (∼2–10 psi) are applied to the pipette; dye is injected to fill the ganglion. Total injected volume is ∼500 nl. The cervical wound is closed and animals are returned to their home cage for 2–5 d to allow anterograde transport of CGD to central vagal varicosities.
Brainstem slice preparation.
After anterograde transport, animals are reanesthetized with urethane (ethyl carbamate, 1.5 g/kg i.p.; Sigma-Aldrich); this anesthesia readily washes out of tissue and does not have long-term effects on the activity of neurons in these slices (Hara and Harris, 2002). After decapitation and swift removal of the brainstem to cold, carbogenated, cutting solution (∼4°C; 95% O2; 5% CO2; see recipe below, In vitro drugs and solutions), 300-μm-thick slices are cut coronally through the medulla with a sapphire knife on a vibrating microtome (Leica model VT1200). Slices are then transferred to a scintillation vial containing carbogenated normal Krebs (see recipe below, In vitro drugs and solutions) at 29°C for ∼1 h before conducting experiments.
Perfusion chamber.
Individual brain slices are transferred to the temperature-regulated perfusion chamber (Delta T4 culture dish, Bioptechs) maintained at 33°C with a solution flow rate of 3 ml/min. The confocal depth of field of 1–2 μm is just sufficient to image terminal varicosities and fibers that are, themselves, no more than 2–3 μm in diameter (Rogers et al., 2006a). Therefore, no drift can be tolerated. To solve this problem, we support the recording chamber on a large fixed stage (Gibralter platform; Burley); this fixed stage supports and moves the microscope in the X-Y plane. To maximize slice stability, we have redesigned the chambers such that the brainstem slices are supported by nylon hosiery mesh while being perfused. Additionally, slices placed in the chamber are held fast with a harp-type pressor foot made from 22 gauge gold wire and individual nylon hosiery fibers (Figure 1). Slices rapidly settle into the mesh and are then essentially immobilized for the duration of the experiment. This chamber redesign has qualitatively improved slice stability and viability.
Figure 1.
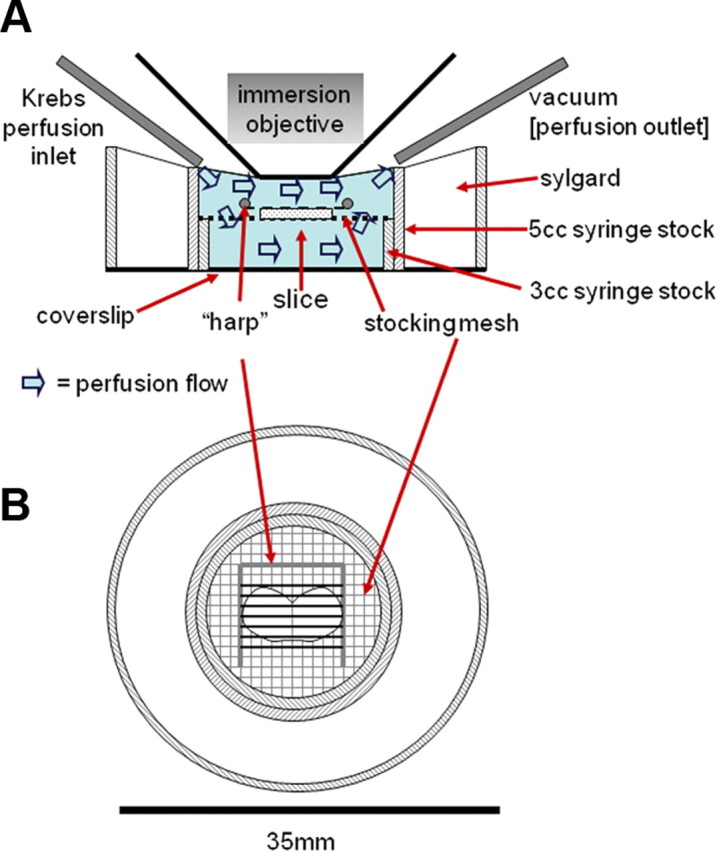
Cross-sectional (A) and top view (B) drawings of recording chamber. Stocking mesh and harp arrangement in this chamber design qualitatively increased the stability of the slice when compared with the conventional chambers with coverslip floors. This design also improves viability of the cells by allowing both subfusion and superfusion of the slice (A).
In vitro drugs and solutions.
The cutting solution contained (in mm): 110 choline chloride, 25 NaHCO3, 2.5 KCl, 7 MgSO4-7H2O, 1.25 NaH2PO4; 10 glucose; 0.5 CaCl2-2H2O; bubbled with 95% O2/5% CO2 during the entire cutting process. Normal Krebs contained (in mm): 124 NaCl, 25 NaHCO3, 3.0 KCl, 1 MgSO4-7H2O, 1.5 NaH2PO4; 10 glucose; 1.5 CaCl2-2H2O; bubbled with 95% O2/5% CO2, continuously; osmolality was 300 ± 10 mOsm; pH = 7.3.
Where specified in the Experimental Design section below, one of the following drugs was added to the Normal Krebs (note: specified concentrations of reagents are based on references cited for similar in vitro studies): 2 nm TNF (comparable to the circulating concentration following a significant immune challenge (Peprotech; Hermann et al., 2002); 500 nm WIN 52212-2 (WIN2, CB1 agonist; Sigma-Aldrich; Derbenev et al., 2004); 500 nm WIN 52212-3 (WIN3, inactive enantiomer of WIN2; Sigma-Aldrich; Derbenev et al., 2004); 1 μM H89 (PKA antagonist; Sigma-Aldrich; Rogers and Hermann, 2008); combination of TNF plus WIN2; combination of TNF plus WIN3; combination of TNF plus H89.
Imaging instrumentation.
Time-lapse laser confocal calcium imaging was performed with a Nikon Fast Scan laser confocal head equipped with a Luca EMCCD camera (Andor Technology). Imaging equipment was coupled to a Nikon F1 fixed stage upright microscope equipped with 10× and 40× water-immersion objectives. At 400× total magnification, full frames of view represented ∼200 × 150 μm of brainstem slice area. CGD preloaded fibers were visualized using a 488 nm excitation/509 nm long pass emission filter arrangement. Changes in intracellular calcium levels (fluorescence) were collected at a rate of three frames per second; confocal fluorescent signal was processed by Nikon Elements AR software.
Electrical stimulation of the solitary tract.
A concentric bipolar electrode (tip diameter = 50 μm) was placed in the solitary tract (ST) using a Burleigh three-axis microdrive. Stimulation current trains (four pulses, 200 μA, 0.5 ms, 20 Hz) were generated by a WPI A310 stimulator (World Precision Instruments) coupled to a stimulus isolation unit. These stimulation parameters are sufficient to activate both A- and C-fiber primary vagal afferents (Andresen and Yang, 1995; McDougall et al., 2009; McDougal et al., 2011). Delivered current was continuously monitored on an oscilloscope by measuring the voltage drop across a 1000 Ω resistance in line with the positive (center) electrode.
In vitro calcium signal analysis.
Relative changes in cytoplasmic calcium in response to agonist solutions are expressed as changes in fluorescence: (ΔF/F)%, where F is the fluorescence intensity within the ROI (e.g., the outline of a vagal afferent fiber or varicosity) before stimulation; ΔF is the change from this value during neuronal activity (Helmchen, 2000). Background fluorescence (i.e., noninvolved areas adjacent to the area of interest) is subtracted from both ΔF and F. A responsive fiber or varicosity was classified as one in which a stimulus pulse train delivered to the ST-evoked calcium signal elevation (ΔF/F) of 3% or greater (example in Figure 2).
Figure 2.

Response of CGD-prelabeled vagal afferent varicosities in the NST to ST electrical stimulation and exposure to TNF. A, Field of varicosities at rest, i.e., before ST stimulation or exposure to TNF. (Circled varicosity is same in all images and was used to generate the relative calcium plot in D.) B, Same field at the peak of the effect of ST stimulation alone. C, Same field responding to second ST stimulation (i.e., after exposure to TNF). D, Plot of the effect of ST stimulation on relative terminal calcium. Red, plot of change in fluorescence in response to ST stimulation before TNF; black, response plot after TNF. (Note: Although responses of other varicosities in the field were not plotted here, they show similar response patterns.)
Experimental design and analysis.
As seen previously (Rogers et al., 2006a,b; Kline et al., 2009; Austgen et al., 2011), electrical stimulation of the ST produces a brisk and highly repeatable increase in intracellular calcium in vagal afferent terminals. Thus, we can rely on comparing “before” and “after” responses of individual varicosities or fibers under the various experimental conditions. Therefore, the basic experimental design consisted of two sets of electrical stimulation pulses delivered to the ST. The initial train of stimulation occurred under normal Krebs perfusion of the slice to establish the basic response profile of the prelabeled vagal afferent terminals under observation. A 10 min “rest period” followed during which the slice was perfused with either normal Krebs (i.e., “time control”) or one of the experimental conditions [i.e., TNF (alone), WIN2 (alone), WIN3 (alone), H89 (alone), or combinations of TNF plus one of the antagonists (as listed above)]. At the end of the 10 min time/drug treatment period, a second stimulation train was again applied to the ST. The magnitudes of responses to these two stimulation trains were compared.
Thus, each fiber or varicosity was exposed to the same electrical stimulation twice (i.e., before and after exposure to a drug treatment condition) and could serve as its own control. Differences in response between the first (control) and second (treatment) stimulations were calculated; data are presented as percentage response after experimental condition relative to response after initial stimulation. Results across treatments were subjected to one-way ANOVA followed by Tukey multiple-comparison post-tests; values of p < 0.05 were considered statistically significant.
Results
Averaged relative responses of prelabeled vagal afferent fibers and varicosities to electrical stimulation are presented in Figure 3. As previously observed, electrical stimulation of the ST produced a brisk and highly repeatable increase in intracellular calcium in vagal afferent terminals. Treatment of the medullary slice preparation with TNF produced a significant increase in the evoked release of intracellular calcium relative to that evoked under normal Krebs' conditions (TNF: 179.5 ± 19.0% versus Control: 99.5 ± 4.0%). The CB1 cannabinoid agonist, WIN2, caused a significant reduction of the ST-evoked calcium release (56.7 ± 5.3%) while the inactive analog, WIN3, had no such effect (98.6 ± 5.5%). The combination of WIN2 plus TNF markedly reduced the ST-evoked response relative to TNF alone (78.0 ± 7.2% versus 179.5 ± 19.0%). In contrast, WIN3 had no effect on the TNF amplification of stimulus-evoked calcium levels in vagal afferent terminals (172.0 ± 10.0%). Additionally, while the selective PKA antagonist (H89) blocked the effect of TNF to augment vagal terminal responsiveness to stimulation (69.3 ± 7.2%) alone, it did not have an effect (82.0 ± 5.7%) to diminish the vagal terminal increase in intracellular calcium in response to ST stimulation by itself.
Figure 3.

TNF amplification of vagal terminal calcium responses are suppressed by CB1 agonist (WIN2) and a PKA antagonist (H89). Responses from individual prelabeled vagal terminals to two exposures of ST stimulation (spaced 10 min apart) were evaluated. The first (control) stimulation was used as a basis of comparison for the results of the second ST stimulation. Thus, results are reported as percentage responses relative to the first ST stimulation. ANOVA F(7,243) = 28; p < 0.0001; Tukey multiple-comparison post-tests. Time control: second stimulation evoked a change in fluorescence that averaged 99.5 ± 4.0% of the response evoked during the first stimulation. TNF alone: 2 nm TNF amplified the afferent response to ST stimulation by an average of 179.5 ± 19.0% relative to the first (control) ST stimulation (statistically larger than time control responses; *p < 0.05). WIN2 alone: 500 nm CB1 agonist by itself depressed the relative calcium response to ST stimulation (statistically smaller than time control responses; *p < 0.05). WIN3 alone (inactive enantiomer of WIN2) and H89 alone (PKA antagonist) had no effect on the ST stimulated calcium signal. WIN2 + TNF: CB1 agonist blocked the effects of TNF to amplify vagal terminal responses to ST stimulation (WIN2 + TNF: 78.0 ± 7.2% response relative to first ST stimulation versus TNF alone: 179.5 ± 19.0%; statistically significantly smaller than TNF alone responses; #p < 0.05). WIN3 + TNF: inactive CB1 antagonist had no effect on TNF amplification of afferent input. H89 + TNF: suppression of PKA activity (see Fig. 4) also blocked the effects of TNF to amplify vagal terminal responses to ST stimulation (statistically smaller than TNF alone responses; #p < 0.05).
Discussion
Cannabinoids appear to be effective in ameliorating the visceral malaise common to cancer and other chronic disease processes (Kalant, 2001; Hall et al., 2005; Parker et al., 2011). Cannabinoids are also useful in suppressing neuropathic pain associated with chronic inflammation (Thaler et al., 2011). Visceral malaise and pain associated with chronic disease are, at least in part, due to the activation of TNF synthesis and release (Hermann et al., 2005; Hermann and Rogers, 2008). These symptoms are probably due to TNF effects on presynaptic release mechanisms used by both pain and visceral afferent pathways (Pickering et al., 2005; Hermann and Rogers, 2008; Youn et al., 2008).
The dorsal vagal complex contains the circuitry for vago-vagal reflex control of gastrointestinal function (e.g., gastric motility, tone, and acid secretion). This medullary region is composed of the sensory NST, the area postrema, and their interconnections with the dorsal motor nucleus of the vagus (Rogers et al., 1995). Most vago-vagal control of gastric motility and tone is inhibitory, i.e., activation of visceral vagal afferents results in a marked reduction in gastric motility and tone by initiating gastric vago-vagal reflexes (Rogers and Hermann, 2012). Gastroinhibition (i.e., gastroparesis) is perceived as nausea and serves as an essential prodrome to emesis (Syed et al., 2005). Indeed, this region of the caudal hindbrain is also essential for the generation of the emetic motor program (Blessing, 1997). TNF sensitizes glutamatergic afferent transmitter release via its action on the regulation of vagal afferent terminal calcium levels (Pickering et al., 2005; Rogers et al., 2006b; Hermann and Rogers, 2008). TNF is also effective in amplifying glutamatergic synaptic strength by upregulating postsynaptic AMPA receptor numbers (Stellwagen et al., 2005). TNF induces gastroparesis and visceral malaise by amplifying vagal afferent signaling to vago-vagal reflex mechanisms; cannabinoids may ameliorate these symptoms by directly opposing these cellular effects of TNF.
Cytoplasmic calcium concentration is the dominant factor in determining the amount of transmitter released from nerve terminals (Katz and Miledi, 1967). Regulation of availability of cytoplasmic calcium to drive vesicular release underlies several synaptic plasticity mechanisms including long-term potentiation (LTP; Katchman and Hershkowitz, 1993; Collin et al., 2005). Vagal and spinal pain, touch, and temperature afferents demonstrate LTP when exposed to TNF (Emch et al., 2000; Liu et al., 2007). TNF amplifies transmitter release by activating ryanodine channels in the endoplasmic reticulum (Rogers et al., 2006b) through the activation of CD38 and the synthesis of cyclic ADP-ribose; a potent ryanodine agonist. In turn, this causes an increase in CICR and potentiation of neurotransmitter release (Pollock et al., 2002; Lelli et al., 2003).
Studies in cultured hippocampal neurons suggested that activation of the CB1 receptor initiates a chain of transduction events leading to downregulation of ryanodine channel activity through the withdrawal of PKA-mediated phosphorylation of the channel (Zhuang et al., 2005). Thus, CB1 agonists should inhibit ryanodine-mediated CICR and oppose the action of TNF. As expected, the active CB1 agonist WIN2 (but not WIN3) reduced stimulus-evoked terminal calcium (expected as a function of its effects on voltage-gated calcium channels; Huang et al., 2001; Hoddah et al., 2009; Fig. 4). The suppression of TNF signaling by H89 (Figs. 3, 4) is consistent with the hypothesis that the CB1 receptor can act through PKA. However, it is certainly possible that a generalized suppression of PKA could block TNF signaling through mechanisms other than inhibition of CICR.
Figure 4.

Summary diagram explaining present results in context with previous observations of TNF and CB1 effects on excitability. Our previous results (Rogers et al., 2006b) showed that TNF has a potent effect to amplify calcium release from ER stores, ultimately via sensitization of the ER ryanodine receptor. This effect is obtained through upregulation of the CD38-catalyzed production of cADPR, a potent ryanodine receptor agonist. Stimulation of CD38, as well as the ryanodine mechanism itself, can be initiated through PKA phosphorylation. Activation of PKA through Gs-adenylcyclase is one of many transduction mechanisms “adapted” to the TNFR1 receptor. The ryanodine-ER calcium release CICR “amplifier” is also activated by calcium entry through voltage-gated calcium channels (VGCC) in the terminal membrane. Activation of the CB1 receptor initiates a Gi mechanism, opposing the transduction events set in motion by the TNFR1 receptor. Additionally, activation of the CB1 receptor damps calcium flux through VGCC's.
We believe that this is the first demonstration of the likely intracellular mechanism used by CB1 analogs to block the effects of TNF on neurotransmitter mechanisms that cause pain and visceral malaise. These results may explain how cannabinoids can be effective in the treatment of the allodynia and visceral malaise of chronic disease (Walker and Hohmann, 2005; Davis et al., 2007; Slatkin, 2007).
Footnotes
This work was supported by Grants NS52142, DK56373, and NS60664 from the National Institutes of Health.
The authors declare no competing financial interests.
References
- Andresen MC, Yang M. Dynamics of sensory afferent synaptic transmission in aortic baroreceptor regions on nucleus tractus solitarius. J Neurophysiol. 1995;74:1518–1528. doi: 10.1152/jn.1995.74.4.1518. [DOI] [PubMed] [Google Scholar]
- Austgen JR, Hermann GE, Dantzler HA, Rogers RC, Kline DD. Hydrogen sulfide augments synaptic neurotransmission in the nucleus of the solitary tract. J Neurophysiol. 2011;106:1822–1832. doi: 10.1152/jn.00463.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16:585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- Bashir MM, Sharma MR, Werth VP. TNF-alpha production in the skin. Arch Dermatol Res. 2009;301:87–91. doi: 10.1007/s00403-008-0893-7. [DOI] [PubMed] [Google Scholar]
- Ben Amar M. Cannabinoids in medicine: A review of their therapeutic potential. J Ethnopharmacol. 2006;105:1–25. doi: 10.1016/j.jep.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Blessing WW. The lower brainstem and bodily homeostasis. Oxford: Oxford UP; 1997. Eating and metabolism; pp. 323–372. [Google Scholar]
- Collin T, Marty A, Llano I. Presynaptic calcium stores and synaptic transmission. Curr Opin Neurobiol. 2005;15:275–281. doi: 10.1016/j.conb.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Davis M, Maida V, Daeninck P, Pergolizzi J. The emerging role of cannabinoid neuromodulators in symptom management. Support Care Cancer. 2007;15:63–71. doi: 10.1007/s00520-006-0180-0. [DOI] [PubMed] [Google Scholar]
- Derbenev AV, Stuart TC, Smith BN. Cannabinoids suppress synaptic input to neurones of the rat dorsal motor nucleus of the vagus nerve. J Physiol. 2004;559:923–938. doi: 10.1113/jphysiol.2004.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande DA, Walseth TF, Panettieri RA, Kannan MS. CD38/cyclic ADP-ribose-mediated Ca2+ signaling contributes to airway smooth muscle hyper-responsiveness. FASEB J. 2003;17:452–454. doi: 10.1096/fj.02-0450fje. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. TNF-alpha activates solitary nucleus neurons responsive to gastric distension. Am J Physiol Gastrointest Liver Physiol. 2000;279:G582–G586. doi: 10.1152/ajpgi.2000.279.3.G582. [DOI] [PubMed] [Google Scholar]
- Gorter RW. Cancer cachexia and cannabinoids. Forsch Komplementarmed. 1999;6(Suppl 3):21–22. doi: 10.1159/000057152. [DOI] [PubMed] [Google Scholar]
- Green EC. Anatomy of the rat. New York: Hafner Publishing; 1935. [Google Scholar]
- Hall W, Christie M, Currow D. Cannabinoids and cancer: causation, remediation, and palliation. Lancet Oncol. 2005;6:35–42. doi: 10.1016/S1470-2045(04)01711-5. [DOI] [PubMed] [Google Scholar]
- Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. table of contents. [DOI] [PubMed] [Google Scholar]
- Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev. 2010;15:543–562. doi: 10.1007/s10741-010-9168-4. [DOI] [PubMed] [Google Scholar]
- Helmchen F. Calibration of fluorescent calcium indicators. In: Yuste R, Lanni F, Konnerth A, editors. Imaging neurons: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2000. pp. 32.31–32.39. [Google Scholar]
- Hermann GE, Rogers RC. TNFalpha: a trigger of autonomic dysfunction. Neuroscientist. 2008;14:53–67. doi: 10.1177/1073858407305725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC. TNF activates astrocytes and catecholaminergic neurons in the solitary nucleus: implications for autonomic control. Brain Res. 2009;1273:72–82. doi: 10.1016/j.brainres.2009.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Tovar CA, Rogers RC. LPS-induced suppression of gastric motility relieved by TNFR:Fc construct in dorsal vagal complex. Am J Physiol Gastrointest Liver Physiol. 2002;283:G634–G639. doi: 10.1152/ajpgi.00412.2001. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Holmes GM, Rogers RC. TNF(alpha) modulation of visceral and spinal sensory processing. Curr Pharm Des. 2005;11:1391–1409. doi: 10.2174/1381612053507828. [DOI] [PubMed] [Google Scholar]
- Hoddah H, Marcantoni A, Comunanza V, Carabelli V, Carbone E. L-type channel inhibition by CB1 cannabinoid receptors is mediated by PTX-sensitive G proteins and cAMP/PKA in GT1–7 hypothalamic neurons. Cell Calcium. 2009;46:303–312. doi: 10.1016/j.ceca.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Huang CC, Lo SW, Hsu KS. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol. 2001;532:731–748. doi: 10.1111/j.1469-7793.2001.0731e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalant H. Medicinal use of cannabis: history and current status. Pain Res Manag. 2001;6:80–91. doi: 10.1155/2001/469629. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Hershkowitz N. Early anoxia-induced vesicular glutamate release results from mobilization of calcium from intracellular stores. J Neurophysiol. 1993;70:1–7. doi: 10.1152/jn.1993.70.1.1. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. A study of synaptic transmission in the absence of nerve impulses. J Physiol. 1967;192:407–436. doi: 10.1113/jphysiol.1967.sp008307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Hendricks G, Hermann G, Rogers RC, Kunze DL. Dopamine inhibits N-type channels in visceral afferents to reduce synaptic transmitter release under normoxic and chronic intermittent hypoxic conditions. J Neurophysiol. 2009;101:2270–2278. doi: 10.1152/jn.91304.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli A, Perin P, Martini M, Ciubotaru CD, Prigioni I, Valli P, Rossi ML, Mammano F. Presynaptic calcium stores modulate afferent release in vestibular hair cells. J Neurosci. 2003;23:6894–6903. doi: 10.1523/JNEUROSCI.23-17-06894.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YL, Zhou LJ, Hu NW, Xu JT, Wu CY, Zhang T, Li YY, Liu XG. Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52:708–715. doi: 10.1016/j.neuropharm.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Machado Rocha FC, Stéfano SC, De Cássia Haiek R, Rosa Oliveira LM, Da Silveira DX. Therapeutic use of Cannabis sativa on chemotherapy-induced nausea and vomiting among cancer patients: systematic review and meta-analysis. Eur J Cancer Care (Engl) 2008;17:431–443. doi: 10.1111/j.1365-2354.2008.00917.x. [DOI] [PubMed] [Google Scholar]
- Maenhaut N, Van de Voorde J. Regulation of vascular tone by adipocytes. BMC Med. 2011;9:25. doi: 10.1186/1741-7015-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal DH, Hermann GE, Rogers RC. Vagal afferent stimulation activates astrocytes in the nucleus of the solitary tract via AMPA receptors: evidence of an atypical neural-glial interaction in the brainstem. J Neurosci. 2011;31:14037–14045. doi: 10.1523/JNEUROSCI.2855-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougall SJ, Peters JH, Andresen MC. Convergence of cranial visceral afferents within the solitary tract nucleus. J Neurosci. 2009;29:12886–12895. doi: 10.1523/JNEUROSCI.3491-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W, Zadina JE, Harlan RE, Weber JT, Banks WA, Kastin AJ. Tumor necrosis factor-alpha: a neuromodulator in the CNS. Neurosci Biobehav Rev. 1997;21:603–613. doi: 10.1016/s0149-7634(96)00047-4. [DOI] [PubMed] [Google Scholar]
- Park KM, Bowers WJ. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal. 2010;22:977–983. doi: 10.1016/j.cellsig.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker LA, Rock EM, Limebeer CL. Regulation of nausea and vomiting by cannabinoids. Br J Pharmacol. 2011;163:1411–1422. doi: 10.1111/j.1476-5381.2010.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering M, Cumiskey D, O'Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–670. doi: 10.1113/expphysiol.2005.030734. [DOI] [PubMed] [Google Scholar]
- Pogrebniak HW, Matthews W, Pass HI. Chemotherapy amplifies production of tumor necrosis factor. Surgery. 1991;110:231–237. [PubMed] [Google Scholar]
- Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH. TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology. 2002;42:93–106. doi: 10.1016/s0028-3908(01)00163-0. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Mechanisms of action of CCK to activate central vagal afferent terminals. Peptides. 2008;29:1716–1725. doi: 10.1016/j.peptides.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, Hermann GE. Brainstem control of gastric function. In: Johnson LR, editor. Physiology of the gastrointestinal tract. Ed 4. Burlington, MA: Elsevier Academic; 2012. pp. 861–892. [Google Scholar]
- Rogers RC, McTigue DM, Hermann GE. Vagovagal reflex control of digestion: afferent modulation by neural and “endoneurocrine” factors. Am J Physiol. 1995;268:G1–G10. doi: 10.1152/ajpgi.1995.268.1.G1. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Nasse JS, Hermann GE. Live-cell imaging methods for the study of vagal afferents within the nucleus of the solitary tract. J Neurosci Methods. 2006a;150:47–58. doi: 10.1016/j.jneumeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Van Meter MJ, Hermann GE. Tumor necrosis factor potentiates central vagal afferent signaling by modulating ryanodine channels. J Neurosci. 2006b;26:12642–12646. doi: 10.1523/JNEUROSCI.3530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin NE. Cannabinoids in the treatment of chemotherapy-induced nausea and vomiting: beyond prevention of acute emesis. J Support Oncol. 2007;5(Suppl 3):1–9. [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed AA, Rattansingh A, Furtado SD. Current perspectives on the management of gastroparesis. J Postgrad Med. 2005;51:54–60. [PubMed] [Google Scholar]
- Thaler A, Gupta A, Cohen SP. Cannabinoids for pain management. Adv Psychosom Med. 2011;30:125–138. doi: 10.1159/000324070. [DOI] [PubMed] [Google Scholar]
- Walker JM, Hohmann AG. Cannabinoid mechanisms of pain suppression. Handb Exp Pharmacol. 2005;168:509–554. doi: 10.1007/3-540-26573-2_17. [DOI] [PubMed] [Google Scholar]
- Youn DH, Wang H, Jeong SJ. Exogenous tumor necrosis factor-alpha rapidly alters synaptic and sensory transmission in the adult rat spinal cord dorsal horn. J Neurosci Res. 2008;86:2867–2875. doi: 10.1002/jnr.21726. [DOI] [PubMed] [Google Scholar]
- Zhuang SY, Bridges D, Grigorenko E, McCloud S, Boon A, Hampson RE, Deadwyler SA. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology. 2005;48:1086–1096. doi: 10.1016/j.neuropharm.2005.01.005. [DOI] [PubMed] [Google Scholar]