

Abstract
3,5-Dibromo-4-(3,4-dimethoxyphenyl)-1H-pyrrole-2-carboxylic acid ethyl ester is a promising antitubulin lead agent that targets the colchicine site of tubulin. C-2 analogues were synthesized and tested for microtubule depolymerizing and antiproliferative activity. Molecular modeling studies using both GOLD docking and HINT (Hydropathic INTeraction) scoring revealed two distinct binding modes that explain the structure–activity relationships and are in accord with the structural basis of colchicine binding to tubulin. The binding mode of higher activity compounds is buried deeper in the site and overlaps well with rings A and C of colchicine, while the lower activity binding mode shows fewer critical contacts with tubulin. The model distinguishes highly active compounds from those with weaker activities and provides novel insights into the colchicine site and compound design.
Keywords: Antitubulin, hydropathic interactions, docking, multifunctional pyrroles, structure−activity relationship
Microtubules are major cytoskeletal components in eukaryotic cells and participate in a variety of cell functions including maintenance of cell shape, intracellular transport, and forming mitotic spindles for segregating chromosomes during mitosis. Microtubules assemble and disassemble by a reversible process called dynamic instability involving discrete α/β tubulin heterodimers.1 Diverse agents suppress microtubule dynamics; in rapidly dividing cells, they induce mitotic arrest and initiate apoptosis.2 Compounds that target microtubules bind at four major binding sites: the taxane and the laulimalide/peloruside A sites for microtubule-stabilizing agents and the vinca and colchicine sites for microtubule-destabilizing agents.2,3 Taxanes and vinca alkaloids have achieved notable success in cancer chemotherapy, but no colchicine site agents have been approved for systemic use against cancer.4
Recent studies of one family of colchicine site agents, analogues of combretastatin A4 (CA4), have reported antivascular actions leading to the rapid collapse of tumor vasculature.5 A number of CA4 analogues are in clinical trials refueling the search for novel colchicine site agents. Emerging drug resistance due to the expression of the βIII-tubulin isotype has compromised the clinical use of taxanes and vinca alkaloids.6 Resistance to different types of microtubule targeting agents was recently suggested to be related to their binding sites, and that βIII-tubulin mediated drug resistance might be circumvented by colchicine site agents.7 Natural and synthetic compounds, for example, podophyllotoxins, arylindoles, sulfonamides, 2-methoxyestradiols, and flavonoids, bind within the colchicine site.8 This structural diversity provides many possibilities for optimization and new scaffold design. The colchicine site has been characterized with X-ray crystallography by cocrystallization of the protein with DAMA-colchicine;9 the site is at the interface between α- and β-tubulin. Complexation with other agents has documented the flexibility of this pocket.10 To understand the structural basis for ligand binding at the colchicine site, a common pharmacophore model was built by Nguyen et al. based on 15 structurally diverse colchicine site inhibitors.11
We previously showed that 1 [3,5-dibromo-4-(3,4-dimethoxyphenyl)-1H-pyrrole-2-carboxylic acid ethyl ester, JG-03-14, Figure 1] is a potent microtubule-destabilizing agent.12 Because 1 inhibited the binding of [3H]colchicine and COMPARE analysis, which evaluates the similarity between two compounds with respect to the NCI 60-cell line assay,13 showed correlation between 1 and colchicine, it is likely that 1 also binds at this site.14 Although 1 does not structurally resemble other classes of agents, it more or less fits the previous pharmacophore model.14 In addition, 1 and an unfocused set of analogues produced a quantitative linear quantitative structure–activity relationship (QSAR) relationship between the IC50 and the HINT15 binding score.14 This scoring model, which considers hydrophobic and polar interactions as well as entropic effects, has been shown to correlate with binding free energy for small molecule–biomacromolecular complexes.16
Figure 1.

Structures of colchicine and lead compound JG-03-14 (1).
Further investigations on autophagic cell death, polyploidy, senescence, and effects on endothelial cell functions for 1 suggest that it is a viable lead candidate for optimization as a new colchicine site anticancer agent.17−19 Of particular note is that there is considerable synthetic flexibility for 1 and analogues, such that each of the five atoms of the pyrrole ring can be differentially probed, elaborated, and optimized for SAR. Here, we report the synthesis, physical properties, and microtubule inhibitory effects for C-2 analogues of 1. Modeling studies indicate that two distinct binding modalities are required to explain the observed SAR.
In this study, we retain the 3,4-dimethoxylphenyl at C-4 and the two bromine groups at C-3 and C-5 of 1 and focus on modifications to the ester at the C-2 position of the pyrrole core. We have previously reported17 the synthesis of 1 (JG-03-14) and have utilized a similar sequence of reactions as outlined in Scheme 1 to prepare the new analogues listed in Table 1. 3,4-Dimethoxyphenylacetic acid (2) was converted to the corresponding vinamidinium salt (3) using Vilsmeier–Haack–Arnold conditions. Compound 3 was condensed with glycinate esters to give either pyrrole ethyl ester (4a) or pyrrole t-butyl ester (4b). Compound 4a was hydrolyzed with base in aqueous ethanol to produce the corresponding pyrrole acid (5), which served as the key building block for the majority of the analogs. The various pyrrole esters (6a–i) were constructed with the appropriate alcohol, carbonyldiimidazole, DMF, and DBU. The final step involving dibromination was accomplished with dibromodimethylhydantoin in refluxing chloroform. The only exception was converting the pyrrole t-butyl ester (4b) directly to the corresponding dibromopyrrole (7) with dibromodimethylhydantoin. All reactions gave product yields in excess of 65%.
Scheme 1. Preparation of JG-03-14 Analogues with Modifications at the C-2 Position; See Table 1 for Identities of Various R Groups.

Reagents: (a) POCl3, DMF and heat, followed by H2O/NaPF6. (b) Glycine ethyl ester or glycine t-butyl ester and NaOt-Bu, DMF, and heat. (c) NaOH, EtOH/H2O, and heat. (d) ROH, 1,1′-carbonyldiimidazole, DBU, and DMF. (e) Dibromodimethylhydantoin, CHCl3, and heat.
Table 1. Structures, Biological Activity, and Properties of Pyrrole Compounds 1 and 7a–l.
| compd | R | antiproliferationa IC50 (μM) | cellular microtubule lossb | binding mode | HINT scorec | HINT log P | ALOGPsd |
|---|---|---|---|---|---|---|---|
| colchicine | 0.016 ± 0.002 | 100% loss at 0.5 μM | 549 | 3.24 | 1.59 | ||
| 1 | ethyl | 0.036 ± 0.002e | 100% loss at 0.5 μM | I | 418 | 2.60 | 4.44 |
| 7a | methyl | 0.618 ± 0.07 | 50% loss at 5 μM | I | 524 | 2.06 | 3.87 |
| 7b | n-propyl | 0.067 ± 0.002 | 75% loss at 5 μM | I | 157 | 3.14 | 4.74 |
| 7c | i-propyl | 0.109 ± 0.008 | 70% loss at 5 μM | I | –179 | 3.14 | 4.70 |
| 7d | t-butyl | 1.82 ± 0.3 | no loss up to 10 μM | II | 187 | 3.24 | 5.02 |
| 7e | n-butyl | 1.30 ± 0.04 | 15% loss at 10 μM | II | 530 | 3.68 | 5.05 |
| 7f | n-hexyl | 3.3 ± 0.3 | 35% loss at 10 μM | II | 256 | 4.76 | 5.83 |
| 7g | benzyl | 5.3 ± 0.3 | no loss up to 10 μM | II | 713 | 3.61 | 5.39 |
| 7h | –(CH2)3NMe2 | 4.6 ± 0.2 | 10% loss at 10 μM | II | 293 | 2.52 | 4.10 |
| 7i | –(CH2)2NMe2 | 5.2 ± 0.3 | 10% loss at 10 μM | II | 358 | 2.57 | 3.82 |
| 7j | –(CH2)3NMe2H+Cl– | 8.0 ± 0.3 | no loss up to 10 μM | II | 631 | 0.27 | 0.39 |
| 7k | –(CH2)2NMe2H+Cl– | 10.7 ± 0.4 | no loss up to 10 μM | II | 774 | 0.78 | 0.27 |
| 7l | 4-methoxylphenyl | 18.3 ± 2.7 | no loss up to 10 μM | II | 957 | 4.37 | 5.48 |
Experiments were performed using human MDA-MB-435 cancer cells.
Loss of interphase microtubules was evaluated in A-10 cells.
515 HINT score units ≈ 1 kcal mol–1 (ref (15)).
ALOGPs were calculated at Virtual Computational Chemistry Laboratory, http://www.vcclab.org.
Ref (12).
Antiproliferative activities were measured in MDA-MB-435 cancer cells using the sulforhodamine B assay, and effects on cellular microtubules were evaluated in A-10 cells using immunofluorescence as previously described.12 Results are presented in Table 1.
For this study, the SAR was analyzed with respect to the antiproliferative activities of compounds 1 and 7a–l. The antitubulin activity generally trends with antiproliferative activity. Compound 1 remains the most active compound (36 nM).12 As compared to 1, 7a had a 17-fold decrease in activity likely due to its one-carbon shorter ester. Similarly, the longer and bulkier alkyl substitutions n-propyl (7b) and i-propyl (7c) decreased antiproliferative activity. Larger groups, t-butyl (7d), n-butyl (7e), or n-hexyl (7f), were tolerated but with a significant activity loss of at least 36-fold. A dramatic loss was also observed for aromatic substitutions (7g and 7l). The incorporation of a comparatively polar amine did not increase the activity significantly (7h–k), suggesting that the activity drop is truly related to sterics and not solubility.
The observation that the protonated amines (7j and 7k) had a further 2-fold drop in activity as compared to their free base analogues (7h and 7i) may be due to their weaker ability to penetrate the cell membrane. Moreover, no microtubule effects were observed up to 10 μM for the amine derivatives, suggesting that a different mechanism of action of antiproliferation might be at play. The SAR suggests that only the properly sized group would be favorable for activity, and the ethyl group of 1 provides that optimum.
To rationalize the SAR from a structure-based perspective, we performed docking studies with the X-ray crystal structure of DAMA-colchicine/tubulin (PDB ID: 1sa0).9 It should be noted that the resolution of the 1sa0 structure for αβ-tubulin is poor (3.58 Å), and resulting modeling studies have a higher degree of uncertainty than in other systems. The colchicine site is mostly buried in the β-subunit surrounded by helices H7 and H8, loop T7, and strands S8 and S9. The T5 loop of the α-subunit also contributes to the pocket (see Figure 2). DAMA-colchicine occupies the pocket such that ring A fits deep within a subpocket close to H7, ring C fits into another subpocket close to T5, ring B is centered within the main pocket, and the DAMA chain is pointing to the pocket's entrance. For convenience, we will refer to the subpockets where rings A and C bind as subpockets A and C. Compound 1 and its analogues 7a–l were docked in the colchicine site with poses generated by the docking program GOLD20 and rescored with HINT.15,16 The compounds can be divided into two sets based on their computationally predicted binding modes (Table 1 and Figure 2). In both modes, the dimethoxyphenyl ring locates in the subpocket A, overlapping the trimethoxyphenyl ring (ring A) of DAMA-colchicine. The positions of the C-2 ester chain differ between the two modes. In mode I, the R group of the ester has “acceptable” size (i.e., 1 and 7a–c) and fits within subpocket C and thus overlaps well with ring C of DAMA-colchicine, while in mode II, the bulkier 7d–l R groups extend out from the main pocket toward its opening.
Figure 2.
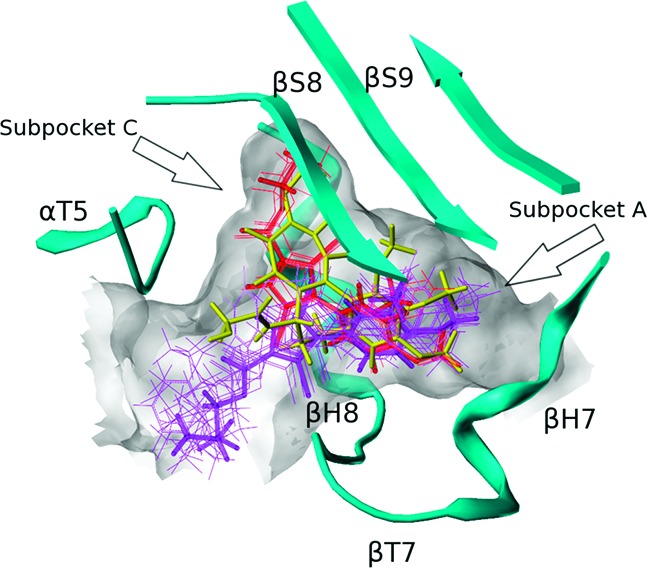
Colchicine (yellow) and binding modes of pyrrole-based C-2 analogues (mode I, red; mode II, purple). The extents of the colchicine site, as illustrated by MOLCAD, are shown in grayish white.
To illustrate the specific interactions between the ligands and the site, we calculated intermolecular HINT interaction maps21 using 1 as representing mode I binding (Figure 3A) and 7e representing mode II (Figure 3B). First, subpocket A, which fits the dimethoxyphenyl ring in both modes, is quite hydrophobic. In both modes, the four-carbon side chains of Leu248β and Leu255β clamp the phenyl ring in place, while deeper in the pocket other residues lock the ligand's methoxys. Polar interactions also play a part as Cys241β is in proximity to these two methoxys, with distances between the cysteine's sulfur and the oxygens of 3.06 and 3.45 Å, thus likely forming at least one hydrogen bond to support the binding. Also in both modes, there is a favorable interaction in the main pocket between the backbone oxygen of Asn258β and the ligand's pyrrole nitrogen.
Figure 3.
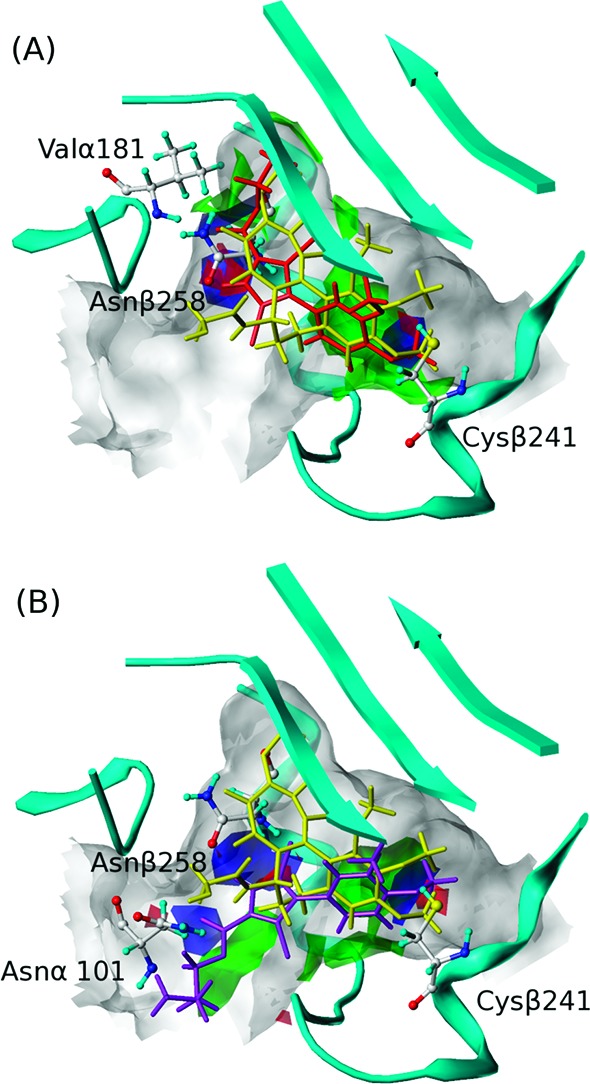
HINT interaction maps of (A) 1 (binding mode I) and (B) 7e (binding mode II). For 1 and 7e, green contours represent favorable hydrophobic interactions; blue contours represent favorable polar interactions (hydrogen bonds, acid/base, Coulombic); red contours represent unfavorable polar interactions. Compound 1 is shown in red, 7e is shown in purple, and colchicine is shown in yellow.
Both hydrophobic and polar residues characterize subpocket C, which fits the esters in mode I binding. The alkyl ends reach the hydrophobic bottom, while the carboxyl oxygens anchor the ester by forming hydrogen bonds with the backbone nitrogen of Val181α. The main pocket includes its funnel opening and is much more spacious than subpocket C. It easily tolerates the size of the longer esters binding with mode II by flipping the pyrrole core, thus exposing the ester tail to the solvent while keeping the dimethoxylphenyl ring in subpocket A. Our models suggest that a new hydrogen bond, stabilizing the ester tail in mode II, is formed between the amide nitrogen of Asn101α and the ligand's carbonyl oxygen. The interactions for the various R groups of 7d–l are poorly defined as the pocket entrance broadens and has a large solvent exposure.
The compounds in mode I displayed notably higher antiproliferative activity and antitubulin activity than the compounds in mode II. It is clearly important to effectively occupy both subpockets A and C in the colchicine site. The SAR within the mode I set is size related: The methyl of 7a, the n-propyl of 7b, and the i-propyl of 7c may not position the ester carbonyl (hydrogen bonded to Val181β) as well as the ethyl of 1. In contrast, in the mode II set, the ester R extends from the pocket into (and possibly out of) the pocket's entrance. The SAR simply may not be interpretable as these tails are highly flexible and thus subject to interactions with a wide array of residues as well as solvent.
It is also instructive to compare, in detail, the binding of colchicine and the pyrrole-based compounds 1 and 7a–l: (1) depletion of ring B of colchicine retains activity, while rings A and C, which adopt a similar conformation as in mode I, are necessary for high affinity binding;22 (2) residues Cys241β (subpocket A) and Val181α (subpocket C) appear to be important for antitubulin activity since the removal of any A ring methoxy group close to Cys241β weakens the binding to tubulin and microtubule inhibition.23 Also, isocolchicine, whose structural difference to colchicine is in the C ring (methoxy at C-9 and keto at C-10) binds weakly and only poorly inhibits microtubule assembly,24 probably because of a loss of hydrogen bonding to Val181α. Both residues anchor the ligand in the more active mode I, while only Cys241β does so in the less active mode II. This may largely explain the difference in activity between the binding modes.
The HINT scores of Table 1 were poor in distinguishing between binding in modes I and II. The reasons for this failure are instructive. First, the poor resolution of the tubulin crystal structure and the flexibility of the pocket,10 especially the T5 and T7 loops, are a partial explanation. However, the binding modes themselves and the nature of the pocket are larger factors. Table 2 lists the HINT scores in terms of two fragments—the common dimethoxyphenyl plus pyrrole (ring) and the ester. Interaction types further differentiate the latter. The total ring score is largely invariant (580 ± 70), excluding 7b and 7c, where it is lower by >200. The ester's HHH for mode I (750 ± 130) is much higher than for mode II (280 ± 90). Interestingly, HHH is highest for 7b and 7c, but accommodation of these longer esters was penalized by poorer ring interactions. For 1 and 7a–c, hydrophobic binding quality in subpocket C is key. Although the esters of mode II compounds appear to make productive contacts, these are in the very open funnel-like entrance of the pocket where dynamic solvent effects that can disrupt polar interactions must be assumed.
Table 2. HINT Scores by Fragment and Interaction Type.
| HINT
scorea |
||||||
|---|---|---|---|---|---|---|
| ring | ester |
|||||
| compd | mode | HTOTAL | HHB + HAB | HHH | HAA + HBB | HHP |
| 1 | I | 492 | 858 | 711 | –499 | –1328 |
| 7a | I | 518 | 680 | 583 | –427 | –1048 |
| 7b | I | 363 | 1069 | 855 | –593 | –1681 |
| 7c | I | 321 | 766 | 833 | –478 | –1821 |
| 7d | II | 570 | 778 | 226 | –398 | –1169 |
| 7e | II | 626 | 781 | 225 | –364 | –1015 |
| 7f | II | 435 | 665 | 387 | –369 | –1046 |
| 7g | II | 589 | 812 | 284 | –341 | –868 |
| 7h | II | 557 | 549 | 268 | –336 | –1071 |
| 7i | II | 581 | 726 | 237 | –400 | –1027 |
| 7j | II | 650 | 375 | 213 | –236 | –634 |
| 7k | II | 596 | 847 | 196 | –403 | –755 |
| 7l | II | 587 | 827 | 494 | –240 | –924 |
Interaction types: favorable polar (hydrogen bond, HHB, and acid/base, HAB), hydrophobic (HHH), unfavorable polar (acid/acid, HAA, and base/base, HAB), and unfavorable hydrophobic polar (HHP).
In summary, mode I is a new binding motif observed for pyrrole compounds based on JG-03-14 (1) that is different from previously reported binding modes.11,14 The ester chain in mode I overlaps with the C-10 substituents of colchicine and the SAR of colchicine C-10 analogues also shows that increasing length of the alkyl chain causes a concomitant decrease in activity.25 We propose that the deeper burial of mode I ligands is more disruptive to the association of α- and β-tubulin subunits than is binding with mode II. We are continuing design and development of additional JG-03-14 (1) analogues by focusing on other positions of the pyrrole core as we attempt to gain a full view of the SAR.
Acknowledgments
The excellent technical assistance of Lyda Robb, Cara Westbrook, and Nicholas Dbydal-Hargreaves is gratefully acknowledged.
Supporting Information Available
Experimental details for the synthesis and characterization of reported compounds, procedures for the assays, and additional information on the computational methods utilized. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was partially supported by NIH R01 CA135043 (to D. A. Gewirtz, VCU), NIH R15 CA067236 (to J.T.G.), and the President's Council Research Excellence Award (to S.L.M).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jordan M. A.; Wilson L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-Binding Agents, A Dynamic Field of Cancer Therapeutics. Nat. Rev. Drug Discovery 2010, 9, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett M. J.; Barakat K.; Huzil J. T.; Tuszynski J.; Schriemer D. C. Discovery and Characterization of the Laulimalide-Microtubule Binding Mode by Mass Shift Perturbation Mapping. Chem. Biol. 2010, 17, 725–734. [DOI] [PubMed] [Google Scholar]
- Stanton R. A.; Gernert K. M.; Nettles J. H.; Aneja R. Drugs That Target Dynamic Microtubules: A New Molecular Perspective. Med. Res. Rev. 2011, 31, 443–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemann D. W. The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seve P.; Dumontet C. Is Class III Beta-tubulin a Predictive Factor in Patients Receiving Tubulin-binding Agents?. Lancet Oncol. 2008, 9, 168–175. [DOI] [PubMed] [Google Scholar]
- Stengel C.; Newman S. P.; Leese M. P.; Potter B. V.; Reed M. J.; Purohit A. Class III Beta-tubulin Expression and in vitro Resistance to Microtubule Targeting Agents. Br. J. Cancer 2010, 102, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Liu T.; Dong X.; Hu Y. Recent Development and SAR Analysis of Colchicine Binding Site Inhibitors. Mini-Rev. Med. Chem. 2009, 9, 1174–1190. [DOI] [PubMed] [Google Scholar]
- Ravelli R. B.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into Tubulin Regulation from a Complex with Colchicine and a Stathmin-like Domain. Nature 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
- Dorleans A.; Gigant B.; Ravelli R. B.; Mailliet P.; Mikol V.; Knossow M. Variations in the Colchicines-binding Domain Provide Insight into Structural Switch of Tubulin. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13775–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. L.; McGrath. C.; Hermone A. R.; Burnett J. C.; Zaharevitz D. W.; Day B. W.; Wipf. P.; Hamel E.; Gussio R. A Common Pharmacophore for a Diverse Set of Colchicine Site Inhibitors Using a Structure-Based Approach. J. Med. Chem. 2005, 48, 6107–6116. [DOI] [PubMed] [Google Scholar]
- Mooberry S. L.; Weiderhold K. N.; Dakshanamurthy S.; Hamel E.; Banner E. J.; Kharlamova A.; Hempel J.; Gupton J. T.; Brown M. L. Identification and Characterization of a New Tubulin-Binding Tetrasubstituted Brominated Pyrrole. Mol. Pharmacol. 2007, 72, 132–140. [DOI] [PubMed] [Google Scholar]
- Cleaveland E. S.; Monks A.; Vaigro-Wolff A.; Zaharevitz D. W.; Paull K.; Ardalan K.; Cooney D. A.; Ford H. Jr. Site of Action of Two Novel Pyrimidine Biosynthesis Inhibitors Accurately Predicted by the COMPARE Program. Biochem. Pharmacol. 1995, 49, 947–954. [DOI] [PubMed] [Google Scholar]
- Tripathi A.; Fornabaio M.; Kellogg G. E.; Gupton J. T.; Gewirtz D. A.; Yeudall W. A.; Vega N. E.; Mooberry S. L. Docking and Hydrophobic Scoring of Polysubstituted Pyrrole Compounds with Antitubulin Activity. Bioorg. Med. Chem. 2008, 16, 2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg G. E.; Abraham D. J. Hydrophobicity: Is LogPo/w More than the Sum of Its Parts?. Eur. J. Med. Chem. 2000, 35, 651–661. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Amadasi A.; Fornabaio M.; Abraham D. J.; Mozzarelli A.; Kellogg G. E.; Cozzini P. The Consequences of Scoring Docked Ligand Conformations using Free Energy Correlations. Eur. J. Med. Chem. 2007, 42, 921–933. [DOI] [PubMed] [Google Scholar]
- Gupton J.; Burnham B.; Krumpe K.; Du K.; Sikorski J.; Warren A.; Barnes C.; Hall I. Synthesis and Cytotoxicity of 2,4-Disubstituted and 2,3,4-Trisubstituted Brominated Pyrroles in Murine and Human Cultured Tumor Cells. Arch. Pharm. Pharm. Med. Chem. 2000, 333, 3–9. [DOI] [PubMed] [Google Scholar]
- Dalyot-Herman N.; Delgado-Lopez F.; Gewirtz D. A.; Gupton J. T.; Schwartz E. L. Interference with Endothelial Cell Function by JG-03–14, An Agent that Binds to the Colchicine Site on Microtubules. Biochem. Pharmacol. 2009, 78, 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur C. R.; Gupton J. T.; Kellogg G. E.; Yeudall W. A.; Cabot M. C.; Newsham I. F.; Gewirtz D. A. Autophagic Cell Death, Polyploidy and Senescence Induced in Breast Tumor Cells by the Substituted Pyrrole JG-03–14, a Novel Microtubule Posion. Biochem. Pharmacol. 2007, 74, 981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G; Willett P; Glen R. Molecular Recognition of Receptor Sites Using a Genetic Algorithm with a Description of Desolvation. J. Mol. Biol. 1995, 245, 43–53. [DOI] [PubMed] [Google Scholar]
- Wireko F. C.; Kellogg G. E.; Abraham D. J. Allosteric Modifiers of Hemoglobin. 2. Crystallographically Determined Binding Sites and Hydrophobic Binding/Interaction Analysis of Novel Hemoglobin Oxygen Effectors. J. Med. Chem. 1991, 34, 758–767. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya B.; Panda D.; Gupta S.; Banerjee M. Anti-Mitotic Activity of Colchicine and the Structural Basis for Its Interaction with Tubulin. Med. Res. Rev. 2008, 28, 155–183. [DOI] [PubMed] [Google Scholar]
- Andreu J. M.; Perez-Ramirez B.; Gorbunoff M. J.; Ayala D.; Timasheff S. N. Role of the Colchicine Ring A and Its Methoxy Groups in the Binding to Tubulin and Microtubule Inhibition. Biochemistry 1998, 37, 8356–8368. [DOI] [PubMed] [Google Scholar]
- Hastie S. B.; Williams R. C. Jr; Puett D.; Macdonald T. L. The Binding of Isocolchicine to Tubulin. Mechanisms of Ligand Association with Tubulin. J. Biol. Chem. 1989, 264, 6682–6688. [PubMed] [Google Scholar]
- Staretz M. E.; Hastie S. B. Synthesis and Tubulin Binding of Novel C-10 Analogues of Colchicine. J. Med. Chem. 1993, 36, 758–764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.