

Abstract
Ligand transport through myoglobin (Mb) has been observed by using optically heterodyne-detected transient grating spectroscopy. Experimental implementation using diffractive optics has provided unprecedented sensitivity for the study of protein motions by enabling the passive phase locking of the four beams that constitute the experiment, and an unambiguous separation of the Real and Imaginary parts of the signal. Ligand photodissociation of carboxymyoglobin (MbCO) induces a sequence of events involving the relaxation of the protein structure to accommodate ligand escape. These motions show up in the Real part of the signal. The ligand (CO) transport process involves an initial, small amplitude, change in volume, reflecting the transit time of the ligand through the protein, followed by a significantly larger volume change with ligand escape to the surrounding water. The latter process is well described by a single exponential process of 725 ± 15 ns at room temperature. The overall dynamics provide a distinctive signature that can be understood in the context of segmental protein fluctuations that aid ligand escape via a few specific cavities, and they suggest the existence of discrete escape pathways.
The efficient transport of ligands into and out of the heme protein myoglobin (Mb) has been the subject of many experimental (1–7) and theoretical (8–11) investigations. While ligands may be thermodynamically driven to enter proteins by changes in the free energy with changes in ligand concentration, the mechanism of ligand transport through the protein to the active site with respect to the observed kinetics is not at all well understood. For example, from x-ray crystallography measurements (12, 13) it can be shown that the barrier to ligand entry to the protein would be very significant (>420 kJ/mol) (9) if the atoms were confined to thermal fluctuations about the equilibrium structure of deoxymyoglobin (deoxyMb). The barrier is high enough that it would strongly affect the reaction rate. In effect, the ligand could not enter or leave Mb on any reasonable time scale. This statement pertains only to the condition that the atoms remain close to the equilibrium structures of the deoxy state in the case of ligand entry or oxy tertiary structure in the case of ligand escape. Clearly, the ligand moves readily between the exterior and interior of the protein. Thus, there must be a dynamical pathway that connects the oxy and deoxy relaxed structures that permits shuttling of the ligand to and from the active site.
The mechanism by which these fluctuations are correlated to allow ligand diffusion to the active site is an important fundamental issue. Proteins are highly associated, glass-like, systems. How these structures maintain sufficient rigidity to retain structural integrity and function yet allow for fluid-like properties with respect to ligand access to active sites is an intriguing question. In this regard, it has been established that proteins sample many different conformational substates while undergoing relaxation (14–19). Furthermore, a growing body of evidence shows that the escape of a ligand from Mb is guided by a few specific internal cavities. Specifically, the x-ray crystallographic studies of photoinduced intermediates of MbCO trapped by either cryogenic (3) or time-resolved (20) methods observe only a few CO occupation sites that coincide with previously identified xenon pockets (3, 7) within the protein. Fluctuations in the global protein structure direct the ligand migration by acting to open and close these cavities. If this mechanism is correct, the time scale for thermally sampling a dynamic pathway of void spaces should provide a characteristic signature for a hopping process. This information cannot be retrieved directly from studies of protein relaxation. The various probe transitions used in this context are sensitive primarily to the local environment and report with various degrees of sensitivity to all relaxation pathways. Only a small subset of the total conformational distribution will map onto the required ligand diffusion pathway. A direct measurement of ligand motion through the protein is essential to resolve the above issues. Providing the ligand transport through the protein can be distinguished, the concept of a few, discrete, fluctuating cavities makes the important prediction that there should be a dramatic reduction in the number of conformational states that assist the ligand transport. The dynamics should be distinct from the complete conformational relaxation processes and presumably exhibit a more simplified response reflecting the discrete nature of the ligand hopping process.
The structural changes involved in ligand motion through the protein necessarily involve changes in the protein volume (V), or the development of material strain (ΔV/V), i.e., the protein has to deform to accommodate the ligand. In this regard, Phase grating spectroscopy provides an observable that is sensitive to the global volume changes of the protein and is therefore a unique probe of the dynamic coupling between the ligand and protein. Specifically, mass displacements show up as changes in the index of refraction or phase component of a photoinduced grating. Previous studies exploiting this effect focused on the tertiary relaxation processes of Mb that are convolved to the ligand dissociation coordinate (21). To attain better sensitivity, we have implemented heterodyne detection of the phase grating signal that amplifies the signal-to-noise ratio by more than two orders of magnitude over previous related work and enables the separation of Real (phase) and Imaginary (amplitude) parts of the diffracted signal. This advance in spectroscopic method has made it possible to fully resolve ligand escape from a protein in real time. A report of CO escape, based on transient IR spectra at fixed delays, has been made and aids in this assignment (1). The most singular feature of the data is that the ligand escape is characterized by a surprisingly simple functional form. This observation provides evidence that is consistent with the view of cavity-directed ligand migration, and it further suggests that there are a very limited number of escape pathways through the protein, with potentially one dominant pathway.
Materials and Methods
A schematic of the experiment is shown in Fig. 1 and has previously been discussed in detail (21–23). Briefly, the excitation pulses (527 nm, 25 ns full width at half maximum) and probe (1.064 μm continuous wave) beams were coincident and impinged normally on the diffractive optic through one of three gratings to give grating fringe spacings of Λ = 30 μm, 15 μm, and 7.5 μm at the sample. A mask blocked all but the four ±1 diffracted orders to form the two excitation beams, the probe beam and the reference beam (the other diffracted orders and the mask are not shown in the diagram). The resultant IR beams were automatically at the appropriate respective angles for Bragg diffraction (24) upon refocusing at the sample cell by spherical and folding mirrors.
Figure 1.

Optically heterodyned experimental setup. The Inset shows the beam geometry. The dashed black lines are the diffracted signals that are mixed at their respective detectors with the undiffracted probe (solid black) that serves as a reference.
The signal was detected by using a combination of two photodiodes housed in an enclosure with an adjustable iris for the inputs to minimize scatter. The ratio of reference to signal intensity was controlled by the combination of waveplate 1 and polarizer 1, and the heterodyned signal was measured at detector 1 (Thorlabs PDA255; Newton, NJ). A differential detection method was used to further improve the signal-to-noise ratio: detector 2 measured only the reference laser noise for subtraction from the heterodyned signal at detector 1. Unambiguous separation of the Re (Re) and Imaginary (Im) parts of the signal was accomplished by using the two cover slips as described previously (22). The signals were recorded with a 500-MHz digitizer (Textronix 520D; Beaverton, OR).
For measurements up to t = 400 μs, the sample flow rate ensured a new and effectively stationary sample volume for each laser shot. A stopped-flow configuration was used to measure the dynamics occurring past 400 μs. A variation on the differential detection method was used where both +1 and −1 orders of diffraction were detected. The undiffracted beams served as local oscillators for heterodyne detection at detector 1 and detector 2. Because of the sign difference between the relative phase of signal and local oscillator fields at the two detectors, the Re component was automatically isolated by subtraction of the signals at detectors 1 and 2. This method will be described in detail elsewhere. This procedure was used in the stopped-flow configuration because the low repetition rate and reduced signal-to-noise ratio made it difficult to assign the phase component. Experiments were done with sample bleaches of 10%, 5%, and 2.5%, respectively, to verify the absence of multiphoton absorption.
The preparation of the horse heart MbCO and deoxyMb is standard and has been described elsewhere (22). MbCO (deoxyMb) samples were kept under a CO (N2) environment while the experiment was in progress.
Results and Discussion
The ability of this experiment to clearly separate the Re and Im parts of the heterodyne signal is demonstrated for deoxyMb in Fig. 2. The Re part of the grating signal arises from changes in the index of refraction, from processes such as absorption, heat deposition, and volume changes of the protein itself (22). DeoxyMb has an extremely short excited state lifetime (≪1 ps). All of the relevant photophysics involve thermalization of the photon energy on picosecond time scales such that deoxyMb serves as a control for a purely thermal process (25). Thus, the Re part of the signal shown in Fig. 2 decays exponentially with the expected decay due to thermal diffusion diminishing the grating contrast. The Im signal is sensitive to absorption differences between the initial and final protein states, which for deoxyMb is zero on this time scale, as expected.
Figure 2.

DeoxyMb phase dependence for Λ = 7.5 μm. By varying the relative phase Δφ between signal and reference beams, heterodyne detection allows separation of the Re and Im parts of the signal.
Ligand Dynamics.
In the case of MbCO, optical absorption impulsively induces ligand dissociation that in turn triggers the protein relaxation dynamics that are convolved to the bond dissociation coordinate and ligand escape from the protein. Fig. 3 illustrates the wealth of information contained in the grating components of the MbCO response at 20°C for the full range of time scales studied in this experiment. The Im part is simple and shows the same trends as previous transient absorption studies (26), i.e., essentially a constant offset that decays with CO recombination. In contrast, the Re part of the signal is dramatically different. At early times, the Re signal has a positive amplitude that is consistent with the protein contraction (ΔnMb) and population contribution (Δnex) observed in earlier experiments. A small peak and decay is observed within the first 20 ns that is consistent with the induced structural birefringence and rotational diffusion recently discovered by Goodno et al. (21). Following the birefringence is a barely perceptible plateau region, after which the signal changes sign with a mono-exponential expansion process of 725 ± 15 ns. This latter feature, especially the sign change that is clearly observable with heterodyne detection (e.g., Fig. 3A for t > 500 ns), has important consequences. Previous studies of MbCO relaxation on these time scales have observed small shifts in absorption that exhibit monotonic relaxation toward the fully relaxed spectra that is described by stretched exponential behavior (27). This grating decay component cannot be described by a stretched exponential (see below), and most importantly, the change in sign unambiguously assigns this response to a different process than that driven by the initial bond dissociation—i.e., it is not a monotonic extension of the initial process. This secondary perturbation is ligand escape, as will be elaborated on below. Slower relaxation dynamics that are dominated by thermal diffusion (Δnth) are observable in the time windows t < 10 μs in Fig. 3 B and C. The CO diffusion, and the bimolecular recombination and protein diffusion are even slower and may be discerned for t < 400 μs (Fig. 3D) and t < 10 ms (Fig. 3E), respectively.
Figure 3.

Re (black) and Im (gray) data at 20°C for all timescales. E shows the full time evolution of the Re part of the signal on a log(time) axis. A 3-point smoothing was performed because the data sets A–D were recorded at different sampling rates.
Several control studies facilitated the identification of these contributions to the signal and the isolation of the protein dynamics. Transport processes (such as thermal diffusion and CO and protein diffusion) were identified by means of a fringe spacing dependence and were found to agree well with values expected from known diffusivity constants. Temperature-dependence studies were used to probe the sample at the zero thermal expansion point of water, at which temperature the deposition of thermal energy into the aqueous lattice would not effect a change in sample density (Δnth = 0). These results are shown in Fig. 4. At −1.5°C the thermal component was measured to constitute at most 3% of the total signal (from deoxyMb control), such that this temperature provides a clear observation of the isolated protein dynamics. The elimination of the thermal component from the MbCO dynamics at this temperature is further demonstrated in Fig. 5, where a fringe spacing dependence is shown for MbCO at −1.5°C in comparison to the pure thermal grating dependence at room temperature. The negligible amount of thermal component produces identical data for all three Λ before CO and protein diffusive processes become apparent. After the fast initial rise (protein contraction) in the T = −1.5°C signal is the decay of the protein anisotropy, and the geminate recombination of 4% of the dissociated ligands. The signal at t < 10 μs then takes the form of a delayed-onset exponential—i.e., there is a relatively flat plateau region (region I in Fig. 6), followed by an exponential decay (region II). This unusual dynamical signature is present at all temperatures. In comparing the fringe spacing of MbCO to that of deoxyMb, it is clear that the thermal contribution is negligible at −1.5°C. Only past 7 μs (Fig. 5B) do the data start to show a slight fringe spacing dependence that is too slow to be due to thermal diffusion. An additional transport component identified in the fringe spacing dependence is likewise too fast to be due to translation diffusion of the protein. The fringe spacing dependence was used to determine a diffusion constant (28) of 1.8 ± 0.2 × 10−5 cm2/s at 20°C, which is in reasonable agreement with that expected for CO and has a positive volume contribution also consistent with CO solvation (29). On the basis of the magnitude of the observed diffusion constant, this component can be definitely assigned to solvated CO and is measured to be ≈35% of the initial signal amplitude.
Figure 4.

The 7.5-μm temperature dependent data with the following temperatures from bottom to top: 20°C, 15°C, 10°C, 5°C, 0°C, and −1.5°C. The suppression of the thermal feature in going from room temperature to −1.5°C is most apparent in the 1− to 10-μs window. In addition, the relatively flat feature of the data, clearly visible at T = −1.5°C from t = 0 to t ≈ 400 ns becomes shorter in going to higher temperatures.
Figure 5.

(A) MbCO data at −1.5°C for all Λ. Note that the dynamics are independent of Λ until 7 μs. The Inset (B) shows a magnified view of the dynamics after 7 μs (grayscale for Λ as in C), where the CO diffusion effects begin to become apparent. (C) DeoxyMb data at room temperature for all Λ, indicating the strong dependence of the thermal phase grating signal on fringe spacing on this same time scale.
Figure 6.

(A) Re signal showing the plateau region for t < 400 ns at −1.5°C and 7.5-μm fringe spacing. (B) Fits and data for t < 10 μs and 7.5 μm at the following temperatures (from bottom to top): 20°C, 15°C, 10°C, 5°C, 0°C, −1.5°C.
The interpretation of the delayed-onset exponential was aided by a careful examination of its temperature dependence. The exponential decay time, which is 725 ns at 20°C, slows to 2.86 μs at −1.5°C, while the delay of the onset goes from 50 ns at 20°C to 390 ns at −1.5°C, as can be seen in Fig. 4. These data were found to be well described by a four-state model for ligand escape proposed by Frauenfelder and co-workers in ref. 15. This model emerged from flash-photolysis studies of MbCO rebinding rates for a wide range of viscosities and temperatures. The various dependencies suggested that the CO was rebinding from four distinct sites as characterized by fairly well defined barriers to the rebinding, with the largest barrier assigned to solvent-separated CO. The results were modeled with a modified Kramers equation (30). The CO ligand escape was proposed to follow
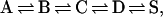 |
where A is the bound state, B is the CO unbound and in the heme pocket, C and D are two intermediate cavities, and S represents CO escape to the solvent. The passage between states is either opened or closed to the CO, with kij denoting the rate coefficient for the transition from state i to state j. The effect of the protein on the effective solvent viscosity was included phenomenologically in the Kramers equation through a parameter κ such that
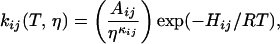 |
1 |
where Hij are the barrier heights, T is thermodynamic temperature, R is the molar gas constant, and Aij/ηκij describes the interaction of the protein with the solvent.
This earlier work was not able to directly follow the CO escape from the protein, and the effect was most discernable at low temperatures and relatively high viscosities. The present work provides a rigorous test of the primary features of the four-state model for ligand escape. A fitting routine that incorporated this model for ligand behavior and the known components of the Re signal was used to describe the data as a function of temperature, viscosity, and fringe spacing. Specifically, the changes in index associated with movement of the ligand from C → D (ΔnCD) and D → S (ΔnDS) were fit to the data. With sufficiently high time resolution, the total protein signal is described as ΔnMb = ΔnAB + ΔnBC + ΔnCD + ΔnDS. However, ΔnAB and ΔnBC were too fast for the time resolution of this experiment [expected time constants < 2 ps and 150 ps for aqueous solution at 20°C (15), respectively] and contribute only to the t = 0 protein contraction of the data. The fits to the data were accomplished by using a Levenberg–Marquardt fitting algorithm in ORIGIN 6.0 (Microcal Software, Northampton, MA).
The values for τij (= 1/kij) obtained with this algorithm for all temperatures were in excellent agreement with the rate constants derived from the model of Beece et al. (15). At 20°C, the average τCD = 49 ns ± 10 ns, and the average τDS = 725 ns ± 15 ns. These results should be compared with the expected time constants of τCD = 42 ns, with an amplitude of −0.131 and τDS = 420 ns, with an amplitude of 0.92, using the four-state model at this temperature and viscosity. Note that the amplitudes are quoted as the proportion of the initial protein contraction. The activation barriers computed over the 21.5°C temperature range of this experiment were HCD = 27 ± 6 kJ/mol [κCD = 0.5 (15)] and HDS = 31 ± 1 kJ/mol [κDS = 0.6 (15)]. All of the relevant time constants over the temperature range shown in Fig. 6 are within a factor of 2 of the calculated time constants for CO motion coupling sites C and D with the solvent from ref. 15. The barrier heights are also in good agreement, albeit over a limited temperature range. Given the direct access to the CO motion through the protein in the present experimental studies, this degree of agreement is exceptional, as the prior work was inferred from a distribution of rebinding rates—rather than directly observing the CO escape. The model correctly predicts trends over an exhaustive range of temperatures and viscosity. It would seem to have the essential components correctly described.
It should be noted that the exponential decay component assigned to CO escape to the solvent is in excellent agreement with the earlier photoacoustic studies of Peters and his colleagues (τ ≈ 700 ns; refs. 31–33), previous work from this lab (22), and the recent grating studies of Terazima and his colleagues (also ≈700 ns; ref. 34). The use of heterodyne detection and extended dynamic range has enabled the resolution of the CO motion through the protein, notably the distinctive plateau region in the CO escape statistics. This distinctive dynamical feature can be attributed to the CO transport through the protein. Although it is impossible to rule out volume changes caused by the migration of water into the protein, the volume change associated with CO escape to the solvent is quite significant (35% of the signal amplitude), and would have been clearly resolvable during this relaxation phase. The good agreement with the four-state model (15) and work of Anfinrud and his colleagues (1), who found that the CO had escaped to the aqueous phase within 3 μs, support this assignment.
The CO motion represents a significant perturbation to the protein structure. The fairly simple protein response to this motion could be considered to be contrary to concepts of stretched exponential relaxation processes accompanying protein relaxation. The data were therefore fit to this functional form [exp(−[t/τ]β)]. The results are depicted in Fig. 7, which shows fits of the −1.5°C data with the two single exponentials vs. the stretched exponential behavior. The much longer time constant associated with ligand diffusion and rebinding produces an essentially constant offset on this time scale, and is included in the analysis. The superior fit of the biexponential description (Inset A) is obvious from this comparison. Inset B of Fig. 7 shows the residual plot for the stretched exponential using previously reported β = 0.6 (35). In all cases, using β < 1 gave poor fits, with residuals comparable to those shown for a direct comparison. Letting β and τ vary as independent parameters gave best fits with β > 1 and still larger residuals than a two-step relaxation process (rise and decay). No stretched exponential process correctly captures the initial part of the signal. It is apparent that the protein response is not a simple monotonic relaxation from the initial perturbation of ligand dissociation, but rather involves an abrupt relaxation step that changes sign and reflects the CO escape.
Figure 7.

Comparison of fits to the data with a stretched exponential or exponential relaxation processes. (A) Residuals of the fit to the model of ref. 15 (solid line). (B) Residuals of the fit to the data by using a stretched exponential of β = 0.6, optimized to give the best fit (τ = 5.9 μs).
This comparison reflects the difference in the observable of this work in relation to previous studies that observe stretched exponential behavior. It can be concluded that the protein volume changes convolved to the ligand motion are dominated by the activation barriers to the CO sampling process. The protein fluctuations supporting the CO motion are dominated by higher-frequency components than the slow relaxation processes observed in other experiments. In general, experiments reporting stretched exponential relaxation processes involve a probe transition that is most sensitive to changes in its local environment or fine-grain features in the immediate vicinity of the heme. To create void spaces large enough to accommodate the ligand, the protein must undergo more global motions involving highly correlated displacements. In this respect the grating observable is a direct probe of these global motions. The very distinctive two-tier response function observed demonstrates that a discrete number of cavities are involved in ligand transport out of the protein. As a corollary statement, this effect illustrates a pronounced hierarchy in the conformational substates that satisfy this condition.
The most distinctive feature of the data is the magnitude of the protein volume changes associated with the different steps in the CO transport process. The CO migration through the protein (region I) causes only very small changes in protein volume. In contrast, the dynamical feature associated with CO escape (region II) creates nearly an order of magnitude larger effective change in volume in the protein. The larger volume change of the protein with this step has also been noted previously (22, 34), and it is this significant feature that highlights the dramatic two-tier nature of the protein response. The barrier to ligand escape from the protein involves reorganization of both the protein and the surrounding water to accommodate the ligand. The fact that this feature is essentially exponential indicates that either a single channel or at least a dominant channel directs ligand escape and entry into the protein. Because the protein rearrangements contribute significantly to the barrier (9), if there were a multitude of pathways for escape, a distribution of exponentials should have been observed instead of this highly simplified response.
The step-like change in the protein volume with ligand escape is intriguing in another context. By what mechanism could the protein deform to give an abrupt change in volume with ligand exit? This issue is shown schematically in Fig. 8. If the protein behaved as a highly associated fluid or homogeneous glass, the motion of the ligand should be accompanied by a distribution of volume changes and arrival times for the CO in the solvent as the system evolves between the two end points (ligand at the protein active site and ligand free in the solvent). The dynamics do not fit such a diffusive process. Previously, the term “protein quake” was coined to provide a simple mechanical model of protein relaxation (36). The relaxation was envisaged to involve localized motions, relieving stress, in analogy to an earthquake. Localized motions require a definition of relevant length scales; otherwise there is no distinction between solid-like or liquid-like behavior. Within this analogy, the important motions will depend on the nature of the relaxation coordinate. If motions involving one amino acid were sufficient to accommodate the CO motion, the ligand would explore the entire protein and the process would be diffusive. The observed dynamics indicate that this is not the case. The CO motion involves a correlation of displacements over several residues. At the opposite extreme, the entire protein cannot respond uniformly if the process is to be directed and discontinuous in nature (discrete intermediates) as observed. However, if the thermal sampling involves segmental displacements over length scales at least comparable to the exit pathway, then there could be an abrupt change in protein volume with ligand escape—truly a protein quake. This process is shown schematically in Fig. 8, in which the CO volume element keeps the relevant segmental domains apart until it escapes. The previous use of “protein quakes” (36) to describe relief of stress appears more relevant than previously envisaged, as the current observations strongly implicate segmental motions involving correlations over a significant fraction of the relevant pathway.
Figure 8.

Segmental vs. localized protein motions. The “fluid-like” case schematically depicts the random diffusive motion of the ligand if the protein behaved as a highly associated fluid in which localized fluctuations permitted ligand access to the entire protein. There would be a distribution of volume changes and escape times characteristic of a diffusive process. The lower, segmental motions depict fluctuations involving correlations over the length scales required to create a contiguous path to void spaces in the protein and the solvent for ligand escape. The dynamics in this case would reflect a thermally activated hopping process with discrete intermediates that would depend on the number of accessible void spaces in the protein.
Protein Energetics and Strain.
A comprehensive understanding of the protein's functionally important dynamics requires measuring the associated energy changes as well as the dynamics. The amount of energy that is stored in or released by the protein provides a measure of the importance of the corresponding dynamics. The processes that release the most energy define the dominant mechanisms responsible for protein relaxation toward its equilibrium state.
The phase grating observable is sensitive to the release of energy by the protein through the thermal grating component. The temperature dependence enables a separation of the thermal grating components from the protein contribution to the index of refraction changes and thus, a direct determination of the energetics (25, 37). In this regard, deoxyMb is used to calibrate the signal amplitude to dissipated energy. These experiments have measured the internal energy of the dissociated MbCO to be 68 ± 11 kJ/mol by 40 ns, with a small release of energy, 8 ± 4 kJ/mol upon CO solvation, for a total internal energy of 60 ± 12 kJ/mol for t > 1 μs. The volume changes associated with the different stages of CO motion through the protein were found to be ΔVCD = −1.4 ± 0.46 ml/mol and ΔVDS = +9.29 ± 0.52 ml/mol. The energetics agree well with recent studies combining photoacoustic and thermal grating measurements (34), and the volume changes show the same trends. The observations support the previous conclusions of the earlier picosecond studies that found that more than 50% of the protein relaxation energy is released during the inertial collective phase of the protein relaxation bond-breaking event and the magnitude of the initial changes in protein volume (21, 37).
Summary and Conclusion
The implementation of diffractive optics-based nonlinear spectroscopy has provided an unprecedented level of sensitivity for the study of protein dynamics and has made it possible to attain fully resolved real-time observations of ligand motion through proteins. The observed dynamics for the photodissociation of CO from MbCO can be fit extremely well with two single exponentials, one related to CO motion within the protein (ΔnCD) and the other to ligand escape (ΔnDS). The single-exponential nature of the escape for all temperatures signifies that the ligand is escaping to the solvent over an effective single, or at least dominant, activation barrier. Likewise, the fact that the escape occurs abruptly (significantly larger volume change) after a “quiet” period indicates the absence of a distribution of arrival times of the CO to the protein surface. The fit of the four-barrier model of Beece et al. (15) to these two unusual data signatures strongly suggests that the ligand follows a small subset of escape routes, and does so via specific internal cavities in the protein. Given the single-exponential character of the solvent escape step, the ligand motion samples either a dominant pathway or self-similar paths with respect to activation barriers.
The apparently abrupt change in protein volume and simple kinetics provide further support for the role of collective modes in defining the protein's functional response. If the relevant motions involve segmental, as opposed to localized, motions, the escape of CO would lead to a singular change in protein volume, rather than a distribution of volume changes that would depend on the CO position. The whole process could be likened to a quake in which the escape of CO out of the protein leads to a segmental annealing of the stress created by this perturbation. The concept of collective or segmental motions has also been implicated in the initial relaxation convolved to the Fe-ligation coordinate (37). The energetics have now been followed out to much longer time scales and have extended the dynamic range from the previous picosecond to 10-ns window out to the microsecond range. Based on a Fe—CO bond energy of 105 kJ/mol, the relaxation energetics are dominated by the inertial collective phase, i.e., ≈25 kJ/mol on picosecond time scales (37), less than 13 kJ/mol associated with slower relaxation processes (10- to 400-ns window), and 8 kJ/mol involved in ligand escape. In both the bond breaking and ligand transport aspects of functionally relevant protein motions, the primary issue is the enormous conformational phase space accessible to the system. It appears that the strongly damped collective modes of the protein are important in directing both the initial relaxation process and processes on longer time scales and act to effectively coarse-grain the sampling process.
These observations have some interesting consequences with respect to understanding protein function. Mb is often used as a model for understanding the tertiary processes that are involved in the classic molecular cooperativity response exhibited by hemoglobin. In this problem, allosteric regulation of ligand binding seems to depend only on the state of ligation of the different protein subunits (12). This state of ligation is communicated through the protein–protein contact and represents a spatially averaged coordinate comparable to the protein radius. The present study gives a direct observation of how complex systems such as proteins are able to elicit such simplistic functions, over the correct length scale, irrespective of their enormously diverse conformational distributions. In the context of the present observations, the similarity in dynamics for CO escape and the R to T quaternary transition for the molecular cooperativity of hemoglobin (26) should not go unnoticed.
Acknowledgments
We thank M. Sakakura, S. Yamaguchi, N. Hirota, and M. Terazima for preprints of their work. This work was supported by the Natural Sciences and Engineering Research Council of Canada.
Abbreviations
- Mb
myoglobin
- Re
Real
- Im
Imaginary
References
- 1.Jackson T A, Lim M, Anfinrud P A. Seventh International Conference on Time-Resolved Vibrational Spectroscopy. 1995. comps. Dyer, R. B., Martinez, M. A. D., Shreve, A. & Woodruff, W. H. (Los Alamos Natl. Lab., Los Alamos, NM), Report LA-13290-C, pp. 9–13. [Google Scholar]
- 2.Chu K, Vojtchovsky J, McMahon B H, Sweet R M, Berendzen J, Schlichting I. Nature (London) 2000;403:921–923. doi: 10.1038/35002641. [DOI] [PubMed] [Google Scholar]
- 3.Brunori M, Vallone B, Cutruzzola F, Travaglini-Allocatelli C, Berendzen J, Chu K, Sweet R, Schlichting I. Proc Natl Acad Sci USA. 2000;97:2058–2063. doi: 10.1073/pnas.040459697. . (First Published February 11, 2000; 10.1073/pnas.040459697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunori M. Biophys J. 1999;76:1259–1269. doi: 10.1016/S0006-3495(99)77289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunori M. Biophys Chem. 1999;86:221–230. doi: 10.1016/s0301-4622(00)00142-3. [DOI] [PubMed] [Google Scholar]
- 6.Carlson M L, Regan R M, Gibson Q H. Biochemistry. 1996;35:1125–1136. doi: 10.1021/bi951767k. [DOI] [PubMed] [Google Scholar]
- 7.Scott E E, Gibson Q H. Biochemistry. 1997;36:11909–11917. doi: 10.1021/bi970719s. [DOI] [PubMed] [Google Scholar]
- 8.Agmon N, Hopfield J J. J Chem Phys. 1983;79:2042–2053. [Google Scholar]
- 9.Case D A, Karplus M. J Mol Biol. 1979;132:343–368. doi: 10.1016/0022-2836(79)90265-1. [DOI] [PubMed] [Google Scholar]
- 10.Sagnella D E, Straub J E, Jackson T A, Lim M, Anfinrud P A. Proc Natl Acad Sci USA. 1999;96:14324–14329. doi: 10.1073/pnas.96.25.14324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elber R, Karplus M. J Am Chem Soc. 1990;112:9161–9175. [Google Scholar]
- 12.Perutz M F. Nature (London) 1970;228:726–739. doi: 10.1038/228726a0. [DOI] [PubMed] [Google Scholar]
- 13.Frauenfelder H, McMahon B. Proc Natl Acad Sci USA. 1997;95:4795–4797. doi: 10.1073/pnas.95.9.4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Austin R H, Beeson K W, Eisenstein L, Frauenfelder H, Gunsalus I C. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 15.Beece D, Eisenstein L, Frauenfelder H, Good D, Marden M C, Reinisch L, Reynolds A H, Sorenson L B, Yue K T. Biochemistry. 1980;19:5147–5157. doi: 10.1021/bi00564a001. [DOI] [PubMed] [Google Scholar]
- 16.Ansari A, DiIorio E E, Dlott D D, Frauenfelder H, Iben I E T, Langer P, Roder H, Sauke T B, Shyamsunder E. Biochemistry. 1986;25:3139–3146. doi: 10.1021/bi00359a011. [DOI] [PubMed] [Google Scholar]
- 17.Ansari A, Berendzen J, Brauenstein D, Cowen B R, Frauenfelder H, Hong M K, Iben I E T, Johnson J B, Ormos P, Sauke T B, et al. Biophys Chem. 1987;26:337–355. doi: 10.1016/0301-4622(87)80034-0. [DOI] [PubMed] [Google Scholar]
- 18.Ansari A, Jones C M, Henry E R, Hofrichter J, Eaton W A. Biochemistry. 1994;33:5128–5145. doi: 10.1021/bi00183a017. [DOI] [PubMed] [Google Scholar]
- 19.Nienhaus G U, Mourant J R, Frauenfelder H. Proc Natl Acad Sci USA. 1992;89:2902–2906. doi: 10.1073/pnas.89.7.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srajer V, Teng T, Ursby T, Pradervand C, Rev Z, Adachi S, Schildkamp W, Bourgeois D, Wulff M, Moffat K. Science. 1996;274:1726–1729. doi: 10.1126/science.274.5293.1726. [DOI] [PubMed] [Google Scholar]
- 21.Goodno G D, Astinov V, Miller R J D. J Phys Chem B. 1999;103:603–607. [Google Scholar]
- 22.Dadusc G, Goodno G D, Chiu H L, Ogilvie J P, Miller R J D. Is J Chem. 1998;38:191–206. [Google Scholar]
- 23.Goodno G D, Dadusc G, Miller R J D. J Opt Soc Am B. 1998;15:1791–1794. [Google Scholar]
- 24.Rogers J A, Nelson K A. Physica B. 1996;219–220:562–564. [Google Scholar]
- 25.Richard L, Genberg L, Deak J, Chiu H-L, Miller R J D. Biochemistry. 1992;31:10703–10715. doi: 10.1021/bi00159a010. [DOI] [PubMed] [Google Scholar]
- 26.Murray L P, Hofrichter J, Henry E R, Eaton W A. Biophys Chem. 1988;29:63–76. doi: 10.1016/0301-4622(88)87025-x. [DOI] [PubMed] [Google Scholar]
- 27.Jackson T A, Lim M, Anfinrud P A. Chem Phys. 1994;180:131–140. [Google Scholar]
- 28.Terazima M. J Phys Chem A. 1999;103:7401–7407. [Google Scholar]
- 29.Moore J C, Battino R, Rettich T R, Handa Y P, Wilhelm E. J Chem Eng Data. 1982;27:22–24. [Google Scholar]
- 30.Kramers H A. Physica. 1940;7:284–304. [Google Scholar]
- 31.Westrick J A, Goodman J L, Peters K S. Biochemistry. 1987;26:8313–8318. doi: 10.1021/bi00399a043. [DOI] [PubMed] [Google Scholar]
- 32.Westrick J A, Peters K S. Biophys Chem. 1990;37:73–79. doi: 10.1016/0301-4622(90)88008-g. [DOI] [PubMed] [Google Scholar]
- 33.Norris C L, Peters K S. Biophys J. 1993;65:1660–1665. doi: 10.1016/S0006-3495(93)81223-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakakura, M., Yamaguchi, S., Hirota, N. & Terazima, M. (2001) J. Am. Chem. Soc., in press. [DOI] [PubMed]
- 35.Tian W D, Sage J T, Srajer V, Champion P M. Phys Rev Lett. 1992;63:408–411. doi: 10.1103/PhysRevLett.68.408. [DOI] [PubMed] [Google Scholar]
- 36.Ansari A, Berendzen J, Bowne S F, Frauenfelder H, Iben I E T, Sauke T B, Shyamsunder E, Young R D. Proc Natl Acad Sci USA. 1985;82:5000–5004. doi: 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller R J D. Acc Chem Res. 1994;27:145–150. [Google Scholar]