

Abstract
Aim
Abnormal hypermethylation of CpG islands associated with tumor suppressor genes can lead to transcriptional silencing in neoplasia. The aim of this study was to investigate the promoter methylation and expression of E-cadherin gene in gastric cardiac adenocarcinoma (GCA).
Methods
A nested MSP approach, immunohistochemistry method and RT-PCR were used respectively to examine the methylation status of the 5' CpG island of E-cadherin, its protein expression and mRNA expression in tumors and corresponding normal tissues.
Results
E-cadherin was methylated in 63 of 92 (68.5%) tumor specimens, which was significantly higher than that in corresponding normal tissues (P < 0.001). Methylation frequencies of stage III and IV tumor tissues was significantly higher than that in stage I and II tumor tissues (P = 0.01). Methylation status of poor differentiation group was significantly higher than moderate and poor-moderate differentiation groups (P < 0.01). By immunostaining 51 of 92 tumor tisssues demonstrated heterogeneous, positive immunostaining of tumor tissues (44.6%), significantly different from matched normal tissues (P < 0.001). Positive immunostaining of stage III and IV tumor tissues was significantly lower than stage I and II tumor tissues (P < 0.01). Poor differentiation group was also significantly lower than moderate and poor-moderate differentiation groups (P < 0.05). 80 percent of tumor tissues with E-cadherin gene methylated showed inactivated mRNA expression.
Conclusions
High methylation status of the 5' CpG island of E-cadherin gene may be one of the mechanisms in the development of gastric cardiac adenocarcinoma.
Keywords: E-cadherin, methylation, gastric cardiac adenocarcinoma
Introduction
E-cadherin is a Mr 120,000 transmembrane glycoprotein expressed on epithelial cells and is responsible for homophilic, Ca2+-dependent intercellular adhesion that is essential for the maintenance of normal tissue architecture in epithelial tissues [1]. The cytoplasmic domain of E-cadherin binds to α-, β-, and γ-catenins, which are in turn linked to actins, and this interaction is critical for its function [2]. Cell-cell and cell-matrix interactions are crucially involved in neoplastic transformation and metastasis [3,4]. Defective cell adhesion may contribute to loss of contact inhibition of growth and loss of cell adhesion may account for the ability of cancer cells to cross normal tissue boundaries and metastasis [5]. The importance of E-cadherin in maintaining cell adhesion implies that its dysfunction may play an important role in tumorigenesis. Loss of E-cadherin expression occurs in a variety of human tumors and is hypothesized to be an important step in the progression from tumor formation to invasion and metastasis [6].
In recent years, there are several studies on the critical role of E-cadherin in tumorigenesis. It has been shown that germ-line mutations of the E-cadherin gene is related to familial gastric and colorectal cancer [7,8]. Furthermore, somatic mutations of E-cadherin were also found in gastric carcinoma and allelic loss of the E-cadherin locus at 16q22.1 has been reported in different epithelial tumors such as breast, ovarian, endometrial, and prostate carcinomas [9,10]. Except for this genetic alterations, epigenetic alteration include hypermethylation of the 5' CpG island within the promoter of E-cadherin is also responsible for transcriptional repression of the gene [11]. It is known that abnormal hypermethylation of CpG islands associated with tumor suppressor genes can lead to transcriptional silencing in neoplasia [12]. Indeed, methylation-associated silencing of E-cadherin represents the most common cause for its inactivation in several cancers such as liver, prostate, breast and esophageal [13-15].
Gastric cardiac adenocarcinoma (GCA), which was formerly registered as esophageal cancer or gastric cancer, has been diagnosed independently in very recent years, due to the improvement in early endoscopic screening and pathologic diagnosis. China is a country with high incidence regions of GCA, Especially in Taihang mountain of North China. Exogenous factors including nutrition deficiency, unhealthy living habits, consumption of alcohol and tobacco, pathogenic infections are generally considered as the risk factors for developing GCA in China [16-18]. However, only a subset of individuals exposed to the above listed exogenous risk factors would develop GCA, suggesting that multiple genetic and epigenetic events may contribute to the progression of GCA. It is now increasingly recognized that epigenetic silencing of gene expression by promoter CpG hypermethylation is an important alternative mechanism in inactivating tumour suppressor genes and tumour associated genes in cancers [19]. Mutations of the E-cadherin gene are rare in GCA, thus, in this study, we evaluated the role of methylation of the 5' CpG island of E-cadherin and its correlation with reduced E-cadherin expression in GCA.
Methods
Patients and specimens
Tumor and paired normal tissue specimens were obtained from 92 patients. These tissues were divided into two parallel parts, one part was frozen and stored at -80°C until RNA was extracted, the other part was formalin-fixed and paraffin-embedded. The cases were all inpatients for surgical treatment in the Fourth Affiliated Hospital, Hebei Medical University between 2004 and 2006. Histological tumor typing was carried out on the basis of resected specimens in the Department of Pathology of the same hospital. All gastric cardiac carcinomas were adenocarcinomas with their epicenters at the gastroesophageal junction, i.e. from 1 cm above until 2 cm below the junction between the end of the tubular esophagus and the beginning of the saccular stomach [20]. Information on TNM staging was available from hospital recordings and pathological diagnosis. The study was approved by the Ethics Committee of Hebei Cancer Institute and informed consent was obtained from all recruited subjects.
Methylation specific polymerase chain reaction (MSP) for E-cadherin promoter methylation
Genomic DNA from gastric cardiac adenocarcinomas and adjacent nonmalignant sections was isolated from paraffin-embedded tissue slides by standard methods using a simplified proteinase K digestion method. To examine the DNA methylation patterns, we treated genomic DNA with sodium bisulfite, as described previously [21]. In brief, 2 μg of DNA were denatured with 2 M NaOH at 37°C for 10 minutes, followed by incubation with 3 M sodium bisulphite, pH5.0, at 50°C for 16 hours. Bisulphite treated DNA was then purified (DNA Cleanup Kit; Promega, Madison, Wisconsin, USA), incubated with 3 M NaOH at room temperature for five minutes, precipitated with 10 M ammonium acetate and 100% ethanol, washed with 70% ethanol, and resuspended in 20 μl of distilled water.
A nested PCR approach was used to determine the methylation status within the E-cadherin CpG island in Exon 1 (sequence -126 bp to +144 bp relative to transcription start, GenBank accession number D49685) that has been published previously [15]. In the first round of PCR, 100 ng of bisulfite-treated DNA were amplified. The sequencing primers were 5'-GTTTA GTTTTGGGGAGGGGTT-3' (sense) and 5'-ACTAC TACTCCAAAAACCCATAACTAA-3' (antisense), and the cycling conditions were one cycle of 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 50°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min. The size of the product after this initial PCR reaction was 270 bp. For the second round of PCR, this product was diluted 1: 50 in water, and 2 μl of the dilution were used for MSP. Nested primer sequences for E-cadherin for the methylated reaction were 5'-TG TAGTTACGTATTTATTTTTAGTGGCGTC-3' (sense) and 5'-CGAATACGATCGAATCGAACCG-3' (antisense), and primer sequences for the unmethylated reaction were 5'-TGGTTGTAGTTATGTATTTATT TTTAGTGGTGTT-3' (sense) and 5'-ACACCAAATA CAATCAAATCAAACCAAA-3' (antisense). PCR parameters were as listed above, except that the annealing temperatures for the methylated and unmethylated reactions were 64°C and 62°C, respectively. The product sizes of the methylated and unmethylated reactions were 112 and 120 bp, respectively. The breast cancer cell line MB-MDA-231, which demonstrates methylation and silencing of E-cadherin and reagent blanks were used as positive and negative controls.
Immunohistochemical staining for E-cadherin
E-cadherin expression was determined by immunostaining using the avidin-biotin complex immunoperoxidase method, which was performed on parallel histopathological sections from paraffin-embedded tumor section and paired normal tissue. Endogenous peroxidase was blocked with 3% hydrogen peroxide for 10 minutes, followed by microwave antigen retrieval for nine minutes at 98°C in 10 mM sodium citrate buffer (pH 6.0) and incubated in 2% normal horse serum to minimize non-specific binding. The slides were sequentially incubated with primary monoclonal, mouse anti-E-cadherin antibody (1: 100 dilution in phosphate buffered saline, Santa cruz, sc-8426) overnight at 4°C, biotinylated secondary antibody for 30 min at 37°C and ABC reagent for 45 min at 37°C. 0.5% 3,3'-Diaminobenzidine (Sigma, St Louis, MO) was used as the chromagen. For a negative control, the primary antibody was replaced with mouse IgG. Slides with normal gastric mucosa were used as a positive control.
Measurement of mRNA expression of E-cadherin
RNA was extracted from frozen section tissues by standard methods using Trizol (Invitrogen, USA). cDNA was synthesized using the Advantage RT-for-PCR kit (Clontech, Palo Alto, CA) with oligo (dT) priming as recommended in the protocol provided. The GAPDH gene was used as a control. Primer sequences of E-cadherin were 5°‰-CGACCCAACC CAAGAATCTA-3°‰(sense) and 5°‰-AATGGCAG GAATTTGCAATC-3°‰(antisense), primer sequence of GAPDH were 5°‰-GGGAAACTGTGGCGT GAT-3°‰(sense) and 5°‰-GTGGTCGTTGAGGG CAAT-3°‰(antisense), product size were 202 bp and 342 bp, respectively. PCR products were resolved on 2% agarose gels and signal intensities were quantified using a computer imaging system. The levels of gene transcripts were quantified as the ratio of the intensity of the E-cadherin signal to the intensity of β-actin. Inactivated expression was scored when expression of an E-cadherin gene in the tumor sample was < 25% of its expression in the corresponding normal sample.
Statistical analysis
Statistical analysis was performed using SPSS10.0 software package (SPSS Company, Chicago, Illinois, USA). Fisher's exact test and Chi-square test were used to assess statistical significance of differences and compare categorical associations. Two-sided tests were used to determine significance, and P values less than 0.05 were regarded as statistically significant for all statistic tests.
Results
Subject characteristics
As shown in Table 1, 92 GCA patients were obtained in this research, including 73 male and 19 female, age ranged from 38~76, mean age 56.9. All of the cases were classified into 4 tumor-node-metastasis (TNM) stages according to UICC standard, 8 of stage I (8.7%), 32 of stage II (34.8%), 38 of stage III (41.3%), 14 of stage IV (15.2%). According to the pathological phases, the cases were classified into 3 groups, 42 (45.7%) of moderate group, 34 (36.9%) of poor-moderate group and 16 (17.4%) of poor group.
Table 1.
Clinical and pathological characteristics of GCA patients
| Groups | n (%) |
|---|---|
| Sex | |
| Male | 73 (79.3) |
| Female | 19 (20.7) |
| Mean age in years (SD) | 56.9 (8.86) |
| TNM stage | |
| I | 8 (8.7) |
| II | 32 (34.8) |
| III | 38 (41.3) |
| IV | 14 (15.2) |
| Pathological differentiation of tumor | |
| moderate | 42 (45.7) |
| poor-moderate | 34 (36.9) |
| poor | 16 (17.4) |
Methylation analysis of E-cadherin gene
The methylation analysis was successfully performed in all tumor and paired normal tissue specimens (Figure 1). For 10% of samples, methylation analysis was repeated for quality control. In 63 (68.5%) of 92 GCA tumors E-cadherin methylation was detected, while only in 10 (10.9%) of paired normal tissues E-cadherin methylation was detected. The frequency of E-cadherin methylation of tumor tissues was significantly higher than paired normal tissues (P < 0.001). When stratified for TNM stages, Frequency of E-cadherin gene methylation of GCA patients with stage III and stage IV (78.8%) were significantly higher than GCA patients with stage I and stage II (55%) (χ2 = 5.96, P = 0.01). When stratified for pathological stages, the E-cadherin gene methylation frequencies of moderate, poor-moderate and poor group were 52.4%, 73.5% and 100%, frequency of E-cadherin gene methylation of poor group significantly higher than that in moderate and poor-moderate groups (χ2 = 8.92, P = 0.003) (Table 2).
Figure 1.
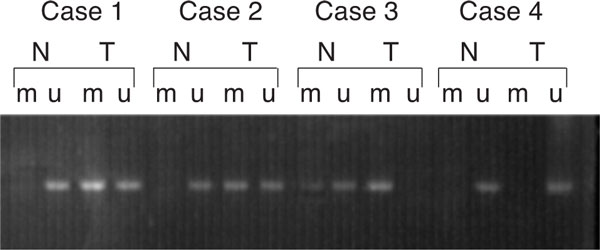
Methylation analysis of E-cadherin in tumor tissue (T) and corresponding normal tissue (N). u: indicates the presence of unmethylated genes; m: indicates the presence of methylated genes. Cases 1 and 2: tumor-specific methylation; Case 3: the tumor is fully methylated, whereas the corresponding normal tissue has a very faint band demonstrating methylation; Case 4: both of tumor and corresponding normal tissue unmethylated.
Table 2.
E-cadherin methylation and immunohistochemical staining characteristics of GCA patients
| Group | methylation status | P | Immunohistochemical staining | P | ||
|---|---|---|---|---|---|---|
| M | U | - | + | |||
| TNM stage | ||||||
| I | 4 | 4 | 2 | 6 | ||
| II | 18 | 14 | 13 | 9 | ||
| III | 29 | 9 | 26 | 12 | ||
| IV | 12 | 2 | 0.015a | 10 | 4 | 0.002a |
| Pathological differentiation of tumor | ||||||
| moderate | 22 | 20 | 20 | 2 | ||
| poor-moderate | 25 | 9 | 18 | 16 | ||
| poor | 16 | 0 | 0.003b | 13 | 3 | 0.022b |
a P value of stage III and IV patients against stage I and II patients, b P value of poor differentiation group against moderate and poor-moderate groups
Immunostaining of E-cadherin gene
As shown in Table 2, the staining was heterogeneous in 51 tumor tissues, tumor cells with decreased membranous E-cadherin staining were mixed with tumor cells showing strong membranous staining (Figure 2). Frequency of protein expression was 44.6% in tumor tissues, while paired normal tissue specimens all demonstrated diffuse strong membranous E-cadherin staining. Frequency of protein expression was significantly different between tumor and paired normal tissues (P < 0.001). When stratified for TNM stages, Frequency of E-cadherin gene protein expression of stage III and IV tumor tissues (30.8%) was significantly lower than that in stage I and II tumor tissues (62.5%) (χ2 = 9.21, P = 0.002). Frequency of E-cadherin gene protein expression of poor group significantly lower than that in moderate and poor-moderate groups (χ2 = 5.23, P = 0.022).
Figure 2.
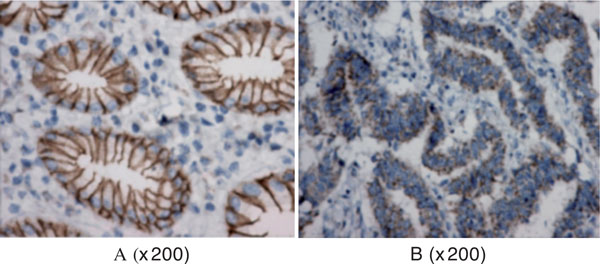
E-cadherin immunostain in GCA tissue. A: positive staining (normal tissue). B: negative staining (GCA tissue).
mRNA expression for the E-cadherin gene
Levels of the transcripts were determined in the 32 selected frozen GCA samples by RT-PCR analysis (Figure 3). The 32 GCA samples including 2 of stage I, 2 of stage II, 8 of stage III and 8 of stage IV of cases in which the tumor was methylated and 12 cases (2 of stage I, 4 of stages II, 4 of stage III and 2 of stage IV) with unmethylated E-cadherin gene. A 202 bp fragment of the E-cadherin gene transcript was generated, with a 342 bp GAPDH fragment of the transcript as a control. 16 (1 of stage II, 7 of stage III and 8 of stage IV) cases (80%) of inactivated mRNA expression were observed in 20 tumor samples with E-cadherin gene methylated, the other 4 (2 of stage I, 1 of stage II, 1 of stage III) cases showed positive expression. 12 tumor samples with E-cadherin gene unmethylated all showed positive expression of E-cadherin gene.
Figure 3.
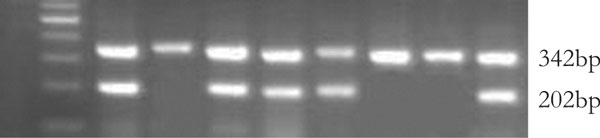
mRNA analysis of E-cadherin in tumor tissues. 1: 100 bp DNA marker 2,4,5,6,9: positive mRNA expression 3,7,8: negative mRNA expression.
Discussion
China is a country with high incidence of digestive tract cancer which including esophageal carcinoma, gastric cancer and gastric cardiac adenocarcinoma (GCA). GCA, which was formerly registered as esophageal cancer or gastric cancer, has been diagnosed independently in very recent years, due to the improvement in early endoscopic screening and pathologic diagnosis. It has been suggested by several epidemiological data that the incidence of GCA is increasing in recent years. Some research has shown that the mechanism and clinical symptom of GCA is different from gastric cancer but similar to esophageal cancer. The exact mechanism of the occurrence of GCA remains unclear for the moment. It is generally accepted that the hereditary factor that irritating the occurrence of tumor including two mechanism, genetics and epigenetics mechanism. Genetic abnormalities of proto-oncogenes and tumor suppressor genes are well-known changes that are frequently involved in cancer pathogenesis. However, Epigenetic inactivation of certain tumor suppressor genes by aberrant promoter methylation is frequently observed in several cancers and may play a pivotal role in tumorigenesis. While genetic abnormalities are associated with changes in DNA sequence, epigenetic events may lead to changes in gene expression that occur without changes in DNA sequence. If tumor suppressor genes are affected, it results usually in transcriptional silencing and, hence, inactivation of that gene. It may then confer growth advantages to these cells that favor cancer development [22].
As a member of cell adhesion molecular, E-cadherin play an important role in maintaining cell adhesion and its dysfunction may result in tumorigenesis.6 The biological consequences of its dysfunction include disruption of intercellular adhesion and impairment of ß-catenin-mediated transactivation [23]. To date, however, the regulatory mechanisms responsible for altered levels of E-cadherin proteins in GCA have not been elucidated. It has been reported that hypermethylation of E-cadherin were associated with gastric cancer and esophageal adenocarcinoma [21,24]. However, there are no other report about the relationship between Ecadherin methylation and tumorigenesis of GCA. In this study, we showed that hypermethylation of the 5' CpG island of the E-cadherin promoter occured frequently in GCA tissues (68.5%)and that this methylation change was associated with reduced expression of E-cadherin protein. Our data suggested that epigenetic silencing of the E-cadherin promoter via hypermethylation may be one of the critical mechanism for inactivation of this gene in GCA. Gene silencing associated with hypermethylation is mediated by methyl-binding proteins that bind to methylated cytosines and recruit a complex of proteins that repress transcription, including histone deacetylases [25].
In our study, we found that in the majority of the cases we examined, E-cadherin methylation was tumor specific. however, 10 cases in which the methylation change was present in both tumor and paired normal tissues from the same patient. Given the sensitivity of nested MSP, it is possible that normal-appearing specimens contained a rare cancer cell that was undetectable by histomorphology. Due to the limitation of specimen, we didn't study the methylation of E-cadherin in normal cardia tissues. However, normal esophageal tissue didn't show aberrant E-cadherin methylation in some studies [21] and in previous studies investigating E-cadherin methylation in different tumor types, normal tissues from bone marrow, breast, thyroid, and oral mucosa were unmethylated [14,26]. These data strongly suggested that the E-cadherin promoter methylation is an aberrant event. The fact that we only detected methylation in paired normal tissues from patients in whom the corresponding tumor was also methylated is consistent with the hypothesis that the cancer in these individuals arose from a methylated clonal precursor. In a study of neoplastic progression in Barrett's esophagus, hypermethylation of the tumor suppressor gene p16 was detected in pathologically normal-appearing specimens obtained from a patient who later developed dysplasia [27]. Therefore, epigenetic inactivation of tumor suppressor genes may be an early feature of tumorigenesis.
Our results showed that protein expression of Ecadherin in tumor tissues significantly lower than that in paired normal tissues, however, immunohistochemical staining also showed normal membranous staining of E-cadherin in some tumor samples with E-cadherin methylation. There are several possible reasons for the events. First, This was probably due to the fact that immunohistochemical staining was not as sensitive as PCR in detecting subpopulations of cells with gene methylation and hence downregulation of E-cadherin. Second, tumor tissues may mingle some normal tissues and may showed normal membranous staining of E-cadherin. Third, gene heterogenic methylation or an allele methylation may be an important reason. In our studies, we found that the methylation status of the tumor tissues that both showed positive protein expression and hypermethylation were incomplete Ecadherin methylation. Furthermore, it has been demonstrated that DNA methylation which suppressed gene expression mainly in transcriptional level and the density of CpG island methylation was related to the suppressed degree of transcription [28]. Dicky promoter can be completely suppressed by lower density methylation, however, when promoter was enhanced by enhanser, function of transcription will be retrieved. In our study, we also found positive mRNA expression in some tumor samples with E-cadherin gene methylated. It partly due to the fact that the extent of promoter methylation was insufficent to suppress E-cadherin transcription. In all, Our study suggested that epigenetic silencing of the E-cadherin promoter via hypermethylation may be one of the mechanism for inactivation of E-cadherin in GCA.
Acknowledgements
We thank the patients and control individuals for taking part in this study.
Supported by Grants from the considerable distinctive subjects foundation of Hebei province.
References
- Takeichi M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol. 1995;7:619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- Grunwald GB. The structural and functional analysis of cadherin calcium-dependent cell adhesion molecules. Curr Opin Cell Biol. 1993;5:797–805. doi: 10.1016/0955-0674(93)90028-O. [DOI] [PubMed] [Google Scholar]
- Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. AmJ Pathol. 1998;153:333–339. doi: 10.1016/S0002-9440(10)65575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenberg S, Yarden A, Kam Z. et al. p27 is involved in N-cadherin-mediated contact inhibition of cell growth and S-phase entry. Oncogene. 1999;18:869–876. doi: 10.1038/sj.onc.1202396. [DOI] [PubMed] [Google Scholar]
- Wijnhoven BP, Pignatelli M. E-cadherin-catenin: more than a "sticky" molecular complex. Lancet. 1999;354:356–357. doi: 10.1016/S0140-6736(99)90055-7. [DOI] [PubMed] [Google Scholar]
- Katagiri A, Watanabe R, Tomita Y. E-cadherin expression in renal cell cancer and its significance in metastasis and survival. Br J Cancer. 1995;71:376–379. doi: 10.1038/bjc.1995.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayther SA, Gorringe KL, Ramus SJ. et al. Identification of germ-line E-cadherin mutations in gastric cancer families of European origin. Cancer Res. 1998;58:4086–9. [PubMed] [Google Scholar]
- Richards FM, McKee SA, Rajpar MH. et al. Germline Ecadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999;8:607–610. doi: 10.1093/hmg/8.4.607. [DOI] [PubMed] [Google Scholar]
- Becker KF, Atkinson MJ, Reich U. et al. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res. 1994;54:3845–52. [PubMed] [Google Scholar]
- Berx G, Becker KF, Hofler H. et al. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12:226–237. doi: 10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Yoshiura K, Kanai Y, Ochiai A. et al. Silencing of the Ecadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA. 1995;92:7416–7419. doi: 10.1073/pnas.92.16.7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB, Herman JG, Graff JR. et al. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- Kanai Y, Ushijima S, Hui AM. et al. The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer. 1997;71:355–359. doi: 10.1002/(SICI)1097-0215(19970502)71:3<355::AID-IJC8>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Graff JR, Herman JG, Lapidus RG. et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–5199. [PubMed] [Google Scholar]
- Corn PG, Heath EI, Heitmiller R. et al. Frequent hypermethylation of the 5' CpG island of E-cadherin in esophageal adenocarcinoma. Clin Cancer Res. 2001;7:2765–9. [PubMed] [Google Scholar]
- Yang CS. Vitamin nutrition and gastroesophageal cancer. J Nutr. 2000;130:338S–339S. doi: 10.1093/jn/130.2.338S. [DOI] [PubMed] [Google Scholar]
- Yokokawa Y, Ohta S, Hou J. et al. Ecological study on the risks of esophageal cancer in Ci-Xian, China: the importance of nutritional status and the use of well water. Int J Cancer. 1999;83:620–624. doi: 10.1002/(SICI)1097-0215(19991126)83:5<620::AID-IJC9>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Launoy G, Milan CH, Faivre J. et al. Alcohol, tobacco and oesophageal cancer: effects of the duration of consumption, mean intake and current and former consumption. Br J Cancer. 1997;75:1389–1396. doi: 10.1038/bjc.1997.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Buckley JD. The role of DNA methylation in cancer. Adv Cancer Res. 1990;54:1–23. doi: 10.1016/s0065-230x(08)60806-4. [DOI] [PubMed] [Google Scholar]
- Siewert JR, Stein HJ. Classification of adenocarcinoma of the oesophagogastric junction. Br J Surg. 1998;85:1457–1459. doi: 10.1046/j.1365-2168.1998.00940.x. [DOI] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S. et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathdee G, Brown R. Aberrant DNA methylation in cancer: potential clinical interventions. Expert Rev Mol Med. 2002;4:1–17. doi: 10.1017/S1462399402004222. [DOI] [PubMed] [Google Scholar]
- Sadot E, Simcha I, Shtutman M. et al. Inhibition of betacatenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci USA. 1998;95:15339–15344. doi: 10.1073/pnas.95.26.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AO, Lam SK, Wong BC. et al. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut. 2003;52:502–506. doi: 10.1136/gut.52.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- Graff JR, Greenberg VE, Herman JG. et al. Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle's cell, and poorly differentiated human thyroid carcinoma. Cancer Res. 1998;58:2063–2066. [PubMed] [Google Scholar]
- Klump B, Hsieh CJ, Holzmann K. et al. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett's esophagus. Gastroenterology. 1998;115:1381–1386. doi: 10.1016/S0016-5085(98)70016-2. [DOI] [PubMed] [Google Scholar]
- Bird A. Molecular biology. Methylation talk between histones and DNA. Science. 2001;294:2113–2115. doi: 10.1126/science.1066726. [DOI] [PubMed] [Google Scholar]