

Abstract
The immune response to infection or injury coordinates host defense and tissue repair, but also has the capacity to damage host tissues. Recent advances in understanding protective mechanisms have found neural circuits that suppress release of damaging cytokines. Stimulation of the vagus nerve protects from excessive cytokine production and ameliorates experimental inflammatory disease. This mechanism, the inflammatory reflex, requires the α7 nicotinic acetylcholine receptor (α7nAChR), a ligand-gated ion channel expressed on macrophages, lymphocytes, neurons and other cells. To investigate cell-specific function of α7nAChR in the inflammatory reflex, we created chimeric mice by cross-transferring bone marrow between wild-type (WT) and α7nAChR-deficient mice. Deficiency of α7nAChR in bone marrow–derived cells significantly impaired vagus nerve–mediated regulation of tumor necrosis factor (TNF), whereas α7nAChR deficiency in neurons and other cells had no significant effect. In agreement with recent work, the inflammatory reflex was not functional in nude mice, because functional T cells are required for the integrity of the pathway. To investigate the role of T-cell α7nAChR, we adoptively transferred α7nAChR-deficient or WT T cells to nude mice. Transfer of WT and α7nAChR-deficient T cells restored function, indicating that α7nAChR expression on T cells is not necessary for this pathway. Together, these results indicate that α7nAChR expression in bone marrow–derived non–T cells is required for the integrity of the inflammatory reflex.
INTRODUCTION
The immune response to infection or injury coordinates host defense and tissue repair, but it also has the inherent capacity to significantly damage host tissues. The release and activity of tumor necrosis factor (TNF), interleukin (IL)-1 and other potentially damaging cyto kines is controlled at multiple levels to prevent unrestrained collateral tissue damage that can disable, or even kill, the host (1). Humoral mechanisms that restrain or inhibit these damaging responses include glucocorticoid hormones, soluble cytokine receptors, IL-10, transforming growth factor (TGF)-β and other antiinflammatory cytokines. Activation of cholinergic receptors is also known to regulate immune system activity (2–9). Recent insights in protective mechanisms have revealed that neural circuits suppress release of damaging cytokines and that neural regulation of immune cell activation is an ancient mechanism dating back to nematode worms, a primitive animal with rudimentary nervous and immune systems (10–12).
A prototypical antiinflammatory neural mechanism is the inflammatory reflex (11,13–15). Action potentials arising in the brain stem are transmitted in the cholinergic vagus nerve to terminate in the celiac ganglion, the site of origin of the adrenergic splenic nerve. Signals through the splenic nerve terminate on specialized T cells that respond to norepinephrine by producing acetylcholine, the terminal neurotransmitter in the circuit. Acetylcholine interacts with cyto kine producing macrophages in the red pulp and marginal zone to suppress TNF release (15). The cytokine-suppressing mechanism of the inflammatory reflex requires the α7 nicotinic acetylcholine receptor (α7nAChR), as evidenced by the observation that the inflammatory reflex is impaired in α7nAChR-deficient mice (16). Furthermore, deleting α7nAChR from isolated macrophages impairs the ability of acetylcholine to suppress TNF and other cytokines.
Although these data imply an importance of α7nAChR in cytokine-producing cells of the innate immune system (5,8,16,17), α7nAChR is also expressed by other cells, including neurons, glial cells and T cells. It remained theoretically possible that the impaired inflammatory reflex in the α7nAChR-deficient mice was attributable to altered neuronal functions. To address this question, we created chimeric mice by cross-transferring bone marrow (BM) derived from wild-type (WT) and α7nAChR-deficient mice and evaluated the competence of the inflammatory reflex. This study demonstrates that α7nAChR expression on BM-derived non–T cells, not on neurons, is required for the integrity of the inflammatory reflex.
MATERIALS AND METHODS
Animals
α7nAChR knockout (KO) B6.129S7-Chrna7tm1Bay/J, WT C57BL/6 and WT B6.SJL-Ptprca Pepcb/BoyJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and B6.Cg/NTac-Foxn1nu NE10 (nude) mice were obtained from Taconic (Albany, NY, USA). Experimental animals were obtained by breeding female heterozygous with male homozygous α7nAChR KO mice. Animal experiments were approved by the local Institutional Animal Care and Use Committee (IACUC).
Bone Marrow Transfer
BM donor mice, either B6.SJL-Ptprca Pepcb/BoyJ (WT mice expressing CD45.1) or B6.129S7-Chrna7tm1Bay/J (α7nAChR-KO mice expressing CD45.2), were euthanized by CO2 asphyxiation and BM harvested. Harvested BM was passed through a 70-μm cell strainer (Fisher Scientific, Pittsburg, PA, USA) into a 50-mL tube and pelleted. Lymphocytes were isolated by using Lympholyte-M (Cedarlane, Burlington, NC, USA) and resuspended in phosphate-buffered saline (PBS) at 1.5–2.5 × 106 BM cells per 100 μL PBS.
Recipient mice were anesthetized by intramuscular injection of ketamine (100 mg/kg) and xylazine (8 mg/kg) and irradiated with 9 Gy by using an AECL 1000 Irradiator (Gammacell, Ottawa, ON, Canada). Twenty-four hours later, the mice were anesthetized again and injected with 3–5 × 106 BM cells via retro-orbital venous sinus injections. Animals were rested for 10 wks before experiments.
T-Cell Isolation and Transfer
T cells were isolated from spleens of WT and α7nAChR KO donor mice using the Pan T-Cell Isolation Kit II (Miltenyi Biotec, Auburn, CA, USA) with a purity of approximately 95%. Recipient C57BL/6 nude mice were anesthetized as described above, and 4–6 × 106 T cells in 200 μL PBS were transferred via retro-orbital venous sinus injections. Five days after the transfer, mice were subjected to either vagus nerve stimulation or sham surgery. For coculture experiments, spleen T cells and T cell–depleted splenocytes were enriched by negative selection using the Pan T-cell Isolation Kit (negative selection of T cells) and CD90.2 microbeads (positive selection of T cells for depletion; Miltenyi Biotec), respectively, and were cocultured in 96-well plates in OptiMEM (Invitrogen, Grand Island, NY, USA) at 106 cells per well, incubated for 20 h and stimulated with 100 ng/mL lipopolysaccharide (LPS). Supernatants were saved for analysis.
Creation of Chimera by Adoptive Transfer of Bone Marrow
Chimeric mice with α7nAChR deficiency in BM-derived cells (BMα70 mice) or non-BM-derived cells (nBMα70 mice) were created by adoptive transfer of WT or α7nAChR-deficient BM cells to α7nAChR-deficient or WT mice (Table 1). In addition, WT or α7nAChR-deficient CD4+ T cells were transferred to nude mice to create Twt and Tα70 nude mice, respectively (Table 1). For BM transplantation, CD45.1+ WT and CD45.2+ α7nAChR-deficient mice were used as donors.
Table 1.
Designation of chimeras produced by adoptive transfer.
| Transfer setup | Resulting chimera | |||
|---|---|---|---|---|
|
|
|
|||
| Designation | Recipient genotype | Transferred cells | Nervous system | Immune system |
| WT | WT | — | WT | WT |
| BMα70 | WT | α7nAChR−/− CD45.2 BM | WT | α7nAChR−/− |
| nBMα70 | α7nAChR−/− | WT CD45.1 BM | α7nAChR−/− | WT |
| Nude | Nude | — | Nude | Nude |
| Twt Nude | Nude | WT CD45.1 CD4+ T cells | Nude | Nude with WT CD4+ T cells |
| Tα70 Nude | Nude | α7nAChR−/− CD45.2 CD4+ T cells | Nude | Nude with α7nAChR−/− CD4+ T cells |
Vagus Nerve Stimulation
Mice were anesthetized as described above. A midline cervical incision was made and the left carotid sheath, which contains the left cervical branch of the vagus nerve, was isolated. A bipolar cuff electrode (Microprobes, Gaithersburg, MD, USA) was secured around the carotid sheath, and 1 mA was applied at 10 Hz for 1 min (Setpoint Medical Stimulator, SetPoint Medical, Valencia, CA, USA). In sham-operated animals, a cervical incision was made, but the electrode was not applied. Subsequently, the incision was stapled closed, and mice recovered in a cage on top of a heating pad. Three hours later, 5 mg/kg endotoxin (LPS from Escherichia coli, 0111:B4; Sigma, St. Louis, MO, USA) was injected intraperitoneally, and mice were euthanized via CO2 asphyxiation 90 min there-after. Blood was obtained via cardiac puncture and spleens were removed.
Enzyme-Linked Immunosorbent Assay (ELISA)
Serum and supernatant TNF were measured using the TNF ELISA kit (R&D Systems).
Flow Cytometry
Splenocytes were isolated as previously described (15) and stained with rat anti-mouse F4/80 (AbD Serotec, Raleigh, NC, USA), rat anti-mouse CD3 (eBio-science, San Diego, CA, USA), mouse anti-mouse CD45.1 (BD Pharmingen, San Diego, CA, USA) and mouse anti-mouse CD45.2 (BD Pharmingen) antibodies. Cells were then washed and resuspended in PBS containing 2% fetal bovine serum and 0.09% sodium azide, acquired in a BD LSR II flow cytometer and analyzed using FlowJo v9.3.1. Purity of T-cell and non–T-cell isolates was determined by using an anti-T cell receptor β (anti-TCRβ) antibody (BD Pharmingen).
Statistics
Data are expressed as the median (lower to upper quartile) because of the asymmetric distribution. Differences between groups were analyzed using the Mann-Whitney U test. P < 0.05 was considered significant.
RESULTS
α7nAChR in BM-Derived Cells Is Required for Vagus Nerve–Mediated Inhibition of TNF
nBMα70 mice were created by adoptive transfer of WT BM to irradiated α7nAChR KO mice to obtain mice that express α7nAChR in BM-derived cells, including T cells and macrophages, but not in neurons and other tissues (Table 1). Vagus nerve stimulation of these chimeric animals significantly reduced serum levels of endotoxin-induced TNF (Figure 1A). Because the BM transfer does not restore α7nAChR expression in neurons, these observations indicate that neural expression of α7nAChR is not required for the integrity of the inflammatory reflex. Subsequently, BMα70 mice were created by adoptive transfer of α7nAChR-deficient BM to irradiated WT mice to obtain animals with WT expression of α7nAChR in neurons and all other tissues, but not in BM-derived cells (Table 1). Vagus nerve stimulation failed to inhibit serum TNF levels in these BMα70 animals (Figure 1B). Flow cytometry analysis of splenocytes from chimeric mice 10 wks after transfer showed that ≥84% of macrophages and ≥70% of T cells were derived from donor mice and cell fractions were similar between the chimeras (Table 2). Together, these results indicate that α7nAChR expression in BM-derived cells, but not neurons, is required for the functional integrity of the inflammatory reflex.
Figure 1.
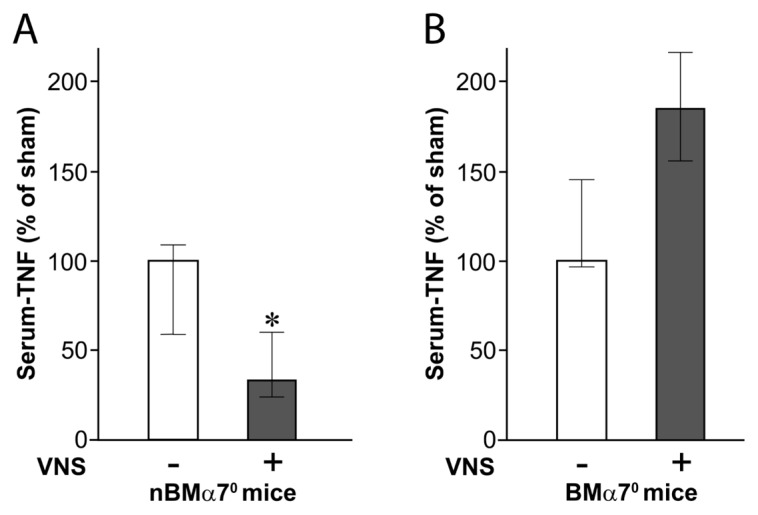
Effect of α7nAChR deficiency in non–BM-derived cells and BM-derived cells on vagus nerve–mediated TNF suppression in endotoxemia. Mice with α7nAChR deficiency either in non–BM-derived (nBMα70) (n = 6) (A) or BM-derived cells (BMα70) (n = 5) (B) were subjected to vagus nerve stimulation followed by intraperitoneal endotoxin injection. Serum TNF was measured 90 min after endotoxin administration. Median TNF levels in pg/mL (lower quartile–upper quartile) were Sham, 476 (279–521), and vagus nerve stimulation (VNS), 159 (114–287), in nBMα70 mice and Sham, 275 (265–400), and VNS, 508 (428–594), in BMα70 mice. Results in diagrams are expressed as the median TNF as percent of sham (lower quartile–upper quartile). *P < 0.05.
Table 2.
Repopulation after adoptive transfer of BM.
| Macrophages | T cells | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Sham | VNS | P | Sham | VNS | P | |
| BMα70 CD45.1+ | 11 ± 0.9 | 13 ± 2.4 | 0.42 | 30 ± 1.6 | 29 ± 2.9 | 0.80 |
| BMα70 CD45.2+ | 86 ± 1.1 | 84 ± 2.9 | 0.83 | 70 ± 1.5 | 71 ± 2.7 | 0.49 |
| nBMα70 CD45.1+ | 92 ± 0.8 | 93 ± 0.6 | 0.23 | 82 ± 1.0 | 82 ± 2.5 | 0.82 |
| nBMα70 CD45.2+ | 4.0 ± 0.6 | 5.0 ± 0.7 | 0.55 | 18 ± 1.0 | 17 ± 2.4 | 0.66 |
Splenocytes from recipient mice were analyzed 10 wks after transfer. Numbers are mean percent ± SEM of CD45.1+ (WT) and CD45.2+ (α7nAChR-deficient) cells of macrophages or T cells as indicated.
α7nAChR Expression on T Cells Does Not Affect Endotoxin-Induced TNF Production
We recently discovered that acetylcholine producing T cells are necessary for the functional integrity of the inflammatory reflex (15). An abundance of data implicates β adrenergic receptors expressed on these T cells as pivotal for their activation to produce acetylcholine, but it remained theoretically possible that T-cell α7nAChR expression (18) is also required for the integrity of the inflammatory reflex. To address this question, we adoptively transferred WT and α7nAChR-deficient T cells to nude mice. Comparable numbers of T cells were observed in the spleen after transfers (Table 3). As expected from previous work (15), vagus nerve stimulation failed to reduce endotoxin-induced serum TNF levels in nude mice, indicating that the inflammatory reflex is functionally impaired in this strain (Figure 2A). Adoptive transfer of WT T cells into nude mice (to create Twt nude mice) restored the ability of vagus nerve stimulation to significantly inhibit TNF (Figure 2B), confirming that T cells are necessary for the inhibition of TNF mediated by vagus nerve stimulation. We then transferred T cells isolated from α7nAChR-deficient mice into nude mice to create Tα70 nude mice. Vagus nerve stimulation inhibited TNF production to a similar degree also in Tα70 nude mice (Figure 2C). Together, these data demonstrate that α7nAChR expression on T cells is not necessary for vagus-nerve mediated TNF suppression in vivo. Furthermore, T cells have been implicated in suppressing innate immune responses (19,20), and it is conceivable that α7nAChR would be required for the effect. To investigate this, T cells were isolated from total splenocytes of α7nAChR-deficient mice and then cocultured with T cell–depleted WT splenocytes. T-cell fractions were ≥94% pure and non–T-cell fractions were ≥97% pure, as assessed by fluorescence-activated cell sorting (Figure 3). Addition of either WT or α7nAChR-deficient T cells to cultures of T cell–depleted splenocytes significantly inhibited endotoxin-induced TNF production (P < 0.05, Figure 4). The magnitude of inhibition was similar after addition of either group of T cells. Collectively, these findings indicate that α7nAChR expression on T cells is not required for T cell–mediated inhibition of TNF.
Table 3.
Transfer of T cells to nude mice.
| Sham | VNS | P | |
|---|---|---|---|
| Twt nude mice | 3.6 ± 0.2 | 3.0 ± 0.3 | 0.13 |
| Tα70 nude mice | 4.8 ± 0.5 | 5.0 ± 0.3 | 0.77 |
Numbers are mean percent T cells of total splenocytes ± SEM.
Figure 2.

Effect of transfer of α7nAChR-deficient T cells to nude mice on vagus nerve–mediated TNF suppression in endotoxemia. Nude mice were subjected to vagus nerve stimulation (VNS) followed by endotoxemia in the presence or absence of functional T cells. TNF was measured in serum 90 min after endotoxin administration. (A) Nude mice (n = 4). Median TNF levels in pg/mL [lower quartile–upper quartile]) were Sham, 324 (170–483), and VNS, 621 (512–816) (P = 0.08). (B) Nude mice after transfer of WT T cells (Twt) (n = 5 sham and n = 6 VNS). TNF levels were Sham, 1,120 (1,070–1,290), and VNS, 630 (589–974) (P = 0.03). (C) Nude mice after transfer of α7nAChR-deficient T cells (Tα7°) (n = 6). TNF levels were Sham, 831 (715–1,030), and VNS, 556 (346–665) (P = 0.04). Results in diagrams are expressed as median TNF as percent of sham (lower quartile–upper quartile). *P < 0.05.
Figure 3.
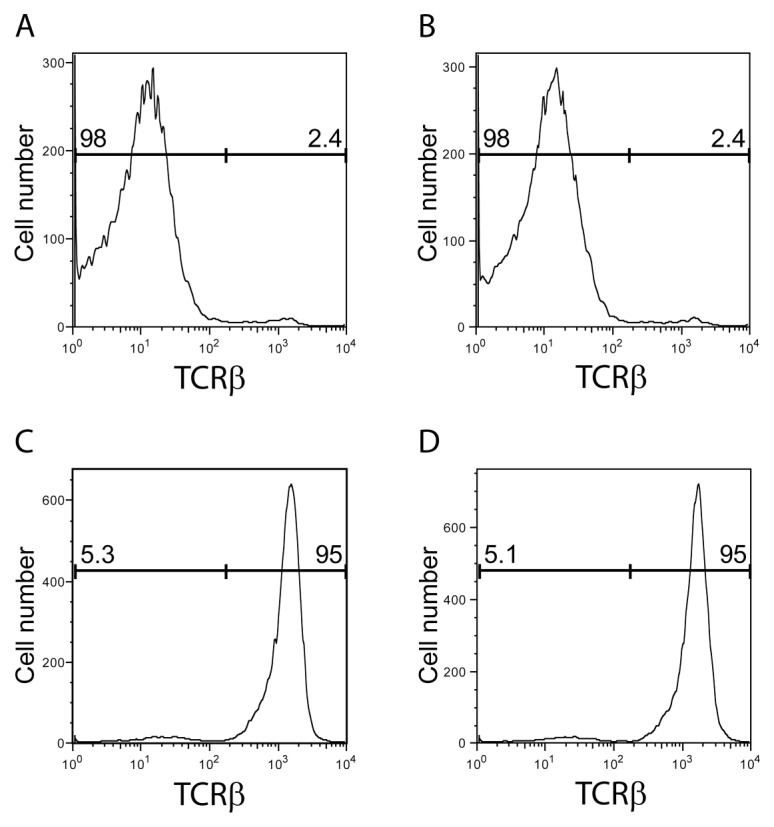
Purity of isolated murine spleno-cyte fractions. Cells were stained with an anti-TCRβ antibody and analyzed by flow cytometry. T-cell–depleted splenocytes from WT (A) and α7nAChR KO mice (B) are shown. T-cell fraction from WT mice (C) and α7nAChR KO mice (D) are shown. The numbers in the graph show fractions of TCRβ−and TCRβ+ cells.
Figure 4.
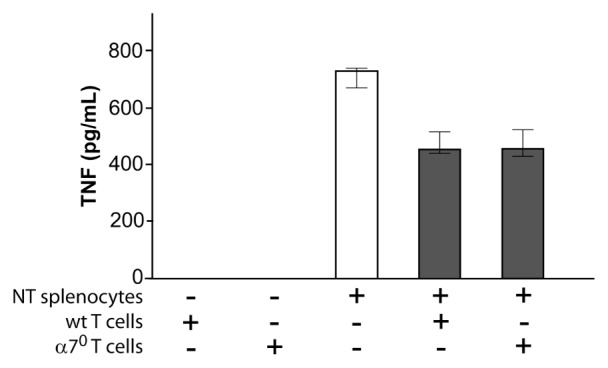
Effect of T-cell α7nAChR deficiency on suppression of endotoxin-induced TNF production in vitro. T cells from WT or α7nAChR-deficient mice were cocultured with T cell–depleted WT splenocytes, stimulated with endotoxin and TNF measured in the supernatant. Results are shown as median TNF in pg/mL (lower quartile–upper quartile). NT, T cell depleted; α70, α7nAChR KO.
DISCUSSION
This study reveals that α7nAChR expression in BM-derived non-T cells is necessary for the function of the inflammatory reflex. α7nAChR in neurons and other non-BM derived cells and on T cells is expendable in vagus nerve stimulation-mediated inhibition of cytokine production. The results are consistent with earlier pharmacological data on cytokine producing cells and implicate a role for α7nAChR signaling in mediating inhibition of cell activation.
The efferent arc of the inflammatory reflex, termed the “cholinergic antiinflammatory pathway,” can be stimulated using electrical vagus nerve stimulators to prevent or reverse damage in experimental endotoxemia, sepsis, pancreatitis, arthritis, colitis and other inflammatory syndromes (21). The spleen is a major organ target for the antiinflammatory effects of efferent vagus nerve signals in endotoxemia because the spleen is the major source of TNF during endotoxemia and efferent vagus signals control production of TNF there (22). Administration of α7nAChR agonists also suppresses cytokine release and attenuates tissue damage during inflammation (5,8,17). Deficiency or impairment of α7nAChR signaling, or the cholinergic antiinflammatory pathway, leads to overproduction of cytokines and enhances tissue damage (16,17).
Whereas it seemed likely that functional expression of α7nAChR by macrophages and cytokine-producing cells accounted for the mechanism, it could not be ruled out that α7nAChR expressed in the brain and autonomic ganglia (23,24) was required to complete the cholinergic antiinflammatory circuit. For instance, vagus nerve stimulation in an intact animal elicits both efferent and afferent signals, and it would be conceivable that α7nAChRs in the central nervous system are involved in processing of afferent signals that ultimately result in efferent activity in the contralateral vagus or other nerves. In addition, α7nAChR expressed in neurons in the celiac ganglion might receive acetylcholine signals released from descending vagus or sympathetic nerve endings that terminate there. Further, the splenic nerve is adrenergic, not cholinergic (25), and it has been unclear how signals from the cholinergic vagus nerve could be sensed by α7nAChR in the spleen. We recently demonstrated that vagus nerve stimulation indeed increases splenic acetylcholine levels and that a subset of acetylcholine-producing T cells are required for the efferent signals in the inflammatory reflex (15). Thus, we resolved the question of how signals from the cholinergic vagus nerve could be sensed by α7nAChR on cytokine-producing splenic macrophages. The present study advances mechanistic understanding and shows that α7nAChR expression in immune cells, but not in neurons or T cells, is required for the functional integrity of the inflammatory reflex.
T cells can inhibit innate immune responses (19,20), as confirmed by the coculture experiments in this study. A subset of splenic acetylcholine-producing T cells respond to adrenergic neural signals (15), and it had been suggested that T-cell α7nAChR expression might be important for the cholinergic antiinflammatory pathway (26,27). The present findings indicate that the integrity of the efferent arm of the inflammatory reflex and the antiinflammatory effects of T cells are independent of T-cell α7nAChR. Because nor-epinephrine can increase production of acetylcholine in select T cells, it is conceivable that activation of β-adrenergic or other neurotransmitter receptors other than α7nAChR contributes to the antiinflammatory effects of T cells (15,18).
CONCLUSION
Thus, the inflammatory reflex requires α7nAChR expression on non–T cell, BM-derived immune cells. Together with our recent findings on splenic acetylcholine-producing T cells, these data resolve the mechanism for signal transfer from the vagus nerve to TNF-producing cells in the spleen.
ACKNOWLEDGMENTS
This work was supported in part by grants from National Institute of General Medical Sciences, National Institutes of Health (NIGMS, NIH; GM57226 and 3R01GM057226-10S1) to KJ Tracey and from the Wenner-Gren Foundations in Stockholm to PS Olofsson.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–28. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matsunaga K, Klein TW, Friedman H, Yamamoto Y. Involvement of nicotinic acetylcholine receptors in suppression of antimicrobial activity and cytokine responses of alveolar macrophages to Legionella pneumophila infection by nicotine. J Immunol. 2001;167:6518–24. doi: 10.4049/jimmunol.167.11.6518. [DOI] [PubMed] [Google Scholar]
- 3.Orr-Urtreger A, Kedmi M, Rosner S, Karmeli F, Rachmilewitz D. Increased severity of experimental colitis in alpha 5 nicotinic acetylcholine receptor subunit-deficient mice. Neuro-report. 2005;16:1123–7. doi: 10.1097/00001756-200507130-00018. [DOI] [PubMed] [Google Scholar]
- 4.Giebelen IA, van Westerloo DJ, LaRosa GJ, de Vos AF, van der Poll T. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock. 2007;28:700–3. doi: 10.1097/shk.0b013e318054dd89. [DOI] [PubMed] [Google Scholar]
- 5.Pavlov VA, et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med. 2007;35:1139–44. doi: 10.1097/01.CCM.0000259381.56526.96. [DOI] [PubMed] [Google Scholar]
- 6.Yeboah MM, et al. Cholinergic agonists attenuate renal ischemia-reperfusion injury in rats. Kidney Int. 2008;74:62–9. doi: 10.1038/ki.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Maanen MA, et al. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009;60:114–122. doi: 10.1002/art.24177. [DOI] [PubMed] [Google Scholar]
- 8.Rosas-Ballina M, et al. The selective alpha7 agonist GTS-21 attenuates cytokine production in human whole blood and human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol Med. 2009;15:195–202. doi: 10.2119/molmed.2009.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karimi K, Bienenstock J, Wang L, Forsythe P. The vagus nerve modulates CD4+ T cell activity. Brain Behav Immun. 2010;24:316–23. doi: 10.1016/j.bbi.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 10.Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol. 2004;5:575–81. doi: 10.1038/ni1078. [DOI] [PubMed] [Google Scholar]
- 11.Rosas-Ballina M, Tracey KJ. The neurology of the immune system: neural reflexes regulate immunity. Neuron. 2009;64:28–32. doi: 10.1016/j.neuron.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun J, Singh V, Kajino-Sakamoto R, Aballay A. Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science. 2011;332:729–32. doi: 10.1126/science.1203411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borovikova LV, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–62. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 14.Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–9. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 15.Rosas-Ballina M, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 17.Parrish WR, et al. Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. 2008;14:567–74. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawashima K, Fujii T. Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 2004;9:2063–85. doi: 10.2741/1390. [DOI] [PubMed] [Google Scholar]
- 19.Guarda G, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–73. doi: 10.1038/nature08100. [DOI] [PubMed] [Google Scholar]
- 20.Kim KD, et al. Adaptive immune cells temper initial innate responses. Nat Med. 2007;13:1248–52. doi: 10.1038/nm1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–96. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huston JM, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203:1623–8. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broide RS, Leslie FM. The alpha7 nicotinic acetylcholine receptor in neuronal plasticity. Mol Neurobiol. 1999;20:1–16. doi: 10.1007/BF02741361. [DOI] [PubMed] [Google Scholar]
- 24.Lips KS, Konig P, Schatzle K, et al. Coexpression and spatial association of nicotinic acetylcholine receptor subunits alpha7 and alpha10 in rat sympathetic neurons. J Mol Neurosci. 2006;30:15–6. doi: 10.1385/JMN:30:1:15. [DOI] [PubMed] [Google Scholar]
- 25.Bellinger DL, Lorton D, Hamill RW, Felten SY, Felten DL. Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: lack of evidence for cholinergic innervation. Brain Behav Immun. 1993;7:191–204. doi: 10.1006/brbi.1993.1021. [DOI] [PubMed] [Google Scholar]
- 26.Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med. 2003;9:125–34. [PMC free article] [PubMed] [Google Scholar]
- 27.Bencherif M, Lippiello PM, Lucas R, Marrero MB. Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell Mol Life Sci. 2011;68:931–49. doi: 10.1007/s00018-010-0525-1. [DOI] [PMC free article] [PubMed] [Google Scholar]