

Synopsis
Childhood vasculitis is a challenging and complex group of conditions that are multisystem in nature and often require integrated care from multiple subspecialties including rheumatology, dermatology, cardiology, nephrology, neurology, and gastroenterology. Vasculitis is defined as the presence of inflammation in the blood vessel wall. The site of vessel involvement, size of the affected vessels, extent of vascular injury, and underlying pathology determine the disease phenotype and severity. This review explores the classification and general features of pediatric vasculitis as well as the clinical presentation, diagnostic evaluation, and therapeutic options for the most common vasculitides.
Keywords: vasculitis, pediatrics, review
Childhood vasculitis is a challenging and complex group of conditions that are multisystem in nature and often require integrated care from multiple subspecialties including rheumatology, dermatology, cardiology, nephrology, neurology, and gastroenterology. Vasculitis is defined as the presence of inflammation in the blood vessel wall. The site of vessel involvement, size of the affected vessels, extent of vascular injury, and underlying pathology determine the disease phenotype and severity. Vasculitis can be secondary to infection, malignancy, drug exposure, and other rheumatic conditions such as systemic lupus erythematosus and juvenile dermatomyositis. This review will explore the classification and general features of vasculitis as well as the clinical presentation, diagnostic evaluation, and therapeutic options for the most common primary systemic vasculitides.
Diagnosis
Making the diagnosis of vasculitis is often challenging, as presenting symptoms may be sub-acute, non-specific, and non-diagnostic. Fever, malaise, diffuse pain, and laboratory evidence of elevated acute phase reactants may be the only early symptoms to suggest systemic inflammation. As vessel damage evolves more specific clinical features such as a purpuric rash, evidence of organ involvement like glomerulonephritis, or detection of certain antibodies such as anti-neutrophil cytoplasmic antibodies (ANCA) may heighten the suspicion of vasculitis. The presenting symptoms can vary widely depending upon the size and location of involved vasculature.
If vasculitis is suspected then a thorough history and physical exam are paramount. The history should include recent infections, drug exposure, and a detailed family history. The physical examination should include a four-extremity blood pressure evaluation. Takayasu arteritis (TA) may present with a blood pressure difference of greater than 10 mm Hg between arms and hypertension is common with many of the vasculitides. Additionally, careful auscultation for bruits (carotid, axillary, aortic, renal, and iliac vessels) and palpation of peripheral pulses is essential. Absent peripheral pulses may help identify areas of vessel involvement. A thorough skin examination is also important; the presence of painful nodules, purpura, ulcerations, microinfarctions, or livedo reticularis is common. A neurological exam should evaluate for peripheral neuropathy; polyarteritis nodosa (PAN) is associated with mononeuritis multiplex. A fundoscopic examination and nailfold capillaroscopy are also helpful to visualize small vessel abnormalities.
The laboratory evaluation for vasculitis should include a complete blood count and acute phase reactants such as the erythrocyte sedimentation rate (ESR) and c-reactive protein (CRP), which can be markedly elevated. Liver enzymes, blood urea nitrogen and creatinine, and urinalysis will evaluate for hepatic and renal involvement. Specific antibody testing such as antinuclear antibodies (ANA) and ANCA, and complements should be sent depending on the vasculitis being considered. When clinical suspicion is high, imaging such as CT angiography, MR angiography, or conventional angiography may help detect blood vessel abnormalities. Imaging may demonstrate prototypical patterns of vessel involvement, such as beading and aneurysms in PAN and TA, respectively. Typically, imaging is most useful when there is suspicion for medium or large vessel disease. The diagnostic gold standard for diagnosis, however, is tissue biopsy.
Classification
Primary vasculitis can be classified according to clinical manifestations, size of the affected vessels, or histopathology including the presence or absence of granuloma. In 2005 the European League Against Rheumatism (EULAR) and the Pediatric Rheumatology European Society (PReS) developed the first pediatric-specific classification of vasculitis (Table 1).1 This classification system is primarily based upon size of affected vessels and the presence or absence of granuloma.
Table 1.
EULAR/PReS classification of pediatric vasculitis
| Vasculitis category | |
|---|---|
| Predominately large vessel | Takayasu arteritis |
| Predominately medium vessel | Childhood polyarteritis nodosa |
| Cutaneous polyarteritis | |
| Kawasaki disease | |
| Predominately small vessel | Granulomatous |
| Wegener’s granulomatosis* | |
| Churg-Strauss syndrome | |
| Non-granulomatous | |
| Microscopic polyangiitis | |
| Henoch- Schönlein purpura | |
| Isolated cutaneous leucocytoclastic vasculitis | |
| Hypocomplementemic urticarial vasculitis | |
| Other | Behçet disease |
| Vasculitis secondary to infection, malignancy, drugs | |
| Vasculitis associated with connective tissue disease | |
| Isolated vasculitis of the central nervous system | |
| Cogan syndrome | |
| Unclassified |
Granulomatosis with polyangiitis. Adapted from Ozen et al.1
Epidemiology and Pathogenesis
The annual incidence of primary vasculitis in children and adolescents younger than 17 years old is approximately 23 per 100,000.2 Primary vasculitis accounts for approximately 2–10% of all pediatric conditions evaluated in pediatric rheumatology clinics.3–6 Of the primary vasculitides, Henoch Schönlein purpura (HSP) and Kawasaki disease (KD) are the most common accounting for 49% and 23% of all childhood vasculitis, respectively.7 The prevalence of diseases may be different based on the population studied. For example, the incidence of KD and Behçet’s disease are higher in Asian and Turkish children, respectively, than in other ethnicities.
These ethnic differences in prevalence suggest that genetics and environment may play an important role in disease susceptibility and pathogenesis. Other theories of pathogenesis include humoral factors, as manifest by ANCA-associated vasculitides. Abnormal regulation of immune complex formation may be contributory, as in HSP. Impaired lymphocyte regulation, specifically T-regulatory cell dysfunction, may also be involved. Antecedent infections, particularly streptococcal infections, have been implicated in many of the vasculitides including HSP, granulomatosis with polyangiitis (GPA), and PAN.
Henoch Schönlein Purpura (HSP)
Etiology and epidemiology
HSP is a leukocytoclastic vasculitis that predominantly affects the small blood vessels. It is also known as anaphylactoid purpura or purpura rheumatica. The EULAR/PReS classification criteria are listed in Table 2.8 Among children less than 17 years the annual incidence of HSP is approximately 20 per 100,000 and the peak age of onset is between 4 and 6 years.2 Caucasians have the highest incidence and African Americans have the lowest incidence. Unlike most vasculitides, males are affected more commonly than females, with a ratio of approximately 2 to 1. Certain autoimmune risk factors such as complement deficiencies9 and hereditary fever syndromes10, such as Familial Mediterranean Fever syndrome, may predispose a child to HSP. HSP is most prevalent during the winter and spring. This seasonal distribution supports the hypothesis that an infectious agent triggers this condition.11 Group A β–hemolytic streptococcus, Staphylococcus aureus, influenza, parainfluenza, Epstein-Barr virus, adenovirus, parvovirus, and mycoplasma have all been reported as triggers for HSP.
Table 2.
EULAR/PReS classification criteria for Henoch Schönlein purpura (HSP)8
Purpura or petechiae with lower limb predominance and at least one of the following:
|
Clinical presentation
The classic presentation of HSP includes lower-extremity purpura, arthritis, abdominal pain, and renal disease. The purpuric rash is usually on dependent areas but may be seen on the arms, face, and ears (Figure 1A). The purpura may be preceded by a maculopapular or urticarial rash that usually disappears within 24 hours.12 The rash may appear as bullae, necrotic lesions (Figure 1B), or deep bruising (Figure 1C).
Figure 1. Skin lesions of HSP.

A) Classic lower extremity purpura, B) Necrotic lesions, C) Deep bruising. Courtesy of David D. Sherry, MD, Philadelphia, PA.
Arthritis affects three-quarters of children and the most commonly affected joints are the knees and ankles. The arthritis is usually oligoarticular, self-limited, and non-destructive. It is the presenting symptom in 15% of patients.13
The gastrointestinal (GI) manifestations of HSP affect 50 to 75% of children and may include bleeding, intussusception, and abdominal pain. GI manifestations may precede the purpura by up to 2 weeks in as many as 20% of children.12 Intestinal bleeding, manifested as gross or occult blood per rectum, occurs in approximately one-third of children. Intussusception occurs in 1 to 5% of children and is mostly ileo-ileal in location.14
Renal disease affects 20 to 60% of children and the most common manifestation is microscopic hematuria with or without proteinuria. Renal disease rarely precedes the onset of rash. Children may present with nephritic or nephrotic syndrome, or rarely renal failure. The majority of children who develop renal disease do so within the first 6 weeks and 97% within 6 months.15 In longitudinal studies of unselected patients the risk of chronic renal impairment and end-stage renal disease is 2–15% and less than 1%, respectively.15,16
Unusual clinical manifestations of HSP may include edema of scrotum, eyes, or hands, pulmonary hemorrhage, seizures, stroke, and mental status changes. The mean duration of symptoms is 3 to 4 weeks and up to one-third of children have at least 1 recurrence.17
HSP must be distinguished from other causes of purpura in childhood including acute hemorrhagic edema of infancy, immune thrombocytopenic purpura, acute post-streptococcal glomerulonephritis, hemolytic-uremic syndrome, disseminated intravascular coagulation, infections, and hypersensitivity vasculitis. Hypersensitivity vasculitis is also known as cutaneous leukocytoclastic vasculitis and microscopic polyarteritis. It typically affects the small vessels and is idiopathic or triggered by infection or drug exposure. Clinical manifestations include urticaria, purpura, or a maculopapular rash, arthralgias, hypocomplementemia, and elevated inflammatory markers (Figure 2).
Figure 2. Hypersensitivity vasculitis.
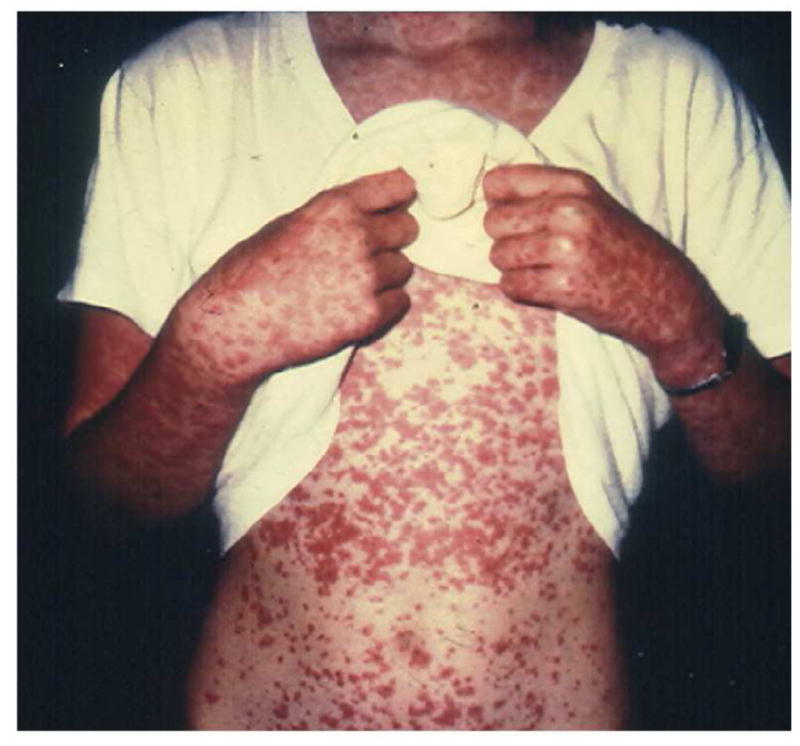
Hypersentivity vasculitis secondary to propylthiouracil exposure. Courtesy of David D. Sherry, MD, Philadelphia, PA.
Treatment
Therapy for mild HSP cases is primarily supportive with analgesics and non-steroidal anti-inflammatory drugs. However, current literature supports the notion that in the hospital setting, early use of corticosteroids for HSP is associated with improved outcomes, particularly GI co-morbidities.18 In more the severe hospitalized cases pulse methylprednisolone (30 mg/kg up to 1g) may be warranted. The optimal dose and duration of corticosteroids has not been well studied. In some cases oral corticosteroid doses of 2 mg/kg/day may be adequate. However, too short a course of corticosteroids or a rapid tapering of corticosteroids may precipitate a flare of symptoms. Corticosteroid treatment for mild cases of HSP remains controversial. A meta-analysis of 15 studies of patients with HSP treated at diagnosis with corticosteroids versus supportive care revealed that corticosteroid treatment significantly reduced the mean resolution time of abdominal pain and reduced the odds of developing persistent renal disease.19 In a 6-month prospective clinical trial of 223 children prednisone was effective in reducing the severity of abdominal and joint pain and in treating renal disease.20 However, in that same study prophylaxis with prednisone did not prevent the development of nephritis.20 In life-threatening cases or acute renal failure, plasmapheresis followed by a more potent immunosuppressive agent such as cyclophosphamide, azathioprine, or cyclosporine, should be considered.
Kawasaki disease (KD)
Etiology and epidemiology
KD is the second most common childhood vasculitis, accounting for 23% of all vasculitides.7 It affects primarily medium-sized blood vessels and is also known as mucocutaneous lymph node syndrome. The EULAR/PReS classification criteria for KD are listed in Table 3. Fewer than 4 criteria are required if there are characteristic coronary artery changes and fever.8 Ninety percent of cases occur in children less than 5 years old. In the US the annual incidence in children less than 5 years old is 20 per 100,000.21 The incidence is higher among children who live in eastern Asia at 100 per 100,000 in Japan22 and 69 per 100,000 in Taiwan.23 Similar to HSP, KD is more common in boys. Children less than 6 months of age are more likely to have atypical features and to develop coronary aneurysms. The cause of KD remains unknown although bacterial and viral infections, superantigens, genetics, and humoral factors such as anti-endothelial cell antibodies, ANCA, and circulating immune complexes may be involved.
Table 3.
EULAR/PReS classification criteria for Kawasaki disease8
Fever that persists for at least 5 days plus at least 4 of the following:
|
Clinical presentation
KD is a tri-phasic disease consisting of an acute febrile period that lasts up to 14 days, a subacute phase of 2–4 weeks, and a convalescent phase that can last months to years. The acute period is characterized by a persistent and high (>38.5° C) fever. The fever is typically minimally responsive to anti-pyretics. The fever is likely related to elevated concentrations of pro-inflammatory cytokines, particularly interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α).24 Conjunctivitis affects 85% of children and is bilateral, non-exudative, and limbic sparing. Other ocular symptoms include anterior uveitis, keratitis, papilledema, vitreous opacities, and subconjunctival hemorrhages (Figure 3A).25 Oral mucosal changes may include dry and cracked lips and strawberry tongue. Cervical adenopathy is the least common of the diagnostic criteria, detectable in 25% of children.26 The adenopathy is usually unilateral and limited to the anterior cervical chain (Figure 3B). Diffuse lymphadenopathy is unusual. The rash associated with KD is typically non-pruritic, macular or target lesions on the trunk and extremities. A perineal rash that desquamates by the end of the first week is common. Early extremity changes include diffuse erythema of the palms and soles and swelling of the dorsum of the hands and feet (Figure 3C); these changes usually last for less than 3 days. Sheet-like desquamation on the fingers and toes occurs towards the end of the acute phase.
Figure 3. Kawasaki disease.

A) Subconjunctival hemorrhage; B) Unliateral cervical lymphadenopathy; c) Pedal edema. Courtesy of Jon M. Burnham, MD MSCE, Philadelphia, PA.
There are also many symptoms of KD that are not part of diagnostic criteria. Gastrointestinal symptoms include diarrhea, vomiting, abdominal pain, and hydrops of the gallbladder. Genitourinary symptoms include scrotal pain and swelling, dysuria, and sterile pyuria. Arthritis affects up to 25% of children27 and the most commonly affected joints are the knees, ankles, and hips. The arthritis can be oligo- or polyarticular and is usually self-limited and non-destructive. The majority of children with KD are very irritable, likely secondary to aspectic meningitis and headache.28
Cardiovascular disease during the acute phase may include valvulitis, myocarditis, and pericarditis. Coronary dilation and aneurysms may be detected during the acute phase, but most develop during the convalescent phase. Up to 10% of children who develop coronary changes do not meet full criteria for KD.29 As many as 20% of untreated children develop aneurysms. Treatment with Intravenous immunoglobulin (IVIG) reduces the incidence of aneurysms by approximately 80%.30 Children younger than 12 months are at the highest risk for coronary aneurysms.
Several conditions can mimic KD including infections (Epstein Barr virus, adenovirus, echovirus, measles), toxin-mediated illnesses (toxic shock syndrome, scarlet fever), inflammatory conditions (systemic juvenile idiopathic arthritis, polyarteritis nodosa), hypersensitivity reactions (mercury), and drug reactions (Stevens Johnson syndrome).
Treatment
The goal of treatment in KD is to reduce inflammation and prevent the formation of coronary aneurysms. The American Heart Association (AHA) recommends treatment with high-dose aspirin (80–100 mg/kg/day) and IVIG (2 g/kg) within the first 10 days of disease.31 Once afebrile for 48 hours the aspirin should be adjusted to an anti-platelet dose of 3–5 mg/kg/day. Although corticosteroids are the cornerstone of therapy for most vasculitides, their use in KD for the primary treatment of KD is controversial.32,33 Approximately 10–15% of patients fail to respond to initial IVIG therapy; failure is defined as persistent fever or recurrence of fever within 36 hours of IVIG therapy. Persistent or recurring fever is concerning because it likely indicates on-going inflammation and is associated with an increased risk of developing coronary aneurysms.34 In a small multicenter randomized prospective trial of a second IVIG infusion versus infliximab, an anti-tumor necrosis (TNF)-α agent (5 mg/kg), for refractory KD there were no statistically significant differences between the two treatment groups in recurrence of fever, coronary artery outcomes, or laboratory markers of inflammation.35 However, current AHA guidelines recommend re-dosing IVIG at least once in the event of IVIG failure.31 If two or more doses of IVIG are ineffective then corticosteroid pulse therapy (30 mg/kg for 1–3 doses) or treatment with infliximab (5 mg/kg) should be considered.31 For uncomplicated KD, an echocardiogram should be performed at diagnosis, at 2 weeks, and then again at 6–8 weeks to assess treatment efficacy for the prevention of aneurysm formation.31
Polyarteritis Nodosa (PAN)
PAN is the third most common childhood vasculitis. PAN is predominantly a medium-sized vasculitis and accounts for 3% of all childhood vasculitides in the US.7 Classification of childhood-onset PAN is listed in Table 4. Similar to HSP, several reports suggest an association with FMF. Peak age of onset in children is 9 years.36 In adults PAN is classically associated with hepatitis B, however this association is less common in childhood. Childhood PAN is associated with fewer relapses and improved survival compared to adult-onset disease.36
Table 4.
EULAR/PReS classification criteria for childhood polyarteritis nodosa (PAN)8
Systemic inflammation with evidence of necrotizing vasculitis or angiographic abnormalities of medium or small-sized arteries plus one of the following:
|
PAN can affect the vascular supply to any organ, although the lungs are typically spared. Vascular insufficiency is most common to the skin, muscles, kidneys, and GI tract. Involvement of the heart, peripheral and central nervous systems are less common. Children may have fever, malaise, weight loss, myalgias, and arthralgias at presentation. Depending upon the distribution of involved vessels, children may have hypertension, ischemic heart disease, testicular pain, abdominal pain, hematuria, or proteinuria. Neurologic involvement may include mononeuritis multiplex with both sensory and motor deficits. Inflammation of the small arteries leads to vasculitic skin rashes including livedo reticularis, purpura, necrosis, and possibly digital gangrene (Figure 4A and B). Painful subcutaneous nodules along affected vessels are also a characteristic feature. Laboratory markers of systemic inflammation are usually quite elevated.
Figure 4. Polyarteritis nodosa.
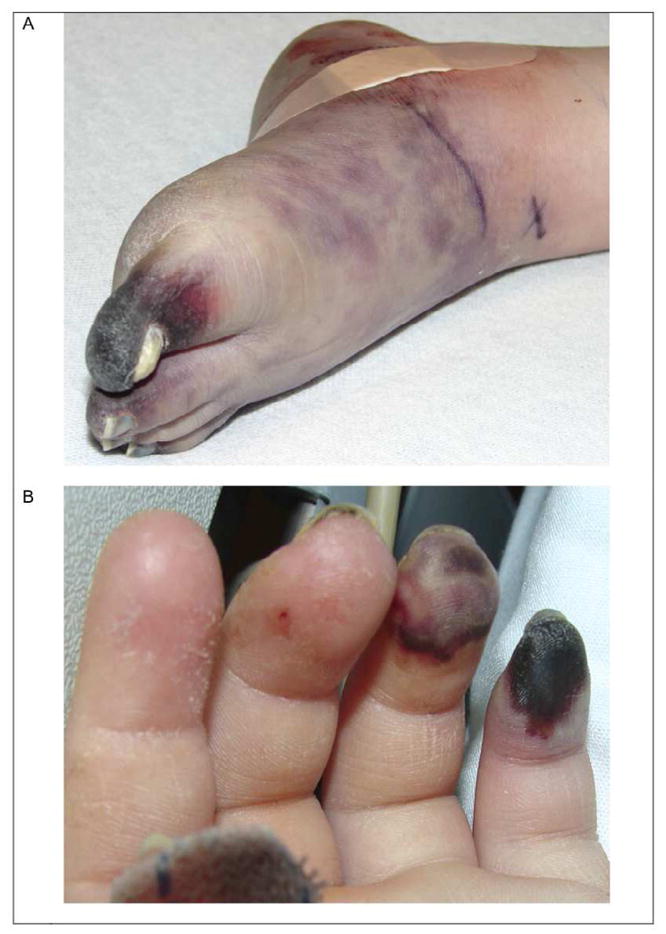
Digital necrosis of toes (A) and fingers (B) from PAN. Courtesy of David D. Sherry, MD, Philadelphia, PA.
There have not been any randomized clinical trials to compare different induction or maintenance therapies for childhood systemic PAN. Therefore, treatment is primarily based upon clinical experience and adult studies. Corticosteroids (1–2 mg/kg/day with or without initial corticosteroid pulses of 30 mg/kg) are the cornerstone of therapy. Additional therapy with cyclophosphamide (oral: 2 mg/kg/day, or IV: 750 mg/m2 monthly) may be warranted during the induction phase. In life-threatening or organ-threatening situations, plasmapheresis is indicated. Maintenance agents include azathioprine, methotrexate, IVIG, and mycophenolate mofetil. More recently, the efficacies of biologic agents and rituximab have been explored.37
Cutaneous PAN is limited to the skin and musculoskeletal system. Characteristic features include fever, painful subcutaneous nodules, purpura, livedo reticularis, myalgias, arthralgias, and non-destructive arthritis. The skin manifestations are usually limited to the lower extremities. Cutaneous PAN is often associated with antecedent streptococcal infection.38 NSAIDs and corticosteroids are the mainstay of therapy. In cases of persistent or relapsing disease steroid-sparing agents such as methotrexate, colchicine, and IVIG have been used. Cutaneous PAN rarely evolves into systemic PAN.
Takayasu Arteritis (TA)
TA is a granulomatous vasculitis that predominantly affects the aorta and its major branches. The EULAR/PReS consensus criteria for childhood TA are listed in Table 5.8 The majority of children are diagnosed during adolescence, with a mean age of 13 years.39 It is more common in females than males (3:1).39 The most commonly involved vessels in children are the aorta, renal, subclavian, and carotid arteries.39
Table 5.
EULAR/PReS classification criteria for childhood-onset Takayasu arteritis (TA)8
Characteristic angiographic abnormalities of the aorta or its main branches and pulmonary arteries plus one of the following:
|
Early diagnosis of TA in children is challenging because the presenting symptoms are usually non-specific. The most common complaints at diagnosis are headache (84%), dizziness (37%), abdominal pain (37%), claudication of the extremities (32%), fever (26%), and weight loss (10%).39 Other early indicators of disease are night sweats, back pain, myalgias, and arthralgias. Hypertension is present in approximately 90% of children at diagnosis.39 If left untreated more specific disease manifestations develop and are determined by the distribution of vessel involvement. Involvement of the aortic arch or its major branches is associated with CNS symptoms, claudication, absent peripheral pulses, and cardiac manifestations. CNS involvement may include headache, ischemic strokes, cerebral aneurysms, and seizures. Cardiac manifestations may include cardiomyopathy, congestive heart disease, and valvular disease. Involvement of the mid-aorta is associated with hypertension, abdominal pain, and lower extremity claudication.
Once TA is suspected imaging often helps to confirm the diagnosis. The gold standard for TA imaging is angiography. However, it is invasive and cannot detect thickened vessel walls, an early sign of inflammation. CT and MR angiograms are less invasive than conventional angiography and can detect luminal diameter changes (Figure 5A) and vessel wall thickening (Figure 5B).40
Figure 5. Takayasu Arteritis.

A) Contrast-enhanced MR angiography demonstrating narrowed right internal carotid and subclavian arteries; B) Thoracoabdominal CT angiography demonstrating thickening of the aortic wall.
Treatment of TA is challenging. Corticosteroids may induce remission in up to 60% of patients, however, half of these patients flare with tapering.41 There are anecdotal reports of treatment with methotrexate, azathioprine, and biologics such as infliximab. Cyclophosphamide should be considered for life-threatening or organ-threatening cases.
Childhood primary central nervous system vasculitis (cPACNS)
Previously healthy children with cPACNS may present with devastating subacute or acute onset neurologic changes and psychiatric symptoms. If the disease is diagnosed early, the inflammation and neurologic damage may be reversible. The Calabrese criteria, designed for adults, requires a new neurological deficit plus angiographic or histologic evidence of CNS vasculitis.42 Two subtypes of cPACNS are recognized: 1) angiography-positive cPACNS that affects the medium and large vessels, and 2) angiography-negative cPACNS which primarily affects the small vessels. Children with angiography-positive cPACNS typically present with focal neurologic symptoms such as unilateral sensory or motor deficits. Systemic inflammatory markers and CSF fluid analysis may be normal.43 The classic MRI finding is a focal area of acute ischemia in a vascular distribution. Diagnosis is confirmed by conventional angiography or MRA that demonstrate large or medium-sized vessel stenosis, tortuosity, beading, or occlusion.
Angiography-positive cPACNS is further defined as progressive or non-progressive based on the appearance of new lesions 3 months after diagnosis. Diffuse neurologic symptoms such as headaches, cognitive dysfunction, and behavioral issues are more common in progressive disease.44 Progressive disease also tends to be multifocal. The treatment of non-progressive cPACNS is controversial but may include anti-coagulation and corticosteroids. Corticosteroids may help prevent recurrence and improve neurologic recovery.45 Treatment of progressive cPACNS includes 6 months of induction therapy with cyclophosphamide (500–750 mg/m2/month) and corticosteroids (2 mg/kg/day initially) followed by maintenance therapy with mycophenolate mofetil or azathioprine for 18 months.46
Angiography-negative cPACNS may present with systemic features including fever and malaise in addition to diffuse or focal neurologic symptoms.44 Systemic inflammatory markers may be normal but CSF fluid analysis typically reveals pleocytosis, elevated protein, or increased opening pressure. MRI findings are usually multifocal and may can be unilateral or bilateral and involve the gray or white matter.44 Leptomeningeal enhancement on imaging can help to distinguish this disease from demyelinating conditions. Conventional angiography, by definition, is negative. If cPACNS is suspected then a brain biopsy must be performed to confirm the diagnosis. Typical histologic findings are a lymphocytic and non-granulomatous vasculitis.47,48 Treatment of angiography-negative cPACNS includes 6 months of induction therapy with cyclophosphamide (500–750 mg/m2) and corticosteroids (2 mg/kg/day initially) followed by maintenance therapy with mycophenolate mofetil or azathioprine for 18 months.46,47
ANCA- vasculitis
The ANCA -associated vasculitides (AAV) are a group of pediatric conditions classified by small- and medium-vessel inflammation, multi-organ system involvement, and potentially life-threatening disease. They are associated with a high frequency of disease- and treatment-related morbidities. The 3 classic vasculitides in the AAV group are Granulomatosus with polyangiitis (GPA, formerly known as Wegener’s granulomatosis), microscopic polyangiitis (MPA), and Churg-Strauss syndrome (CSS). In the acute phase of disease the major co-morbid conditions may include pulmonary hemorrhage, respiratory failure, rapidly progressive glomerular nephritis, and acute renal failure. Untreated AAV follows a severe course, with near 100% mortality49 and a mean survival of 5 months.50 Additionally, renal failure at presentation confers a high risk of end-stage renal disease and death despite potent immunosuppressive therapy.51 Relapses are very common and occur in up to 60% of patients.52,53 In comparison to adults, children are more likely to have multiple organ involvement, renal involvement, and subglottic stenosis.54,55
GPA primarily affects the upper and lower respiratory tracts and the kidneys. The pediatric GPA classification criteria are listed in Table 6.8 The median age of diagnosis in childhood onset disease is 14 years.54 Constitutional symptoms such as fever, malaise, and weight loss are present in 90% of children at diagnosis.54 80% have pulmonary manifestations that may include pulmonary hemorrhage, nodules, infiltrates, pleurisy, oxygen-dependency, and respiratory failure (Figure 6A).54 80% have upper respiratory disease that may include oral or nasal ulcerations, nasal septum perforation (with subsequent saddle nose deformity), recurrent epistaxis, sinusitis, mastoiditis, hearing loss, and subglottic stenosis (Figure 6B).54 75% have renal involvement that may include hematuria, proteinuria, glomerulonephritis, and kidney failure.54 The renal disease is classically necrotizing and pauci-immune on biopsy. 90% of patients with GPA are ANCA positive, and 80–90% of those patients are cytoplasmic-ANCA (c-ANCA)/ pr3-positive.56 Biopsy of inflamed tissue is helpful to confirm the diagnosis.
Table 6.
EULAR/PReS classification criteria for childhood-onset Granulomatosus with polyangiitis (GPA)8
Diagnosis requires 3 of the following 6 criteria:
|
Figure 6. Granulomatosis with polyangiitis.

A) Contrast-enhanced CT of the chest demonstrating pulmonary nodules and infiltrates; B) Contrast-enhanced CT of head showing diffuse sinus disease.
MPA is a necrotizing, non-granulomatous, pauci-immune disease that affects the small vessels. The most common features of MPA include pulmonary capillaritis and necrotizing glomerulonephritis. Almost all children have constitutional symptoms such as fever, malaise, arthralgias, myalgias, and weight loss.57 One-third of children present with renal failure and nearly 100% have renal disease that may include hypertension, hematuria, or proteinuria. 57 Ischemic cerebral insults (30%) and necrotizing vasculitic lesions of the skin (30%) are common.57 MPA is associated with a high titer of c- or peri-nuclear (p-) ANCA.
CSS is a granulomatous vasculitis of small and medium-sized vessels that primarily affects individuals with severe asthma or allergies. Pediatric-specific criteria for CSS do not exist. The American College of Rheumatology (ACR) disease criteria for adult-onset disease are listed in Table 7. CSS usually has an insidious onset over years. The most common features in children at diagnosis are asthma (91%), pulmonary infiltrates (85%), sinusitis (77%), skin involvement (66%), cardiac disease (55%), gastrointestinal symptoms (40%), peripheral neuropathy (39%), and kidney disease (16%).58 Vasculitic skin rashes are also common. The renal disease is usually mild and rarely progresses.59 25% of children are ANCA-positive.
Table 7.
ACR classification criteria for adult-onset Churg-Strauss syndrome (CSS)
Diagnosis requires 4 of the following:
|
Treatment regimens for AAV depend upon the specific type of vasculitis and the end-organ manifestations. Almost all knowledge about the optimal treatment and outcomes of children with AAV have been adapted from adult studies or come from a small collection of case series. Standard induction therapy for adults with severe systemic AAV includes corticosteroids and cyclophosphamide (oral or IV).60 This regimen, however, is suboptimal as there are not only significant treatment-related toxicities including serious infection, hemorrhagic cystitis, infertility, and malignancy, but additionally a high risk of relapse.52,53 Recent studies in adults, therefore, have examined the role of biologic medications and plasmapheresis (PP) as adjunct therapy in an effort to find strategies with comparable efficacy, lower toxicity, and fewer relapses. Methotrexate and corticosteroids are used for induction in milder cases. Maintenance therapy is typically with mycophenolate mofetil or azathioprine for 18 to 24 months. Infliximab (5 mg/kg twice a month), rituximab (375 mg/m2/week for 4 weeks), and IVIG (2g/kg/month) are options for refractory disease.
Summary
Pediatric vasculitis is a challenging and complex group of conditions. The site of vessel involvement, size of the affected vessels, extent of vascular injury, and underlying pathology determine the disease phenotype and severity. The most common vasculitides are HSP and KD. Almost all knowledge about the optimal treatment and outcomes of children with vasculitis, with the exception of HSP and KD, have been adapted from adult studies or come from a small collection of case series. Early diagnosis, which requires a high level of clinical suspicion, may help to improve outcomes. Pediatricians should consider vasculitis as part of the differential diagnosis in children with evidence of systemic inflammation and multisystem disease that cannot be otherwise explained.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936–41. doi: 10.1136/ard.2005.046300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet. 2002;360:1197–202. doi: 10.1016/S0140-6736(02)11279-7. [DOI] [PubMed] [Google Scholar]
- 3.Natter M, Winsor J, Fox K, et al. Rheumatology ACo. The Childhood Arthritis & Rheumatology Research Alliance Network Registry: Demographics and Characteristics of the Initial 6-Month Cohort. Pediatric Rheumatology Symposium (PRSYM); Miami, Florida. 2011. pp. 43–4. [Google Scholar]
- 4.Denardo BA, Tucker LB, Miller LC, Szer IS, Schaller JG. Demography of a regional pediatric rheumatology patient population. Affiliated Children’s Arthritis Centers of New England. J Rheumatol. 1994;21:1553–61. [PubMed] [Google Scholar]
- 5.Malleson PN, Fung MY, Rosenberg AM. The incidence of pediatric rheumatic diseases: results from the Canadian Pediatric Rheumatology Association Disease Registry. The Journal of rheumatology. 1996;23:1981–7. [PubMed] [Google Scholar]
- 6.Bowyer S, Roettcher P. Pediatric rheumatology clinic populations in the United States: results of a 3 year survey. Pediatric Rheumatology Database Research Group. J Rheumatol. 1996;23:1968. [PubMed] [Google Scholar]
- 7.Bowyer S, Roettcher P. Pediatric rheumatology clinic populations in the United States: results of a 3 year survey. Pediatric Rheumatology Database Research Group. J Rheumatol. 1996;23:1968–74. [PubMed] [Google Scholar]
- 8.Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schonlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010;69:798–806. doi: 10.1136/ard.2009.116657. [DOI] [PubMed] [Google Scholar]
- 9.Stefansson Thors V, Kolka R, Sigurdardottir SL, Edvardsson VO, Arason G, Haraldsson A. Increased frequency of C4B*Q0 alleles in patients with Henoch-Schonlein purpura. Scand J Immunol. 2005;61:274–8. doi: 10.1111/j.1365-3083.2005.01533.x. [DOI] [PubMed] [Google Scholar]
- 10.Gershoni-Baruch R, Broza Y, Brik R. Prevalence and significance of mutations in the familial Mediterranean fever gene in Henoch-Schonlein purpura. J Pediatr. 2003;143:658–61. doi: 10.1067/S0022-3476(03)00502-X. [DOI] [PubMed] [Google Scholar]
- 11.Weiss PF, Klink AJ, Luan X, Feudtner C. Temporal association of Streptococcus, Staphylococcus, and parainfluenza pediatric hospitalizations and hospitalized cases of Henoch-Schonlein purpura. The Journal of rheumatology. 2010;37:2587–94. doi: 10.3899/jrheum.100364. [DOI] [PubMed] [Google Scholar]
- 12.Saulsbury FT. Henoch-Schonlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999;78:395–409. doi: 10.1097/00005792-199911000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Trapani S, Micheli A, Grisolia F, et al. Henoch Schonlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum. 2005;35:143–53. doi: 10.1016/j.semarthrit.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Saulsbury FT. Henoch-Schonlein purpura. Current opinion in rheumatology. 2010;22:598–602. doi: 10.1097/BOR.0b013e32833af608. [DOI] [PubMed] [Google Scholar]
- 15.Narchi H. Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child. 2005;90:916–20. doi: 10.1136/adc.2005.074641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart M, Savage JM, Bell B, McCord B. Long term renal prognosis of Henoch-Schonlein purpura in an unselected childhood population. Eur J Pediatr. 1988;147:113–5. doi: 10.1007/BF00442205. [DOI] [PubMed] [Google Scholar]
- 17.Saulsbury FT. Epidemiology of Henoch-Schonlein purpura. Cleve Clin J Med. 2002;69(Suppl 2):SII87–9. doi: 10.3949/ccjm.69.suppl_2.sii87. [DOI] [PubMed] [Google Scholar]
- 18.Weiss PF, Klink AJ, Localio R, et al. Corticosteroids may improve clinical outcomes during hospitalization for Henoch-Schonlein purpura. Pediatrics. 2010;126:674–81. doi: 10.1542/peds.2009-3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss PF, Feinstein JA, Luan X, Burnham JM, Feudtner C. Effects of corticosteroid on Henoch-Schonlein purpura: a systematic review. Pediatrics. 2007;120:1079–87. doi: 10.1542/peds.2007-0667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jauhola O, Ronkainen J, Koskimies O, et al. Renal manifestations of Henoch-Schonlein purpura in a 6-month prospective study of 223 children. Archives of disease in childhood. 2010;95:877–82. doi: 10.1136/adc.2009.182394. [DOI] [PubMed] [Google Scholar]
- 21.Holman RC, Belay ED, Christensen KY, Folkema AM, Steiner CA, Schonberger LB. Hospitalizations for Kawasaki syndrome among children in the United States, 1997–2007. The Pediatric infectious disease journal. 2010;29:483–8. doi: 10.1097/INF.0b013e3181cf8705. [DOI] [PubMed] [Google Scholar]
- 22.Yanagawa H, Nakamura Y, Yashiro M, et al. Results of the nationwide epidemiologic survey of Kawasaki disease in 1995 and 1996 in Japan. Pediatrics. 1998;102:E65. doi: 10.1542/peds.102.6.e65. [DOI] [PubMed] [Google Scholar]
- 23.Huang WC, Huang LM, Chang IS, et al. Epidemiologic features of Kawasaki disease in Taiwan, 2003–2006. Pediatrics. 2009;123:e401–5. doi: 10.1542/peds.2008-2187. [DOI] [PubMed] [Google Scholar]
- 24.Leung DY. The potential role of cytokine-mediated vascular endothelial activation in the pathogenesis of Kawasaki disease. Acta Paediatr Jpn. 1991;33:739–44. doi: 10.1111/j.1442-200x.1991.tb02602.x. [DOI] [PubMed] [Google Scholar]
- 25.Kumagai N, Ohno S. Kawasaki Disease. In: Holland G, Wilhelmus K, editors. Ocular immunity and infection. St. Louis: Mosby; 1996. [Google Scholar]
- 26.Sung RY, Ng YM, Choi KC, Mok GC, Cheng YW, Ho MH. Lack of association of cervical lymphadenopathy and coronary artery complications in Kawasaki disease. The Pediatric infectious disease journal. 2006;25:521–5. doi: 10.1097/01.inf.0000215263.96289.1c. [DOI] [PubMed] [Google Scholar]
- 27.Melish ME. Kawasaki syndrome: a 1986 perspective. Rheumatic diseases clinics of North America. 1987;13:7–17. [PubMed] [Google Scholar]
- 28.Dengler LD, Capparelli EV, Bastian JF, et al. Cerebrospinal fluid profile in patients with acute Kawasaki disease. The Pediatric infectious disease journal. 1998;17:478–81. doi: 10.1097/00006454-199806000-00008. [DOI] [PubMed] [Google Scholar]
- 29.Sundel RP. Update on the treatment of Kawasaki disease in childhood. Curr Rheumatol Rep. 2002;4:474–82. doi: 10.1007/s11926-002-0053-6. [DOI] [PubMed] [Google Scholar]
- 30.Newburger JW, Takahashi M, Burns JC, et al. The treatment of Kawasaki syndrome with intravenous gamma globulin. The New England journal of medicine. 1986;315:341–7. doi: 10.1056/NEJM198608073150601. [DOI] [PubMed] [Google Scholar]
- 31.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114:1708–33. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 32.Newburger JW, Sleeper LA, McCrindle BW, et al. Randomized trial of pulsed corticosteroid therapy for primary treatment of Kawasaki disease. The New England journal of medicine. 2007;356:663–75. doi: 10.1056/NEJMoa061235. [DOI] [PubMed] [Google Scholar]
- 33.Sundel RP, Baker AL, Fulton DR, Newburger JW. Corticosteroids in the initial treatment of Kawasaki disease: report of a randomized trial. The Journal of pediatrics. 2003;142:611–6. doi: 10.1067/mpd.2003.191. [DOI] [PubMed] [Google Scholar]
- 34.Beiser AS, Takahashi M, Baker AL, Sundel RP, Newburger JW. A predictive instrument for coronary artery aneurysms in Kawasaki disease. US Multicenter Kawasaki Disease Study Group. The American journal of cardiology. 1998;81:1116–20. doi: 10.1016/s0002-9149(98)00116-7. [DOI] [PubMed] [Google Scholar]
- 35.Burns JC, Best BM, Mejias A, et al. Infliximab treatment of intravenous immunoglobulin-resistant Kawasaki disease. The Journal of pediatrics. 2008;153:833–8. doi: 10.1016/j.jpeds.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozen S, Anton J, Arisoy N, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. The Journal of pediatrics. 2004;145:517–22. doi: 10.1016/j.jpeds.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 37.Eleftheriou D, Melo M, Marks SD, et al. Biologic therapy in primary systemic vasculitis of the young. Rheumatology. 2009;48:978–86. doi: 10.1093/rheumatology/kep148. [DOI] [PubMed] [Google Scholar]
- 38.David J, Ansell BM, Woo P. Polyarteritis nodosa associated with streptococcus. Archives of disease in childhood. 1993;69:685–8. doi: 10.1136/adc.69.6.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cakar N, Yalcinkaya F, Duzova A, et al. Takayasu arteritis in children. The Journal of rheumatology. 2008;35:913–9. [PubMed] [Google Scholar]
- 40.Kissin EY, Merkel PA. Diagnostic imaging in Takayasu arteritis. Current opinion in rheumatology. 2004;16:31–7. doi: 10.1097/00002281-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 41.Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Annals of internal medicine. 1994;120:919–29. doi: 10.7326/0003-4819-120-11-199406010-00004. [DOI] [PubMed] [Google Scholar]
- 42.Calabrese LH, Furlan AJ, Gragg LA, Ropos TJ. Primary angiitis of the central nervous system: diagnostic criteria and clinical approach. Cleve Clin J Med. 1992;59:293–306. doi: 10.3949/ccjm.59.3.293. [DOI] [PubMed] [Google Scholar]
- 43.Benseler SM, Silverman E, Aviv RI, et al. Primary central nervous system vasculitis in children. Arthritis and rheumatism. 2006;54:1291–7. doi: 10.1002/art.21766. [DOI] [PubMed] [Google Scholar]
- 44.Cellucci T, Benseler SM. Central nervous system vasculitis in children. Current opinion in rheumatology. 2010;22:590–7. doi: 10.1097/BOR.0b013e32833c723d. [DOI] [PubMed] [Google Scholar]
- 45.Soon G, IY, Branson H. Nonprogressive primary CNS vasculitis in children: immunosuppression reduces recurrent ischemic event risk. Arthritis & Rheumatism. 2008;58(S9):s942. [Google Scholar]
- 46.Sen ES, Leone V, Abinun M, et al. Treatment of primary angiitis of the central nervous system in childhood with mycophenolate mofetil. Rheumatology. 2010;49:806–11. doi: 10.1093/rheumatology/kep453. [DOI] [PubMed] [Google Scholar]
- 47.Benseler SM, deVeber G, Hawkins C, et al. Angiography-negative primary central nervous system vasculitis in children: a newly recognized inflammatory central nervous system disease. Arthritis and rheumatism. 2005;52:2159–67. doi: 10.1002/art.21144. [DOI] [PubMed] [Google Scholar]
- 48.Elbers J, Hutchinson C, Halliday W. Brain biopsies in children with smalll-vessel primary CNS vasculitis. Arthritis & Rheumatism. 2008;58(S9):S712. [Google Scholar]
- 49.Cassidy JT, Petty RE, Laxer RM, Lindsley C. Textbook of Pediatric Rheumatology. 6. Philadelphia: Saunders Elsevier; 2011. [Google Scholar]
- 50.Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis) Br Med J. 1958;2:265–70. doi: 10.1136/bmj.2.5091.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jayne DR, Gaskin G, Rasmussen N, et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol. 2007;18:2180–8. doi: 10.1681/ASN.2007010090. [DOI] [PubMed] [Google Scholar]
- 52.Nachman PH, Hogan SL, Jennette JC, Falk RJ. Treatment response and relapse in antineutrophil cytoplasmic autoantibody-associated microscopic polyangiitis and glomerulonephritis. J Am Soc Nephrol. 1996;7:33–9. doi: 10.1681/ASN.V7133. [DOI] [PubMed] [Google Scholar]
- 53.Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352:351–61. doi: 10.1056/NEJMoa041884. [DOI] [PubMed] [Google Scholar]
- 54.Cabral DA, Uribe AG, Benseler S, et al. Classification, presentation, and initial treatment of Wegener’s granulomatosis in childhood. Arthritis and rheumatism. 2009;60:3413–24. doi: 10.1002/art.24876. [DOI] [PubMed] [Google Scholar]
- 55.Akikusa JD, Schneider R, Harvey EA, et al. Clinical features and outcome of pediatric Wegener’s granulomatosis. Arthritis and rheumatism. 2007;57:837–44. doi: 10.1002/art.22774. [DOI] [PubMed] [Google Scholar]
- 56.Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis and rheumatism. 1998;41:1521–37. doi: 10.1002/1529-0131(199809)41:9<1521::AID-ART2>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 57.Peco-Antic A, Bonaci-Nikolic B, Basta-Jovanovic G, et al. Childhood microscopic polyangiitis associated with MPO-ANCA. Pediatric nephrology. 2006;21:46–53. doi: 10.1007/s00467-005-2063-x. [DOI] [PubMed] [Google Scholar]
- 58.Zwerina J, Eger G, Englbrecht M, Manger B, Schett G. Churg-Strauss syndrome in childhood: a systematic literature review and clinical comparison with adult patients. Semin Arthritis Rheum. 2009;39:108–15. doi: 10.1016/j.semarthrit.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Lanham JG, Elkon KB, Pusey CD, Hughes GR. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine. 1984;63:65–81. doi: 10.1097/00005792-198403000-00001. [DOI] [PubMed] [Google Scholar]
- 60.Molloy ES, Langford CA. Advances in the treatment of small vessel vasculitis. Rheum Dis Clin North Am. 2006;32:157–72. x. doi: 10.1016/j.rdc.2005.12.002. [DOI] [PubMed] [Google Scholar]