

Abstract
Honokiol possesses many pharmacological activities including anti-cancer properties. Here in, we designed and synthesized honokiol analogs that block major honokiol metabolic pathway which may enhance their effectiveness. We studied their cytotoxicity in human cancer cells and evaluate possible mechanism of cell cycle arrest. Two analogs, namely 2 and 4, showed much higher growth inhibitory activity in A549 human lung cancer cells and significant increase of cell population in the G0-G1 phase. Further elucidation of the inhibition mechanism on cell cycle showed that analogs 2 and 4 inhibit both CDK11 and cyclin B1protien levels in A549 cells.
Keywords: Honokiol analogs, Cell proliferation, CDK1, Cyclin B1, Human cancer cell
1. Introduction
Honokiol is one of the biphenolic constitute isolated from the stem bark and root of Magnolia officinalis,1 a Chinese medicine used to treat variety of diseases. It is shown to possess many pharmacological activities, including anti-inflammatory,2 anti-thrombosis,3 antioxidant effects,4 antiviral,5,6 and induce differentiation in HL-60 human leukemia cells.7 Studies have demonstrated that honokiol induces apoptosis in several cell types such as human colorectal,8 rat hepatic stellate,9 human squamous lung cancer10 and B cell chronic lymphocytic leukemia cells.11 In addition, honokiol inhibits skin tumor promotion,12 human fibrosarcoma invasiveness in vitro,13 angiogenesis and tumor growth in vivo,14 and overcomes drug resistance in multiple myeloma.15 More recently, honokiol has also been shown to act as chemopreventive agent against UVB induced skin cancer in a hairless mouse model.16 Saibokuto, a traditional Chinese remedy containing honokiol17 as major ingredient, is approved as effective treatment for asthma and bronchitis by Japanese Health Ministry.
Honokiol is reported to induce cell cycle arrest at G0/G1 phase in human prostate cancer cell and vascular muscle smooth cells.18,19 It also suppresses osteoclastogenesis and invasion through modulation of NFκB pathway.20 Honokiol has been shown to interact with cell cycle regulatory proteins such as cyclin-dependent kinases (CDKs) and cyclins;16,20 it down-regulates the levels of cyclins D1, D2, E and associated CDK2, CDK4 and CDK6. In the last decade, there is growing evidence to challenge the classical view of cell cycle regulation.21 Recent investigation on genetic and RNA interference in mammalian cells have reported that CDK2 and CDK4 are not necessary for cell cycle progression, and CDK1 as the only nonredundant cell cycle driver.22,23 Several natural and synthetic small molecule inhibitors have been reported, but cell cycle studies of these molecules are not consistent with specific CDK1 inhibition.24,25 Cyclin B1 is essential for the initiation of mitosis. It accumulates in the S phase of the cell cycle and reaches the maximal level at mitosis, but is absent in G1-phase cells.26 However, recent reports indicated that cyclin B1 was expressed in the G1 phase in several synchronized and asynchronously growing cancer cells.27–31 Furthermore, over expression of cyclin B1has been reported in tumor cells,32–35 thus targeting cyclin B1and its complex partner, namely CDK1 is a promising therapeutic approach for controlling cancer.
There are reports of structure-activity relationship (SAR) studies on honokiol; allyl substituted biphenyl skeleton seems to be common element in these studies.6,14,36 However, the end points of these studies vary widely; therefore no concrete conclusion could be drawn. We were interested in designing novel analogs of honokiol, which could follow a similar mechanism as honokiol but are more potent, by careful manipulation of the functional groups. Our hypothesis for SAR study was that the hydroxyl groups on the honokiol biphenyl skeleton may be subjected to metabolic oxidation by phase I enzymes,37,38 and subsequently excrete out of the system, therefore might reduce its effectiveness. Glucuronidation and sulfation of the free hydroxyl groups have been shown to be the main metabolic pathways for honokiol in rat and human liver leading to a faster excretion and reduced half-life.39 Therefore, we synthesized several honokiol analogs (Figure 1), which have their 4′-hydoxyl group replaced with methoxy group in A-ring, and the substitution pattern of allyl and hydroxyl groups in Bring altered, in order to study their cytotoxicity and mechanism of action.
Figure 1.

Structure of honokiol analogs
In this study, we explore the SAR of honokiol analogs for their cytotoxicity in human lung (A549), melanoma (UACC903) and colon (HT-29) cancer cell lines and determine the underlying mechanism of action by evaluating the cell cycle studies of honokiol analogs 2 and 4, the two best identified analogs based on their potent inhibition of A549 human lung cancer cell growth.
2. Experimental Section
Reagents
All chemicals for the syntheses were obtained from Aldrich Chemical Co. (St. Louis, MO) and used without further purification. Thin-layer chromatography (TLC) was developed on aluminum-supported pre-coated silica gel plates (EM industries, Gibbstown, NJ). Column chromatography was performed on silica gel (70–230 mesh). NMR spectra were recorded on Bruker Avance II 500 MHz instrument and the chemical shifts are given as δ values with reference to tetramethylsilane (TMS) as internal standard. MS spectra were recorded on Applied Biosystems API 3200. Honokiol was supplied by Sigma Aldrich. Stock solution of honokiol and its analogs (final concentration, 10 mM) was prepared in DMSO, stored at −20°C, and diluted with fresh complete medium immediately before use. DMEM was from Mediatech, Inc., trypsin-EDTA solution was from Invitrogen, and fetal bovine serum was from Atlanta biological. Protease inhibitor cocktail was from BD Biosciences, propidium iodide was from Sigma. Polyclonal antibodies to cyclin B1, and cyclin-dependent kinase 1 (CDK1) were from Millipore, protease inhibitor from Thermo Scientific, USA
2.1. Synthesis of honokiol analogs
2.1.1. Syntheses of key components of Suzuki coupling reagents
4-Allylphenol (7)
4-Allylanisole (20 g, 135 mmol) was dissolved in 400 ml of CH2Cl2 and cooled by ice bath under nitrogen atmosphere, BBr3 (37.2 g, 148.5 mmol) was added through double needle. The mixture was stirred for three hours at 0 °C then was allowed to warm up to room temperature over night. The mixture was poured to ice water and organic layer separated. The aqueous layer was extracted with CH2Cl2 (100 ml × 3). The combined organic layer was dried over Na2SO4, solvent was evaporated to yield 19.2 g black oily residue. Crude compound (7.55 g) was purified by column chromatography eluted with hexanes/EtOAc = 20/1 to yield 5.04 g 4-allylphenol (14) as clear oily residue (70.9%).40 1H NMR (CDCl3) δ 7.10 (2H, dt, J=8.50Hz, J=2.0 Hz), 6.82 (2H, dt, J=8.5 Hz, J=2.0 Hz), 6.04~5.96 (1H, m), 5.19 (1H, s), 5.13~5.09 (2H, m), 3.37 (2H, d, J=6.5 Hz). 13C NMR (CDCl3) δ 153.68, 137.87, 132.41, 129.78, 115.53, 115.33, 39.35.
4-Allyl-2-bromo-phenol (8)
4-Allylphenol (7; 1.35 g; 10.06 mmol) dissolved in 300 ml of dry diethyl ether under nitrogen atmosphere was cooled with acetone-dry ice bath. To the reaction mixture, i-propyl magnesium chloride (2.0 M; 5.2 ml; 10.4 mmol) was added dropwise over 3 minutes, and mixture was stirred at −78 °C for 1 hour before 1,3-dibromo-5,5-dimethylhydantoin41 (DBDMH; 1.44 g; 5.04 mmol) was added. The mixture was stirred 20 hours at −78°C then warmed up to room temperature over 1 hour; then 10 ml of 10% NaSO3 was added to the mixture. The aqueous layer was extracted with ether (10 ml × 3), and the combined organic layer was dried over Na2S2O4; then concentrated in vacuo to yield 1.40 g yellowish oil as crude product. The residue was purified by flash column chromatography on silica gel eluted with hexanes/EtOAc = 40/1 to give 9 as colorless oil (0.98 g, 45.7%).40 1H NMR (CDCl3) δ 7.31 (2H, d, J=2.0 Hz), 7.07 (1H, dd, J=8.5 Hz, J=2.0 Hz), 6.98 (1H, d, J=8.50 Hz), 5.98-5.91 (1H, m), 5.45 (1H, s), 5.13-5.08 (2H, m), 3.33 (2H, d, J=7.0 Hz). 13C NMR (CDCl3) δ 150.56, 137.03, 133.74, 131.79, 129.39, 116.17, 115.94, 110.08, 38.96. MS (m/z, Intensity): 214 (M+1+, 25%), 113 (100%), 105 (80%).
4-Allyl-2,6-dibromophenol (9)
4-Allylphenol (7; 1.01g, 7.45 mmol) was dissolved in 25 ml of CHCl3 at room temperature, 1,3-dibromo-5,5-dimethyl-hydantoin41 (DBDMH; 2.24 g, 7.83 mmol) was added in three portion over 5 minutes. The resulting mixture was stirred for 2 hours; the solid was removed by filtration. The organic layer was evaporated in vacuo. The yellow residue was purified by flash column chromatography eluted with hexanes/EtOAc = 20/1 to give 8 as yellowish oil residue (1.1 g, 50.6%). 1H NMR (CDCl3) δ 7.29 (2H, s), 5.95-5.88 (1H, m), 5.78 (1H, s), 5.15-5.09 (2H, m), 3.36 (2H, d, J=6.5 Hz). 13C NMR (CDCl3) δ 147.66, 136.20, 134.68, 132.02, 116.88, 109.67, 38.62. MS (m/z, Intensity): 294 (M+1+, 100%), 213 (5%).
2-Allyl-4-bromophenol (10)
4-Bromophenol was converted to the corresponding allyl 4-bromophenyl ether. The allyl ether was then undergoing thermal rearrangement according procedure described by Sviridov et. al.42 1H NMR (CDCl3) δ 7.62-7.24 (2H, m), 6.72 (1H, dd, J=7.5 Hz, J=1.5 Hz), 6.03-5.97 (1H, m), 5.23-5.18 (2H, m), 4.96 (1H, s), 3.40 (2H, d, J=6.0 Hz). 13C NMR (CDCl3) δ 153.20, 135.46, 132.98, 130.59, 127.65, 117.54, 117.21, 112.92, 34.80. MS (m/z, Intensity): 214 (M+, 100%), 199 (25%), 133 (60%), 118 (32%), 105 (28%).
2-Allyl-4-bromoanisole (11)
2-Allyl-4-bromophenol (10; 1.47 g; 6.89 mmol) was mixed with potassium carbonate (6.0 g) and 30 ml of dry acetone under nitrogen atmosphere. After stirring for 15 minutes, methyl iodide (3.0 ml) was added and the resulting mixture was refluxed for 1.5 hours. The reaction mixture was filtered to remove solid, and then concentrated to give 1.55 g yellowish crude product. Further purification by column chromatography on silica gel eluted with hexanes/EtOAc = 30/1 gave 11 as light yellowish oil (1.27 g, 81.1%). 1H NMR (CDCl3) δ 7.31 (1H, dd, J=8.5 Hz, J=2.5 Hz), 7.27 (1H, d, J=2.5 Hz), 6.75 (1H, d, J=9.0 Hz), 5.98-5.93(1H, m), 5.11-5.07 (2H, m), 3.83 (3H, s), 3.37 (2H, d, J=3.5 Hz). 13C NMR (CDCl3) δ 156.38, 136.03, 132.41, 130.99, 129.88, 116.14, 112.72, 111.97, 55.61, 33.91. MS (m/z, Intensity): 229 (M+, 15%), 201 (100%), 171 (25%), 148 (22%).
4-Methoxy-3-allyl-1-phenylboronic acid (12)
2-Allyl-4-bromoanisole (2.85g, 12.55 mmol) was dissolved in 50 ml of dry THF and cooled to −78 °C under nitrogen atmosphere, n-butyl lithium (2.5M, 5.6 ml, 13.5 mmol) was added dropwise over 5 minutes. The mixture was stirred for 45 minutes, and then tri-isopropyl borate (17.96 g, 95.7 mmol) was added in one portion to the reaction mixture. The mixture was warmed up to room temperature for two hours, then 50 ml of 0.5N HCl was added. The organic layer was separated and aqueous layer was extracted with ether (50 ml × 3). The combined organic layer was dried over MgSO4 and concentrated in vacuo to give pale solid as crude product which was further purified by chromatography on silica gel and eluted with CH2Cl2/MeOH =30/1 to furnish 2.2 g of 12 as pale solid. 1H NMR (CDCl3) δ 8.13 (1H, dd, J=8.0, 1.5Hz), 8.00 (1H, d, J=1.5 Hz), 7.01 (1H, d, J=8.0 Hz), 6.15-6.07 (1H, m), 5.17-5.10 (2H, m), 3.94 (3H, s), 3.51 (2H, d, J=6.4Hz). 13C NMR (DMSO-d6) δ 159.08, 137.56, 136.25, 134.58, 126.82, 115.76, 110.13, 55.67, 34.38.
1-Ally-3-bromo-2-methoxymethoxybenzene (13)
2-Allyl-6-bromophenol was prepared according to procedure reported by Palmer et al.43 In a flask, 2-allyl-6-bromo-phenol (14.6 g) was mixed with 15 g of potassium carbonate in 250 ml of acetone under nitrogen atmosphere, and then chloromethyl methyl ether (6.0 g) was added. The mixture was refluxed overnight. After cooling to room temperature, the solid was filtered, and resulting solution was concentrated. The residue was purified by column chromatography on silica gel eluted with hexanes/EtOAc = 20/1 to yield 13 g of 1-allyl-3-bromo-2-methoxymethoxybenzene (13). 1H NMR (CDCl3) δ 7.45 (1H, dd, J=8.0 Hz, J=1.5 Hz), 7.17 (1H, dd, J=8.0 Hz, J=1.5 Hz), 6.98 (1H, t, J=8.0 Hz), 6.04-5.96 (1H, m), 5.14-5.09 (2H, m), 5.12 (2H, s), 3.67 (3H, s), 3.54 (2H, d, J=6.5 Hz). 13C NMR δ (CDCl3) 152.79, 136.52, 135.81, 131.56, 129.58, 125.65, 117.54, 116.34, 99.91, 57.81, 34.73. MS (m/z, Intensity): 258 (M+, 25%), 225 (97%), 147 (100%).
3-Allyl-2-methoxymethoxy-1-phenylboronic acid (14)
1-Allyl-3-bromo-2-methoxymethoxybenzene (13) (0.99g, 3.9 mmol) was dissolved in 10 ml of dry THF and cooled to −78 °C under nitrogen atmosphere, n-butyl lithium (2.5M, 2.2 ml, 5.5 mmol) was added dropwise over 5 minutes. The mixture was stirred for 45 minutes, and then tri-isopropyl borate (1.8 ml, 8.0 mmol) was added in one portion to the reaction mixture. The mixture was warmed up to room temperature for two hours, then 20 ml of 0.1N HCl was added. The organic layer was separated and aqueous layer was extracted with ether (25 ml × 2). The combined organic was dried over MgSO4 and concentrated in vacuo. The oily residue was purified by chromatography on silica gel eluted with hexane/EtOAc = 4/1, then EtOAc to give 0.93 g of 14 as colorless residue. 1H NMR (CDCl3) δ 7.73 (1H, dd, J=7.5, 2.0Hz), 7.35 (1H, dd, J=7.5, 2.0 Hz), 7.19 (1H, t, J=2.5 Hz), 6.04-5.96 (1H, m), 5.91 (2H, s), 5.16-5.07 (2H, m), 5.06 (2H, s), 3.57 (3H, s), 3.44 (2H, d, J=6.0 Hz). 13C NMR (DMSO-d6) δ 159.03, 137.82, 133.04, 132.04, 131.49, 123.89, 116.27, 100.13, 57.27, 34.33.
2.1.2. General procedure for the synthesis of honokiol analogs
In a 100 ml flask, 3-allyl-4-methoxyphenyl boronic acid (3.9 mmol), Cs2CO3 (11.66 mmol), the corresponding bromo-allyl-phenol (3.54 mmol), and catalyst Pd(Ph3P)4 (0.35 mmol) were mixed in 45 ml of DME and 6 ml of water. The mixture was refluxed under nitrogen overnight. Following standard workup procedure, the product was isolated by chromatography on silica gel. The purities of all the synthetic compounds were >98% according to the proton NMR analysis.
3,5′-Diallyl-2′-hydroxy-4-methoxy-1,1′-biphenyl (1)44,45
The reaction mixture was purified by column chromatography on silica gel eluted with hexanes/EtOAc = 30/1 to yield colorless oil residue. 1H NMR (CDCl3) δ 7.32 (1H, dd, J=8.2 Hz, 2.2 Hz), 7.26 (1H, d, J=2.2 Hz), 7.08 (1H, dd, J=8.13, 2.2 Hz), 7.06 (1H, d, J=2.1 Hz), 6.99 (1H, d, J=8.2 Hz), 6.93 (1h, d, J=8.13 Hz), 6.08-6.97 (2H, m), 5.16 (1H,s), 5.14 (1H, q, J=1.74 Hz), 5.11-5.06 (3H, m), 3.91 (3H, s), 3.47 (2H, d, J=6.63 Hz), 3.38 (2H, d, J= 6.75 Hz). 13C NMR (CDCl3) δ 157.06, 150.84, 137.81, 136.51, 132.17, 130.50, 130.20, 129.80, 129.05, 128.76, 127.90, 127.85, 115.84, 115.56, 115.53, 110.00, 55.56, 39.42, 34.28. MS (m/z; Intensity): 279 (M−-1, 100%), 264 (97%), 249 (32%). Total isolation yield: 52%.
3′-Bromo-3,5′-diallyl-2′-hydroxy-4-methoxy-1,1′-biphenyl (2)
The reaction mixture was purified by column chromatography on silica gel eluted with hexanes/EtOAc = 15/1 to yield the first compound as yellowish oil residue. 1H NMR (CDCl3) δ 7.37 (1H, dd, J=8.5, 2.0 Hz), 7.31 (1H, d, J=2.0 Hz), 7.29 (1H, d, J=2.0 Hz), 7.05 (1H, d, J=2.0 Hz), 6.96 (1H, d, J=8.5 Hz), 6.08~5.92 (2H, m), 5.66 (1H, s), 5.13~5.06 (4H, m), 3.90 (3H, s), 3.45 (2H, d, J=3.5 Hz), 3.35 (2H, d, J=3.0 Hz). MS (m/z, Intensity): 359 (M−, 100%), 344 (20%). Total isolation yield: 15%.
2,6-Di-(4′-methoxy-3′-allylphenyl)-phenol (3)
The same reaction mixture was purified by column chromatography on silica gel eluted with hexanes/ethyl acetate=15/1 to yield the second compound as light yellowish oil residue. 1H NMR (CDCl3) δ 7.41 (2H, dd, J=8.0, 2.0 Hz), 7.34 (2H, d, J=2.0 Hz), 7.06(2H, s), 6.97 (2H, d, J=8.0 Hz), 6.08-5.90 (3H, m), 5.34 (1H, s), 3.90 (6H, s), 3.46 (4H, d, J=6.5 Hz), 3.40 (2H, d, J=6.5 Hz). MS (m/z, Intensity): 425 (M−-1, 100%), 409.8 (30%), 395 (10%). Total isolation yield: 20%.
3,3′-Diallyl-4-methoxy-4′-hydroxy-1,1′-biphenyl (4)
The reaction mixture was purified by column chromatography on silica gel eluted with hexanes/EtOAc = 10/1 to yield colorless oil residue. 1H NMR (CDCl3) δ 7.38 (1H, dd, J=7.0, 2.0 Hz), 7.35-7.31 (3H, m), 6.92 (1H, d, J=8.4 Hz), 6.88 (1H, d, J=7.8 Hz), 6.13-6.03 (2H, m), 5.25-5.19 (2H, m), 5.13-5.07 (2H, m), 3.89 (3H, s), 3.49 (2H,d, J=6.6 Hz), 3.46 (2H, d, J=6.6 Hz). 13C NMR (CDCl3) δ 156.47, 153.23, 136.94, 136.23, 134.08, 133.35, 128.87, 128.85, 128.34, 126.23, 125.46, 125.41, 116.64, 116.15, 115.49, 110.65, 55.58, 35.39, 34.41. MS (m/z; Intensity): 279 (M−-1, 100%), 264 (59%), 249 (11%). Total isolation yield: 12%.
3,3′-Diallyl-2′-hydroxy-4-methoxy-1,1′-biphenyl (5)
The coupling mixture was purified by column chromatography on silica gel eluted with hexanes/ethyl acetate=20/1 to yield colorless oil residue which was further hydrolyzed to 5 by dissolved in 15 ml of THF and 15 ml of 6 N HCl and refluxed for 6 hrs. Reaction mixture was extracted with EtOAc (20 ml × 3) and the combined organic layer was dried over MgSO4. Organic solvent was evaporated to give a colorless residue which was further purified by column chromatography on silica gel eluted with hexanes/EtOAc = 40/1 to give a colorless oil residue. 1H NMR (CDCl3) δ 7.31 (1H, dd, J=7.5, 2.0 Hz), 7.26 (1H, d, J=2.0 Hz), 7.13 (2H, td, J=7.5, 2.0 Hz), 6.98 (1H, d, J=8.5 Hz), 6.94 (1H, t, J=7.5 Hz), 6.13-5.90 (2H, m), 5.36 (1H, s), 5.20-5.06 (4H, m), 3.90 (3H, s), 3.49 (2H, d, J=6.6 Hz), 3.45 (2H, d, J=6.7 Hz). 13C NMR (CDCl3) δ 157.04, 150.49, 136.78, 136.51, 130.66, 129.77, 129.30, 129.16, 128.36, 128.03, 127.99, 126.29, 120.34, 115.86, 115.80, 110.96, 55.55, 34.79, 34.29. MS (m/z; Intensity): 279 (M−-1, 100%), 264 (70%), 249 (30%). Total isolation yield: 53%.
3′,5-Diallyl-2,2′-dihydroxy-1,1′-biphenyl (6)
The reaction mixture was dissolved in 15 ml of THF and refluxed 6 hrs with 15 ml of 6N HCl. After extraction with EtOAc (20 ml × 3), the organic layer was concentrated in vacuo. The residue was purified by column chromatography on silica gel eluted with hexanes/EtOAc = 4/1 to yield white solid. 1H NMR (CDCl3) δ 7.22 (1H, dd, J=7.50, 2.0 Hz), 7.17 (1H, dd, J=8.50, 2.0 Hz), 7.16 (1H, dd, J=7.5, 2.0 Hz), 7.09 (1H, d, J=2.0Hz), 7.01 (1H, t, J=7.5 Hz), 6.99 (1H, d, J=8.5 Hz), 6.11-6.04 (1H, m), 6.02-5.94 (1H, m), 5.21-5.14 (2H, m), 5.13-5.07 (2H, m), 3.51 (2H, d, J=6.50 Hz), 3.38 (2H, d, J=6.51 Hz). 13C NMR (CDCl3) δ 151.40, 151.10, 137.50, 136.47, 133.10, 131.14, 130.53, 130.11, 129.35, 127.19, 123.52, 123.46, 121.32, 116.64, 116.36, 115.85, 39.35, 34.99. MS (m/z; Intensity): 265 (M−-1, 100%), 252 (10%), 247 (8%). Total isolation yield: 21%.
2.2. Cell culture
The human cancer cell lines A549 (lung adenocarcinoma), UACC903 (melanoma) and HT-29 (colon) were purchased from American Type Culture Collection (Manassas, VA). All the cell lines were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 5.6 mmol/l glucose, glutamine at 37 °C in a humidified incubator with 5% CO2.
2.2.1. Cell proliferation and IC50 determination
The culture cell lines UACC 903, A549 and HT-29 were seeded at 5 × 103 cells/well in 96-well plate and after seeding cells were incubated at 37 °C in 5% CO2 for 24 h to allow cell attachment. Each cell line was treated at 6 concentrations (0, 0.5, 1.0, 5, 10, 25 and 50 μM) of honokiol analogs with a non-treatment control for 24 h and 72 h, and the final concentration of DMSO were 0.2%. The cell viability was evaluated through the 3,4-(5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay.46,47 The tetrazolium compound MTS and an electron coupling reagent, phenazine methosulfate (PMS), were used in the MTS assay. Viable cells reduced MTS to formazan, which was measured by determining absorbance at 492 nm using a spectrophotometer. Formazan production is time dependent and proportional to the number of viable cells. After incubation for the indicated time in the appropriate medium, 20 μl of MTS/PMS mixture was added to each well. Then the cells were incubated for 3 h and the formazan product was quantified by measuring absorbance at 492 nm. The background absorbance from the control wells was subtracted from the actual absorbance value. Three duplicate assays were done for each experimental condition. IC50 values were calculated by GraphPad Prism version 4 for Windows (GraphPad Software, San Diego, CA; www.graphpad.com).
2.2.2. Synchronization and cell cycle analysis
The effect of honokiol analogs on cell cycle distribution was determined by flow cytometry after staining the cells with DNA intercalating dye propidium iodide staining for DNA content measurement.18,19 Approximately 5 × 106 A549 cells were seeded into 6 well plate and allowed to attach by overnight incubation. Cells were synchronized by serum deprivation, when cells reached 40–50% cell confluency DMEM medium was added without serum and maintained in medium containing no serum for 48 h. After 48 h restimulated by replacing with fresh medium with 10 % serum containing different (0, 0.5, 2.5 and 10 μM) concentrations of honokiol analogs such as 2 and 4 in similar volume of DMSO was added to the control (final concentration of DMSO 0.2%). Plates were incubated for 48 h at 37 °C. After that floating and adherent A549 cells were collected by treatment with 0.05% trypsin and pelleted, washed with cold PBS. Cells were fixed in 70% ethanol at −20 °C overnight. Following overnight fixation cells were washed twice with ice cold PBS. Afterwards, cells were resuspended 1 ml of 0.1% Triton X-100 in PBS supplemented with 50 μg/ml PI, and 100 μg of DNase free RNase A. Resuspended cells were then stored in the dark at room temperature for 30 min prior to analysis by flow cytometry for the cell cycle specificity of apoptotic cells. Untreated cells were used as the negative controls. Stained cells were then analyzed using FACS (FACScan; BD), and at least 100,000 cells were counted for each sample.
2.2.3. Western blot analysis
Immunoblotting was performed as previously described.18,19 For determination of the total amount of Cdk1 and cyclin B1, A549 cells (5 × 106) were seeded onto 6 well plate and incubated overnight to allow cells attachment and subsequently synchronized by treatment with nocodazole (0.5 μg/ml) for 15 h. Thereafter, the plates were incubated with analogs 2 and 4 (0, 0.5, 1.0, 2.0 and 5.0 μM) or the DMSO for control, the final concentration of DMSO was 0.2% and the plates were incubated for 18 h at 37 °C. The plates were washed with ice cold PBS and then harvested in 600 μl RIPA buffer (50 mM Tris-Hcl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40 and 0.25% sodium deoxycholate), supplemented with a protease inhibitor cocktail (1 mM AEBSF, 800 nM Aprotatin, 50 μM Bestatin, 15 μM E64, 20 μM leupeptin, 10 μM pepstatin A and 3 mM phenylmethylsulfonyl fluoride), and incubated on ice for 30 minutes. Supernatant was recovered after removing cell debris by centrifugation at 12 – 14K rpm for 15 minutes at 4°C. Protein concentration was determined with BCA protein assay reagents. Equal amounts of protein (40 μg) for each sample were resolved on a 12% SDS-polyacrylamide gel and transferred onto a nitrocellulose membrane. The membrane was blocked for 1 h with 5% skim milk in Tris-buffered saline-Tween (TBST) buffer (100 mM Tris, 150 mM NaCl and 0.1% Tween 20) at room temperature. After blocking membrane were incubated with primary antibodies, a rabbit polyclonal CDK1 antibody against human CDK1 and a rabbit polyclonal antibody against human cyclin B1 overnight at 4 °C. The membrane was washed with TBST buffer and incubated with secondary antibody anti-rabbit IgG (H+L) peroxidase conjugate for 2 h at room temperature. Enhanced chemoluminescence reagents were used to visualize the protein bands. Each experiment was performed three times (different cell passages). The relative protein band intensities were determined by using the ImageJ 1.44 software (http://imagej.nih.gov/ij/index.html).
3. Result
3.1.1. Synthesis of honokiol analogs
We prepared honokiol analogs with the phenol group replaced by methoxy group in A-ring and/or by changing the substitution pattern on B-ring of the biphenyl structure. A total of six honokiol analogs were synthesized and their SAR evaluated (Figure 1). Our synthetic approach for honokiol analogs was based on the Pd-catalyzed Myaura-Suzuki coupling reaction36 which provides the flexibility of changing substitution on both rings of biphenyl moiety and also makes synthetic scheme extremely converge.
The syntheses of analogs 1–6 are illustrated in Scheme 1. The important building block for the synthesis are substituted isomers of 4-allyl-2-bromophenol (7) and 4-allyl-2,6-dibromophenol (8) which were prepared independently from same reagent 1,3dibromo-5,5-dimethylhydantoin (DBDMH)48,49 under different reaction condition (Scheme 2). The common reagent for honokiol analogs 1–5 is 3-allyl-4-methoxyphenyl boronic acid (12). Compound 11 was prepared from Claisen Rearrangement of 1-allyloxy-4-bromobenzene50 according to standard procedure43 to give 2-allyl-4-bromophenol 9. Standard methylation with methyl iodide and potassium carbonate yielded 2-allyl-4-bromo-anisole 11. Further treatment of 11 with n-BuLi followed by tri-isopropyl borate43 gave the desire Suzuki coupling reagent 12.
Scheme 1.

Synthetic of honokiol analogs.
Scheme 2.

Syntheses of Suzuki reagents.
Claisen rearrangement of 1-allyloxy-2-bromobenzene to the corresponding 1-ally-3-bromo-2-phenol was carried out according to procedure reported by Palmer et. al.43 under mild condition, the compound was not isolated but further reacted with methyl methoxy chloride (MOMCl) to give excellent yield of 1-ally-3-bromo-2-methoxymethoxybenzene (13). Conversion of compound 13 to the corresponding boronic acid 14 was carried out in the similar fashion as for compound 12 with n-BuLi and tri-iso-propyl borate.43
The Suzuki coupling reaction between the two moieties was carried out by standard conditions using Pd(PPh3)4 and Cs2CO3.43 The yields of this cross-coupling varied depending on different building blocks deployed; we obtained yields from 12% to as high as 52% depending on the substitution pattern on ring B. Synthesis of 66 was carried out with the coupling of 2-bromo-4-allyphenol (13)48,51 and 2-methoxymethyl-3-allylphenyl boronic acid (12).43,48 The MOM group was removed by refluxing the coupled intermediate in 6 N HCl/THF mixtures. The two steps synthesis gave total yield of 21%. All the honokiol analogs were characterized by proton NMR and MS.
3.1.2. Inhibition of human cancer cells growth by honokiol analogs
Literature reports have shown that honokiol inhibit proliferation, suppress tumor growth and induce apoptosis18,19,46 in human cancer cells. We first tested for growth inhibitory activity of the newly synthesized analogs in three different human cancer cell lines UACC903, A549 and HT-29, by MTS assay. The cells were maintained in the presence or absence of different concentration (0–50μM) of honokiol analogs for 24 and 72 h. Cells were treated with increasing concentrations of honokiol analogs. Honokiol analogs significantly inhibited cell viability, as evidenced by a MTS assay, in a time and concentration-dependent manner. The IC50 of honokiol analogs in different cancer cells were examined and the results are shown in Table 1. All new analogs gave relatively low IC50 values as compared to honokiol in all three cancer cell lines, except that analogs 1, 5, and 6 were less effective in UACC903 cells. Analog 2 and 4 were consistently the most cytotoxic than honokiol and other analogs in all three cell lines tested. There were no significant differences in growth inhibition between analog 2 and 4 (Table 1).
Table 1.
IC50 (μM) values of honokiol analogs in human cancer cell lines (24 and 72 h exposure)a.
| UACC903 | A549 | HT-29 | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| analog | 24h | 72h | 24h | 72h | 24h | 72h |
| 1 | 37.68±27.86 | 24.13±19.75 | 1.56±0.88 | 1.24±0.77 | 22.45±17.20 | 14.13±10.84 |
| 2 | 2.98±1.78 | 1.29±0.80 | 1.73±0.88 | 1.04±0.68 | 3.99±2.62 | 1.87±1.23 |
| 3 | 3.94±4.28 | 2.93±1.73 | 1.74±1.10 | 1.25±0.78 | 16.89±12.57 | 11.39±9.14 |
| 4 | 3.05±1.83 | 1.39±0.84 | 2.47±1.32 | 1.99±1.09 | 3.19±1.78 | 2.00±1.27 |
| 5 | 29.04±22.37 | 24.66±18.26 | 11.29±7.78 | 6.28±4.42 | 16.24±13.30 | 11.50±8.92 |
| 6 | 22.28±15.92 | 17.39±12.60 | 2.04±1.15 | 1.08±0.66 | 17.38±14.02 | 12.37±10.31 |
| Honokiol | 7.45±5.28 | 5.10±3.68 | 12.51±7.94 | 7.75±4.96 | 23.85±18.04 | 13.24±10.66 |
Cells were incubated in the presence or absence of honokiol analogs at different concentrations (μM). Effective concentration of honokiol analogs that inhibited cell growth to 50% of control (IC50) in three different cancer cell lines was determined (GraphPad Prism version 4 for Windows) using the MTS assay. Values are given as mean ± SD of three separate assays.
3.1.3. Effects on cell cycle distribution
Honokiol arrests cells in the G1 phase.18,19 We examined whether the new honokiol analogs can regulate the cell cycle similar to honokiol. Therefore, the effect of these analogs on the cell cycle distribution of A549 cells was determined. Flow cytometry was employed to examine the changes in DNA content. Synchronized A549 cells were treated with analog 2 and 4 at various concentrations. We observed that both analog 2 and 4 treated A549 cells induced G0-G1 cell cycle arrest as observed in the case of honokiol (Figure 2A–D). Further analysis indicated that 2 and 4 (2.5 μM and 10 μM) induced the accumulation of significant number of cells in the G0-G1 phase of the cell cycle (Table 2). These results indicated that the observed growth inhibitory effects of 2 and 4 in A549 cells are due to cell cycle arrest, and is accompanied by a decrease in both S phase and G2-M phase cells in a concentration-dependent manner (Table 2).
Figure 2.

Percentage of cell cycle distribution in A549 cells treated for 48 h. A. Percentage of cells distribution by honokiol treatment, B. Percentage of cell distribution by analog 2 treatment and C. Percentage of cell distribution by analog 4 treatment. D. Percentage of cell distribution by DMSO treatment. The results are presented as means (±SD) and similar results were observed in two independent experiments.
Table 2.
Cell cycle distribution (%) with honokiol analogs in A549 cells after 48 h treatment.a
| Compound | Concentration | G0/G1 (%) | S(%) | G2/M (%) |
|---|---|---|---|---|
| DMSO | Control | 40.6±4.6 | 41.2±3.1 | 18.2±2.1 |
| Honokiol | 0.5μM | 50.5±5.3 | 37.9±2.3 | 11.6±1.7 |
| 2.5μM | 61.1±3.1 | 28.1±1.5 | 10.8±4.3 | |
| 10μM | 72.9±5.2 | 21.7±4.2 | 5.4±1.8 | |
| 2 | 0.5μM | 55.8±2.9 | 32.8±2.8 | 11.4±1.8 |
| 2.5μM | 63.1±3.6 | 30.6±1.5 | 6.3±2.0 | |
| 10μM | 76.4±2.6 | 20.7±14.3 | 3.0±0.7 | |
| 4 | 0.5μM | 69.8±1.1 | 23.0±5.2 | 7.2±0.9 |
| 2.5μM | 75.7±3.5 | 20.2±2.7 | 4.2±1.0 | |
| 10μM | 82.8±2.4 | 14.3±3.3 | 2.8±0.6 |
Approximately 5 × 106 A549 cells were starved by serum deprivation, and then incubated in the presence or absence of analog 2 and 4 for 48 h. After incubation, cells were fixed, treated with RNAse A and stained for DNA with propidium iodide. Cell cycle distribution was determined by FACS analysis. Results are presented as a percentage of the total cell population. The whole experiment was repeated twice. Values are given as mean ± SD.
3.1.4. Effects of honokiol analogs on inhibition of CDK 1 and cyclin B1 level in A549 cells
The profound effect of 2 and 4 on cell cycle distribution led to the question of how they interact with cell cycle regulatory machinery, namely CDKs and cyclins. In order to address this question, we investigated CDK1 and cyclin B1, the two important regulatory proteins, as initial targets.21,32,52,53 We examined the ability of analogs 2 and 4 to inhibit the total cellular content of human CDK1 and cyclin B1 proteins in human lung cancer cells (A549). The cells were arrested and released, and then incubated for 18 h with four different concentrations of 2 and 4. The total cellular content of human CDK1 and cyclin B1 proteins was determined by Western blot analysis using polyclonal antibodies. A459 cells treated with analogs 2 and 4 caused a rapid decrease in CDK1and cyclin B1 protein level in a concentration-dependent manner (Figure 3B-C). In comparison, honokiol was less effective (Figure 3A). A significant reduction was observed in the groups treated with 2 and 5 μM of analogs 2 and 4 (Figure 3B-C). The concentration dependent change of CDK1 and cyclin B1 protein level is comparable to the results for honokiol.
Figure 3.

Effects of honokiol analogs on CDK1 and cyclin B1 proteins. A549 cells were treated with honokiol analogs with 0.0, 0.5, 1.0, 2.0 and 5.0 μM for 18 h. After that cell lysates were prepared and the level of CDK1 and cyclin B1 proteins were determined by western blot analysis. A. Cells treated with honokiol, B. Cells treated with 2 and C. Cells treated with 4. The experiments were repeated more than twice using independently prepared lysates with similar results. β-Actin is shown as a loading control. The blots were stripped and reported with anti-actin antibody to normalize for differences in protein loading.
4. Discussion
The SAR studies on honokiol analogs have been reported in the literature with varying success.6,36,54–56 Our goal of using SAR to enhance the activity of honokiol as antitumor agents is based on observed pharmacological properties of honokiol and our hypothesis is that the non-availability of phenol group in the A-ring would enhance honokiol anti-tumor activity. Our investigation is to gain insights into the interaction of newly prepared honokiol analogs and the mechanism of cell cycle regulation using A549 human lung cancer cells as a model. To validate our hypothesis, we prepared six modified honokiol analogs and their cytotoxicity tested in vitro in human cancer cell lines A549, UACC903 and HT-29 for different time and concentration dependent manner. Our results show that treatment of honokiol analogs for 24 and 72 h induces a rapid growth arrest in A549, UACC903 and HT-29 cell lines. This result is comparable with previous studies with honokiol alone.8,9,11,54 The blocking of hydroxyl group by methylation on A-ring analogs, such as analogs 2 and 4 showed higher cytotoxicity for all three cell lines. These results support our hypothesis that making phenol group in A-ring unavailable would enhance activity of honokiol. Specifically, both 2 and 4 showed lower IC50 in A549 cell line compared with other analogs and honokiol. For further investigation we selected 2 and 4 to elucidate underlying mechanisms of growth inhibition, cell cycle arrest and apoptosis. We studied the cell cycle distribution and observed significantly induced a G1 pha se arrest of cells after treatment of 2 and 4 in the synchronized A549 cell line compared to the untreated control cells.
The intracellular events governing the cell division cycle are conserved in many eukaryotic cell types. The kinase activities of CDKs are the driving force for the progression of the cell cycle through the transition check points, due to their ability to activate cyclins, essential components of cyclin and CDK complexes.57 Studies have documented a G1 phase cell cycle arrest by honokiol in human prostate cancer18 and VSMC cells19 treatment at concentrations within the range of analogs 2 and 4 used in the present study. The present study reveals that exposure of A549 cells to pharmacologically achievable concentrations of honokiol analogs 2 and 4 showed growth suppression and G0-G1 phase arrest (Figure 2A–D). The G0-G1 phase cell cycle arrest is modest as evidenced by 10–50% increase in percentage of G0-G1 phase over DMSO treated control (Table 2). The observation is consistent with that reported for honokiol and may suggest that the mechanism of G0-G1 phase arrest for honokiol, 2, and 4 is similar.
To further understand interaction between honokiol analogs and cell cycle regulatory proteins, we determined total protein levels of CDK1 and cyclin B1 after treating A549 cells with honokiol analogs. Our results indicated that analogs 2 and 4 significantly lowered the protein levels of CDK1 and cyclin B1 in a concentration dependent manner (Figure 4). Taken together, the cell cycle distribution studies and the lowering of CDK1 and cyclin B1 may imply that other CDKs are also involved in the cell cycle arrest. The protein kinase CDK1 and cyclin B1 play a crucial role in the onset of mitosis in human cells. The CDK1 and cyclin B1 complex accumulates in the cytoplasm during the G2 phase, being translocated into the nucleus at the beginning of mitosis.58 The decrease in cyclin B1 levels leads to inhibition of cell division owing to low CDK1 activity. Therefore, a sub-cytotoxic concentration of 2 and 4 are likely to inhibit CDK1 activity through a decrease in the levels of cyclin B1. Suppression of CDK1 activity not only inhibits cell division, but also allows the assembly of pre-replication complexes for licensing the DNA for another round of replication.59
Figure 4.
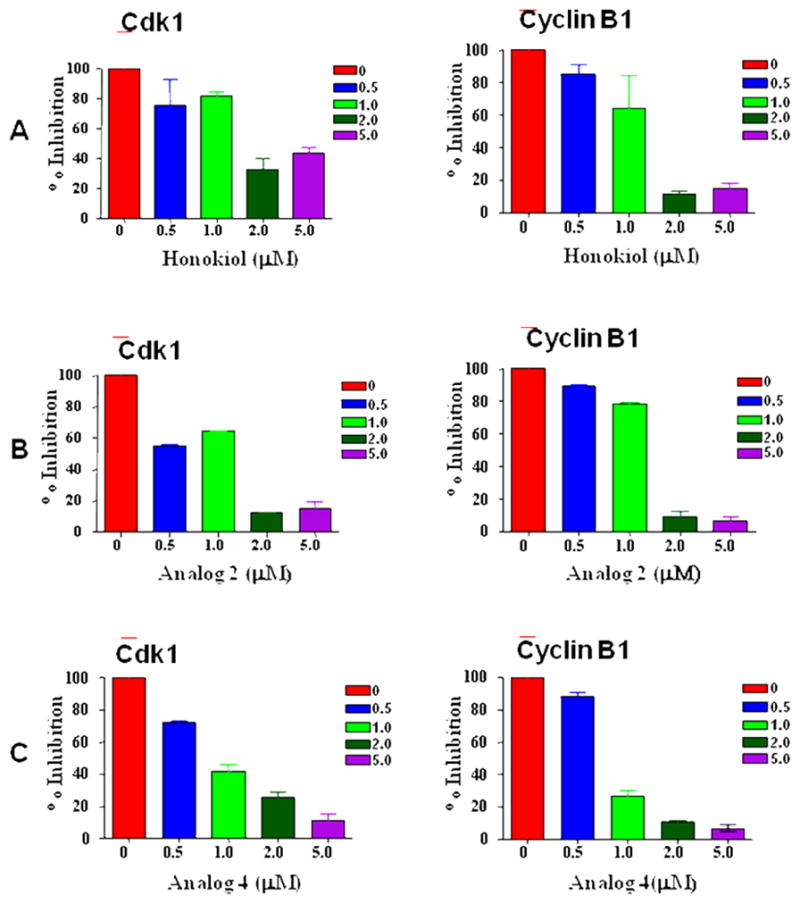
Percentage of CDK1 and cyclin B1 inhibition in A549 cells treated with honokiol analogs. Western blot analysis for CDK1 and cyclin B1 using whole cell lysates from A549 cells treated with honokiol analogs for the indicated concentrations for 18 h. Western blotting for CDK1 and cyclin B1 proteins was done more than two times and the results were consistent. A. Percentage of CDK1 and cyclin B1 inhibition cells treated with honokiol, B. Percentage of CDK1 and cyclin B1 inhibition cells treated with 2 and C. Percentage of CDK1 and cyclin B1 inhibition cells treated with 4.
The introduction of methoxy group on the A-ring of honokiol increases the inhibitory activity in human cancer cell lines. The cell cycle passage in mammalian cells is regulated by a complex system of activating and inhibiting factors. The different cyclin-dependent kinases act in distinct phases of the cell cycle. Thus, potent and selective inhibitors of CDKs might induce different effects on the cell cycle distribution, depending on the respective phase in which the cells are targeted. The results of cell cycle distribution study indicated that analogs 2 and 4 can interfere with cell cycle regulation at G0-G1 phase. In addition, the protein levels of CDK1 and cyclin B1 were reduced significantly upon treatment of 2 and 4 which indicated both analogs can interfere with the regulation of cell cycle at M phase. Taken together, these results suggest that honokiol analogs could exert control at two critical stages of cell cycle, namely, G0-G1 and M phase. It is also interesting to note that recently CDK1 was discovered to be able to drive the mammalian cell cycle.21 Therefore, results of the present study are a significant step forward in understanding the activity of 2 and 4; they also shed new light on the molecular mechanisms underlying the potent effects of the 2 and 4. Honokiol was shown to cause apoptosis in cancer cell lines that have defective p53 gene and activated ras gene.60 Honokiol can also induced cyclophilin D and cause death in cells with wild-type p53.60 Further studies on other human cancer cell lines will be tested in the future.
This investigation confirmed that blocking the possible oxidation of the phenolic hydroxyl group in the biphenyl group skeleton will enhance the anti-tumor activity of honokiol. The newly synthesized analogs are capable of arresting the progress of cell cycle at G0-G1 phase. These studies provide experimental evidence that newly synthesized honokiol analogs are potent tumor cell growth inhibitors and they are also capable of inhibiting CDK1 and cyclin B1, important cell cycle regulatory proteins. In conclusion, honokiol analogs 2 and 4 are potent anti-cancer agents that exhibit strong inhibitory property toward cell growth at lower concentration than the honokiol, and induce cell cycle arrest at G0-G1 phase. Most importantly, both analogs effectively lower CDK1 and cyclin B1 protein levels in the treated cells, which suggest that they interfered with key kinases that promote mitosis. The mechanistic bases for these apparent synergies need further investigation.
Supplementary Material
Acknowledgments
The author thanks the financial support from Penn State Hershey Cancer Institute (J.M.L). This study was supported in part by the NCI Grant R03-CA143999 (A.K.S.). The authors like to thank the Core Research Facilities at College of Medicine, Pennsylvania State University for recording of flow cytometry and NMR facility for NMR spectra. In addition, we appreciate Dr. Kun-Ming Chen for obtaining mass spectra for our compounds.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fujita M, Itokawa H, Sashida Y, Honokiol A. New Phenolic Compound isolated from the Bark of Magnolia obovata Thunb. Chem Pharm Bull (Tokyo) 1972;20(1):212–213. [Google Scholar]
- 2.Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. The anti-inflammatory effect of honokiol on neutrophils: mechanisms in the inhibition of reactive oxygen species production. Eur J Pharmacol. 2003;475(1–3):19–27. doi: 10.1016/s0014-2999(03)02121-6. [DOI] [PubMed] [Google Scholar]
- 3.Teng CM, Chen CC, Ko FN, Lee LG, Huang TF, Chen YP, Hsu HY. Two antiplatelet agents from Magnolia officinalis. Thromb Res. 1988;50(6):757–765. doi: 10.1016/0049-3848(88)90336-2. [DOI] [PubMed] [Google Scholar]
- 4.Lo YC, Teng CM, Chen CF, Chen CC, Hong CY. Magnolol and honokiol isolated from Magnolia officinalis protect rat heart mitochondria against lipid peroxidation. Biochem Pharmacol. 1994;47(3):549–553. doi: 10.1016/0006-2952(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 5.Amblard F, Delinsky D, Arbiser JL, Schinazi RF. Facile purification of honokiol and its antiviral and cytotoxic properties. J Med Chem. 2006;49(11):3426–3427. doi: 10.1021/jm060268m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amblard F, Govindarajan B, Lefkove B, Rapp KL, Detorio M, Arbiser JL, Schinazi RF. Synthesis, cytotoxicity, and antiviral activities of new neolignans related to honokiol and magnolol. Bioorg Med Chem Lett. 2007;17(16):4428–4431. doi: 10.1016/j.bmcl.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fong WF, Tse AK, Poon KH, Wang C. Magnolol and honokiol enhance HL-60 human leukemia cell differentiation induced by 1,25-dihydroxyvitamin D3 and retinoic acid. Int J Biochem Cell Biol. 2005;37(2):427–441. doi: 10.1016/j.biocel.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 8.Wang T, Chen F, Chen Z, Wu YF, Xu XL, Zheng S, Hu X. Honokiol induces apoptosis through p53-independent pathway in human colorectal cell line RKO. World J Gastroenterol. 2004;10(15):2205–2208. doi: 10.3748/wjg.v10.i15.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park EJ, Zhao YZ, Kim YH, Lee BH, Sohn DH. Honokiol induces apoptosis via cytochrome c release and caspase activation in activated rat hepatic stellate cells in vitro. Planta Med. 2005;71(1):82–84. doi: 10.1055/s-2005-837757. [DOI] [PubMed] [Google Scholar]
- 10.Yang SE, Hsieh MT, Tsai TH, Hsu SL. Down-modulation of Bcl-XL, release of cytochrome c and sequential activation of caspases during honokiol-induced apoptosis in human squamous lung cancer CH27 cells. Biochem Pharmacol. 2002;63(9):1641–1651. doi: 10.1016/s0006-2952(02)00894-8. [DOI] [PubMed] [Google Scholar]
- 11.Battle TE, Arbiser J, Frank DA. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood. 2005;106(2):690–697. doi: 10.1182/blood-2004-11-4273. [DOI] [PubMed] [Google Scholar]
- 12.Konoshima T, Kozuka M, Tokuda H, Nishino H, Iwashima A, Haruna M, Ito K, Tanabe M. Studies on inhibitors of skin tumor promotion IX. Neolignans from Magnolia officinalis. J Nat Prod. 1991;54(3):816–822. doi: 10.1021/np50075a010. [DOI] [PubMed] [Google Scholar]
- 13.Nagase H, Ikeda K, Sakai Y. Inhibitory effect of magnolol, honokiol from Magnolia obovata on human fibrosarcoma HT-1080, Invasiveness in vitro. Planta Med. 2001;67(8):705–708. doi: 10.1055/s-2001-18345. [DOI] [PubMed] [Google Scholar]
- 14.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G, Arbiser JL. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278(37):35501–35507. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 15.Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, Shiraishi N, Yasui H, Roccaro AM, Richardson P, Podar K, Le GS, Chauhan D, Tamura K, Arbiser J, Anderson KC. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005;106(5):1794–1800. doi: 10.1182/blood-2005-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaid M, Sharma SD, Katiyar SK. Honokiol, a phytochemical from the Magnolia plant, inhibits photocarcinogenesis by targeting UVB-induced inflammatory mediators and cell cycle regulators: development of topical formulation. Carcinogenesis. 2010;31(11):2004–2011. doi: 10.1093/carcin/bgq186. [DOI] [PubMed] [Google Scholar]
- 17.Maruyama Y, Kuribara H, Morita M, Yuzurihara M, Weintraub ST. Identification of Magnolol and Honokiol as Anxiolytic Agents in Extracts of Saiboku-to, an Oriental Herbal Medicine. Journal of Natural Products. 1998;61(1):135–138. doi: 10.1021/np9702446. [DOI] [PubMed] [Google Scholar]
- 18.Hahm ER, Singh SV. Honokiol causes G0-G1 phase cell cycle arrest in human prostate cancer cells in association with suppression of retinoblastoma protein level/phosphorylation and inhibition of E2F1 transcriptional activity. Mol Cancer Ther. 2007;6(10):2686–2695. doi: 10.1158/1535-7163.MCT-07-0217. [DOI] [PubMed] [Google Scholar]
- 19.Lee B, Kim CH, Moon SK. Honokiol causes the p21WAF1-mediated G(1)-phase arrest of the cell cycle through inducing p38 mitogen activated protein kinase in vascular smooth muscle cells. FEBS Lett. 2006;580(22):5177–5184. doi: 10.1016/j.febslet.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 20.Ahn KS, Sethi G, Shishodia S, Sung B, Arbiser JL, Aggarwal BB. Honokiol potentiates apoptosis, suppresses osteoclastogenesis, and inhibits invasion through modulation of nuclear factor-kappaB activation pathway. Mol Cancer Res. 2006;4(9):621–633. doi: 10.1158/1541-7786.MCR-06-0076. [DOI] [PubMed] [Google Scholar]
- 21.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28(33):2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 22.Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet. 2003;35(1):25–31. doi: 10.1038/ng1232. [DOI] [PubMed] [Google Scholar]
- 23.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3(3):233–245. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 24.Hirai H, Kawanishi N, Iwasawa Y. Recent advances in the development of selective small molecule inhibitors for cyclin-dependent kinases. Curr Top Med Chem. 2005;5(2):167–179. doi: 10.2174/1568026053507688. [DOI] [PubMed] [Google Scholar]
- 25.Knockaert M, Greengard P, Meijer L. Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci. 2002;23(9):417–425. doi: 10.1016/s0165-6147(02)02071-0. [DOI] [PubMed] [Google Scholar]
- 26.Krek W, Nigg EA. Differential phosphorylation of vertebrate p34cdc2 kinase at the G1/S and G2/M transitions of the cell cycle: identification of major phosphorylation sites. EMBO J. 1991;10(2):305–316. doi: 10.1002/j.1460-2075.1991.tb07951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen M, Feng Y, Gao C, Tao D, Hu J, Reed E, Li QQ, Gong J. Detection of cyclin b1 expression in g(1)-phase cancer cell lines and cancer tissues by postsorting Western blot analysis. Cancer Res. 2004;64(5):1607–1610. doi: 10.1158/0008-5472.can-03-3321. [DOI] [PubMed] [Google Scholar]
- 28.Pines J, Hunter T. Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell. 1989;58(5):833–846. doi: 10.1016/0092-8674(89)90936-7. [DOI] [PubMed] [Google Scholar]
- 29.Gong J, Traganos F, Darzynkiewicz Z. Growth imbalance and altered expression of cyclins B1, A, E, and D3 in MOLT-4 cells synchronized in the cell cycle by inhibitors of DNA replication. Cell Growth Differ. 1995;6(11):1485–1493. [PubMed] [Google Scholar]
- 30.Viallard JF, Lacombe F, Dupouy M, Ferry H, Belloc F, Reiffers J. Flow cytometry study of human cyclin B1 and cyclin E expression in leukemic cell lines: cell cycle kinetics and cell localization. Exp Cell Res. 1999;247(1):208–219. doi: 10.1006/excr.1998.4352. [DOI] [PubMed] [Google Scholar]
- 31.Yuan J, Yan R, Kramer A, Eckerdt F, Roller M, Kaufmann M, Strebhardt K. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene. 2004;23(34):5843–5852. doi: 10.1038/sj.onc.1207757. [DOI] [PubMed] [Google Scholar]
- 32.Yuan J, Kramer A, Matthess Y, Yan R, Spankuch B, Gatje R, Knecht R, Kaufmann M, Strebhardt K. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene. 2006;25(12):1753–1762. doi: 10.1038/sj.onc.1209202. [DOI] [PubMed] [Google Scholar]
- 33.Soria JC, Jang SJ, Khuri FR, Hassan K, Liu D, Hong WK, Mao L. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res. 2000;60(15):4000–4004. [PubMed] [Google Scholar]
- 34.Hassan KA, Ang KK, El-Naggar AK, Story MD, Lee JI, Liu D, Hong WK, Mao L. Cyclin B1 overexpression and resistance to radiotherapy in head and neck squamous cell carcinoma. Cancer Res. 2002;62(22):6414–6417. [PubMed] [Google Scholar]
- 35.Yoshida T, Tanaka S, Mogi A, Shitara Y, Kuwano H. The clinical significance of Cyclin B1 and Wee1 expression in non-small-cell lung cancer. Ann Oncol. 2004;15(2):252–256. doi: 10.1093/annonc/mdh073. [DOI] [PubMed] [Google Scholar]
- 36.Esumi T, Makado G, Zhai H, Shimizu Y, Mitsumoto Y, Fukuyama Y. Efficient synthesis and structure-activity relationship of honokiol, a neurotrophic biphenyl-type neolignan. Bioorg Med Chem Lett. 2004;14(10):2621–2625. doi: 10.1016/j.bmcl.2004.02.067. [DOI] [PubMed] [Google Scholar]
- 37.Hughes MF, Hall LL. Disposition of phenol in rat after oral, dermal, intravenous, and intratracheal administration. Xenobiotica. 1995;25(8):873–883. doi: 10.3109/00498259509061900. [DOI] [PubMed] [Google Scholar]
- 38.Eastmond DA, Smith MT, Ruzo LO, Ross D. Metabolic activation of phenol by human myeloperoxidase and horseradish peroxidase. Mol Pharmacol. 1986;30(6):674–679. [PubMed] [Google Scholar]
- 39.Bohmdorfer M, Maier-Salamon A, Taferner B, Reznicek G, Thalhammer T, Hering S, Hufner A, Schuhly W, Jager W. In vitro metabolism and disposition of honokiol in rat and human livers. J Pharm Sci. 2011;100(8):3506–3516. doi: 10.1002/jps.22536. [DOI] [PubMed] [Google Scholar]
- 40.Kwak JH, Cho YA, Jang JY, Seo SY, Lee H, Hong JT, Han SB, Lee K, Kwak YS, Jung JK. Expedient synthesis of 4′-O-methylhonokiol via Suzuki Miyaura cross-coupling. Tetrahedron. 2011;67(48):9401–9404. [Google Scholar]
- 41.Alam A, Takaguchi Y, Ito H, Yoshida T, Tsuboi S. Multi-functionalization of gallic acid towards improved synthesis of alpha and beta-DDB. Tetrahedron. 2005;61(7):1909–1918. [Google Scholar]
- 42.Sviridov SI, Vasil’ev AA, Sergovskaya NL, Chirskaya MV, Shorshnev SV. Azidosubstituted arylboronic acids: synthesis and Suzuki ÇôMiyaura cross-coupling reactions. Tetrahedron. 2006;62(11):2639–2647. [Google Scholar]
- 43.Palmer FN, Lach F, Poriel C, Pepper AG, Bagley MC, Slawin AMZ, Moody CJ. The diazo route to diazonamide A: studies on the tyrosine-derived fragment. Organic & Biomolecular Chemistry. 2005;3(20):3805–3811. doi: 10.1039/b510653b. [DOI] [PubMed] [Google Scholar]
- 44.Denton RM, Scragg JT, Saska J. A concise synthesis of 4′-O-methyl honokiol. Tetrahedron Letters. 2011;52(20):2554–2556. [Google Scholar]
- 45.Taferner B, Schuehly W, Huefner A, Baburin I, Wiesner K, Ecker GF, Hering S. Modulation of GABAA-Receptors by Honokiol and Derivatives: Subtype Selectivity and Structure ÇôActivity Relationship. Journal of Medicinal Chemistry. 2011;54(15):5349–5361. doi: 10.1021/jm200186n. [DOI] [PubMed] [Google Scholar]
- 46.Wu F, Zhang W, Li L, Zheng F, Shao X, Zhou J, Li H. Inhibitory effects of honokiol on lipopolysaccharide-induced cellular responses and signaling events in human renal mesangial cells. Eur J Pharmacol. 2011;654(1):117–121. doi: 10.1016/j.ejphar.2010.11.022. [DOI] [PubMed] [Google Scholar]
- 47.Patterson C, Perrella MA, Endege WO, Yoshizumi M, Lee ME, Haber E. Downregulation of vascular endothelial growth factor receptors by tumor necrosis factor-alpha in cultured human vascular endothelial cells. J Clin Invest. 1996;98(2):490–496. doi: 10.1172/JCI118816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alam A, Takaguchi Y, Ito H, Yoshida T, Tsuboi S. Multi-functionalization of gallic acid towards improved synthesis of [alpha]- and [beta]-DDB. Tetrahedron. 2005;61(7):1909–1918. [Google Scholar]
- 49.Alam A. 1,3-Dibromo-5,5-dimethylhydantoin. Synlett. 2005;2005 (EFirst): 2403; 2404. [Google Scholar]
- 50.Kurosawa W, Kobayashi H, Kan T, Fukuyama T. Total synthesis of (ephedradine A: an efficient construction of optically active dihydrobenzofuran-ring via C-H inserti on reaction. Tetrahedron. 2004;60(43):9615–9628. [Google Scholar]
- 51.Mel’kanovitskaya SG. Allylation of phenols and phenol ethers. Allylation of 2-bromophenol. Uzbekskii Khimicheskii Zhurnal. 1967;11(5):34–36. [Google Scholar]
- 52.Padmakumar VC, Aleem E, Berthet C, Hilton MB, Kaldis P. Cdk2 and Cdk4 activities are dispensable for tumorigenesis caused by the loss of p53. Mol Cell Biol. 2009;29(10):2582–2593. doi: 10.1128/MCB.00952-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matthess Y, Raab M, Sanhaji M, Lavrik IN, Strebhardt K. Cdk1/cyclin B1 controls Fas-mediated apoptosis by regulating caspase-8 activity. Mol Cell Biol. 2010;30(24):5726–5740. doi: 10.1128/MCB.00731-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kong ZL, Tzeng SC, Liu YC. Cytotoxic neolignans: an SAR study. Bioorg Med Chem Lett. 2005;15(1):163–166. doi: 10.1016/j.bmcl.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 55.Zhai H, Nakade K, Oda M, Mitsumoto Y, Akagi M, Sakurai J, Fukuyama Y. Honokiol-induced neurite outgrowth promotion depends on activation of extracellular signal-regulated kinases (ERK1/2) European Journal of Pharmacology. 2005;516(2):112–117. doi: 10.1016/j.ejphar.2005.04.035. [DOI] [PubMed] [Google Scholar]
- 56.Luo Y, Xu Y, Chen L, Hu J, Peng C, Xie D, Shi J, Huang W, Xu G, Peng M, Han J, Li R, Yang S, Wei Y. Semi-synthesis and anti-proliferative activity evaluation of novel analogues of Honokiol. Bioorg Med Chem Lett. 2009;19(16):4702–4705. doi: 10.1016/j.bmcl.2009.06.071. [DOI] [PubMed] [Google Scholar]
- 57.Jacks T, Weinberg RA. Cell-cycle control and its watchman. Nature. 1996;381(6584):643–644. doi: 10.1038/381643a0. [DOI] [PubMed] [Google Scholar]
- 58.Takizawa CG, Weis K, Morgan DO. Ran-independent nuclear import of cyclin B1-Cdc2 by importin beta. Proc Natl Acad Sci U S A. 1999;96(14):7938–7943. doi: 10.1073/pnas.96.14.7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diffley JF. Regulation of early events in chromosome replication. Curr Biol. 2004;14(18):R778–R786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 60.Fried L, Arbiser JL. Honokiol, A Multifunctional Antiangiogenic and Antitumor Agent. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2009.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.