

Abstract
Solid tumors are intrinsically resistant to immune rejection. Abnormal tumor vasculature can act as a barrier for immune cell migration into tumors. We tested whether targeting IFNγ and/or TNFα into pancreatic neuroendocrine tumors can alleviate immune suppression. We found that intratumoral IFNγ causes rapid vessel loss, which does not support anti-tumor immunity. In contrast, low-dose TNFα enhances T-cell infiltration and overall survival, an effect that is exclusively mediated by CD8+ effector cells. Intriguingly, lymphocyte influx does not correlate with increased vessel leakiness. Instead, low-dose TNFα stabilizes the vascular network and improves vessel perfusion. Inflammatory vessel remodeling is, at least in part, mediated by tumor-resident macrophages that are reprogrammed to secrete immune and angiogenic modulators. Moreover, inflammatory vessel remodeling with low-dose TNFα substantially improves antitumor vaccination or adoptive T-cell therapy. Thus, low-dose TNFα promotes both vessel remodeling and antitumor immune responses and acts as a potent adjuvant for active immunotherapy.
Keywords: angiogenesis, vessel normalization, macrophage polarization
The tumor microenvironment is rich in inflammatory cells and cytokines that play a pivotal role in cancer promotion (1). Nevertheless, plasticity of intratumoral immune cells can be exploited therapeutically to foster antitumor immunity (2, 3). We have demonstrated that in the “right” inflammatory context, proangiogenic processes can be reversed to create a tumor environment permissive for immune destruction (4, 5). For instance, in a mouse model of endocrine pancreatic cancer that is intrinsically resistant to immune cell infiltration and destruction, radiation-induced intratumoral inflammation activates tumor endothelia and greatly enhances leukocyte influx (6). Hallmarks of this study were the findings that tumor destruction correlates with (i) remodeling or normalization of the angiogenic vasculature; (ii) strong induction of intratumoral IFNγ and TNFα, and (iii) high level effector T-cell penetration and persistence in the tumor tissue (6). There is now further compelling evidence that vascular remodeling increases the efficacy of immunotherapy (7–9). However, the role of intratumoral cytokines such as IFNγ and TNFα in enhancing effector cell extravasation and/or persistence possibly through inflammatory vessel remodeling remains unclear.
IFNγ and TNFα are well characterized cytokines that exert a plethora of effects. Both cytokines have been shown to promote innate and adaptive antitumor immune responses (10). Moreover, they act directly or indirectly through inflammatory cells on tumor blood vessels (11–13). Most notably, high-dose TNFα disrupts angiogenic vessels and is used in isolated limb perfusion to treat locally advanced melanoma and soft tissue sarcoma (14). More recently, new therapeutic approaches designed to target TNFα selectively into the tumor environment greatly enhanced efficacy of cytotoxic drugs and radiation therapy in preclinical models (15, 16), and clinical trials are underway (17). Furthermore, synergistic actions of IFNγ and TNFα have been reported (10, 18, 19).
Based on our findings (6), we postulated that IFNγ and TNFα, alone or in combination, are effective in altering the vascular bed and alleviating the immunosuppressive tumor environment, thus enhancing antitumor immunotherapy. In the present study, we engineered IFNγ and TNFα with a tumor vasculature-targeting peptide (RGR peptide; ref. 20) to specifically deliver cytokines into mice carrying pancreatic neuroendocrine tumors and assess their adjuvant effects in anticancer immunity. We demonstrate that intratumoral IFNγ predominantly acts as an antivascular agent. In contrast, vascular-targeted, low-dose TNFα greatly enhances anticancer immunotherapy not by destroying angiogenic vessels but instead by increasing vascular functionality.
Results
Distinct Intratumoral Effects of IFNγ and TNFα.
Systemic toxicity of TNFα and IFNγ has limited their clinical use. To focus on their intratumoral effects, recombinant IFNγ and TNFα with N-terminal RGR peptide were synthesized. RGR peptide (CRGRRST) has been shown to specifically bind to highly angiogenic vessels in murine insulinomas (20). Bacterially expressed IFNγ– and TNFα–RGR fusion compounds are biologically active, selectively home into tumors, and are retained in the tumor microenvironment (Fig. S1). RIP1-Tag5 transgenic mice, which express SV40 Large T antigen (Tag) under the control of the rat insulin gene promoter (RIP), develop pancreatic neuroendocrine tumors over time. Vascularized tumors are macroscopically visible at ≈22–23 wk of age. Although there is an initial immune response against Tag protein, solid tumors show little spontaneous infiltration by CD4+ or CD8+ T cells (6). To analyze intratumoral effects of IFNγ, TNFα, and RGR conjugates, 27-wk-old transgenic mice with considerable tumor burden were treated for 2 wk (Fig. 1A). At 29 wk, tumors were analyzed for infiltrating T cells in relation to the vascular bed (Fig. 1B). IFNγ–RGR treatment does not increase the number of extravasating T cells, but instead results in a substantially reduced number of CD31+ blood vessels. In contrast, TNFα–RGR-treated tumors are heavily infiltrated by CD8+ cells while retaining a high degree of vascularization. None of the tumors showed a significant CD4+ T-cell infiltrate. Moreover, intratumoral changes are specific for RGR-tagged compounds (Fig. 1 C and D). Consistent with a reduced vessel count, a higher frequency of apoptotic cells is visible in IFNγ–RGR-treated tumors with clustering of TUNEL+ cells around vascular structures (Fig. 1E); vessel death is not observed in TNFα–RGR-treated tumors (Fig. 1F). Thus, both fusion compounds, IFNγ–RGR and TNFα–RGR, change the tumor environment but have distinct effects on blood vessels and CD8+ T-cell influx.
Fig. 1.

IFNγ and TNFα have distinct effects in the tumor microenvironment. (A) Schematic representation of a short-term treatment regimen in RIP1-Tag5 mice. Arrows indicate four i.v. injections of compounds. Tumors were analyzed at 29 wk. (B) Costaining of control (untreated), IFNγ–RGR and TNFα-RGR treated tumors with specific antibodies: CD8+ T cells, red; CD31+ blood vessels, green. Representative pictures after biweekly i.v. injections of 2 μg of IFNγ–RGR or TNFα-RGR for 2 wk are shown. (Original magnification: 20×) (Scale bar: 100 μm.) (C) Quantification of tumor-infiltrating CD8+ T cells (mean CD8+ T cells per field ± SE, n = 3–9, *P < 0.01 compared with all other groups). (D) Quantification of CD31-positive blood vessels (mean % of CD31-covered area/field ± SE, n = 4–6, *P ≤ 0.01 compared with all other treatment groups). (E) Costaining of CD31-positive blood vessels (red) with TUNEL+, apoptotic cells (green) in IFNγ–RGR treated tumors. (Original magnification: 10×) (Scale bar: 200 μm.) Inset shows clustering of apoptotic cells around a vessel. (Original magnification: 40×) (Scale bar: 50 μm.) (F) Quantification of apoptotic cells in different treatment groups (mean TUNEL+ cells per field ± SE, n = 3–7, *P = 0.02 compared with control and TNFα-RGR treated groups).
TNFα-RGR Monotherapy Effectively Prolongs Survival.
Based on the promising TNFα-RGR immune profile, we hypothesized that local, low-dose TNFα treatment may induce spontaneous antitumor immunity. An in vivo CTL assay was used to analyze the capacity of T cells to lyse splenocytes loaded with a tumor-specific peptide (Tag peptide IV; ref. 21). Tumor-bearing controls or TNFα-treated mice do not mount a tumor-specific immune response. In contrast, TNFα–RGR-treated mice develop anti-Tag CTL activity locally in tumor draining pancreatic lymph nodes (Fig. 2A). Next, long-term survival benefits of TNFα and TNFα-RGR monotherapy was investigated in a therapeutic setting (Fig. 2B). At a dose of 2 μg, toxic side effects are negligible but overall survival is significantly prolonged (Fig. 2C; TNFα-RGR, 34 ± 1 wk; TNFα, 31 ± 0 wk; untreated, 28 ± 2 wk, P = 0.001 TNFα-RGR compared with untreated). To test the functional contribution of CD8+ T cells in therapeutic outcome, RIP1-Tag5 mice were treated in the presence and absence of depleting antibodies. Interestingly, therapeutic efficacy of TNFα-RGR treatment is completely abrogated with CD8+ T-cell depletion (Fig. 2D), indicating that CD8+ T cells are crucial mediators of TNFα-RGR survival benefits. CD4+ T-cell depletion does not change therapeutic outcome. To model current clinical trials, lower TNFα and TNFα-RGR doses (0.2 μg and 0.2 ng) were also assessed (15). At 0.2 μg, some therapeutic efficacy was achieved, however, the selective advantage of TNFα-RGR over TNFα is lost, suggesting peripheral rather than intratumoral effector mechanisms (Fig. S2A; TNFα-RGR, 31 ± 2 wk; TNFα, 32 ± 2 wk). As expected, treatment with a dose of 0.2 ng shows no beneficial effects as monotherapy (Fig. S2B). TNFα-RGR outperforms IFNγ–RGR monotherapy, which is ineffective at 2 μg but moderately efficient at 25 μg when directed into the tumor microenvironment (Fig. S2C, 32 ± 2 wk); this result is consistent with its antiangiogenic function. A combination of TNFα-RGR and IFNγ–RGR (both at 2 μg) is less efficient than TNFα-RGR alone (Fig. S2D). This lack of efficacy is somewhat surprising because synergistic effects of TNFα and a suboptimal IFNγ dose were described earlier (10, 18). Nevertheless, these data imply that although intratumoral effects of TNFα and IFNγ are different, they are not additive. Given the immune-stimulating effects of TNFα alone, our subsequent analyses focused on TNFα-RGR.
Fig. 2.

Long-term survival under TNFα-RGR monotherapy is CD8+ T-cell dependent. (A) Untreated or TNFα/TNFα-RGR treated RIP1-Tag5/F1 mice were assessed for in vivo CTL activity against the Tag-specific peptide IV after 2 wk of treatment. (Left) Combined data for spleen cells (n = 5) and tumor-draining pancreatic lymph nodes (LN; n = 3–5, LNs were pooled in each of two independent experiments). (Right) Representative histograms of percent specific kill of CFSEhigh LN cells from two treatment groups. (B) Long-term treatment scheme: RIP1-Tag5 mice were treated at the age of 22–23 wk with biweekly i.v. injections and survival monitored. Percent survival of RIP1-Tag5 mice treated with 2 μg of TNFα or TNFα-RGR (P = 0.002, TNFα-RGR compared with TNFα; P = 0.001, TNFα-RGR compared with untreated controls (n = 5–7) (C), and 2 μg of TNFα-RGR in the presence (αCD8) and absence (IgG) of CD8+ T-cell depleting antibodies (P = 0.0002, TNFα-RGR plus depletion compared with TNFα-RGR with control IgG, n = 8) (D).
TNFα-RGR Enhances Active Antitumor Immunotherapy.
Intratumoral TNFα induces spontaneous antitumor immunity and lymphocyte access into tumors. Therefore, we asked whether TNFα-RGR was also effective as an adjuvant to active immunotherapy. To test this hypothesis, RIP1-Tag5 mice were treated with a combination of 2 μg TNFα-RGR and an anti-Tag vaccine. Anti-Tag vaccination alone is ineffective once solid tumors arise (Fig. 3A; ref. 5). In contrast, a combination of TNFα-RGR and vaccine substantially enhances survival of transgenic mice compared with TNFα-RGR or vaccination alone (38 ± 5 wk versus 34 ± 2 wk and 31 ± 2 wk, respectively, P = 0.007; TNFα-RGR compared with TNFα-RGR plus vaccine), with 20% of the treated cohort surviving beyond 45 wk. Nanograms of TNFα-RGR (0.2 ng), which confer no survival benefits alone, enhance vaccination efficacy (survival 34 ± 2 wk, Fig. S3). Moreover, survival benefits of up to 60% are achieved in an adoptive transfer protocol that does not rely on endogenous effector cell activation. RIP1-Tag5 mice were treated with ex vivo activated anti-Tag CD4+ and CD8+ T cells alone or in combination with 2 μg of TNFα-RGR, which dramatically increases the efficacy of adoptive transfers (42 ± 4 wk versus 33 ± 3, P ≤ 0.0001) (Fig. 3B). Activated effectors proliferate in tumor-draining, pancreatic lymph nodes in untreated control mice but do not enter tumors in sufficient numbers to impact on growth (Fig. 3C; ref. 21). In contrast, local TNFα treatment facilitates extravasation and accumulation of activated T cells in tumors in correlation with dramatic extension of overall survival.
Fig. 3.

Intratumoral TNFα-RGR enhances efficacy of anticancer immunotherapy. (A) RIP1-Tag5 mice were treated with vaccine alone, 2 μg of TNFα-RGR alone or in combination with anti-Tag vaccine (P = 0.007 single versus combination treatment, n = 8–12). (B) RIP1-Tag5 mice were treated every second week with adoptive transfers (ad T) of preactivated CD4+ and CD8+ Tag-specific T cells alone or in combination with 2 μg of TNFα-RGR, survival was monitored up to 45 wk (P < 0.0001, n = 8–10). (C) Percentage of TagTCR8 T cells in pancreatic lymph nodes (panc LN) or tumors was tracked by FACS analysis for 21 d after adoptive transfer in untreated (Left) or TNFα-RGR treated mice (Right), n = 3.
Intratumoral Low-Dose TNFα Improves Vascular Functionality.
TNFα is best known as an agent that induces endothelial cell apoptosis leading to vessel destruction. However, at lower doses, it improves penetration of anticancer drugs presumably by increasing endothelial permeability (15, 22). Our observation that lymphocyte penetration is increased without concomitant vascular death prompted us to investigate vascular changes in the tumor environment after TNFα-RGR therapy. Staining for the endothelial cell marker CD31 showed comparable vessel numbers but a significant reduction in mean vessel length in TNFα-RGR treatment groups (Fig. 4 A and B; P = 0.01). This effect is mainly caused by a selective loss of large tumor vessels (150–200 μm, Fig. 4C; P = 0.003). Moreover, quality and quantity of pericyte coverage are important parameters for vessel maturation and functionality. In untreated RIP-Tag tumors, vessels are lined with immature PDGFRβ+ pericytes and, to a lesser extent, with more mature αSMA-expressing cells that are located near vessels but mostly detached (Fig. 4D; ref. 8). Strikingly, vascular coverage with PDGFRβ+ pericytes is substantially reduced under TNFα-RGR treatment (Fig. 4E). In contrast, αSMA+ cells are closely attached to vessels, indicating a shift to more mature, stabilized vessels (Fig. 4F). To address the question of whether the observed vascular remodeling also changes vascular permeability, mice were injected with Texas red-labeled dextran followed by saline perfusion. Dextran extravasates into extravascular space through “leaky” tumor vessels in control mice, an effect that is dramatically reduced in TNFα–RGR-treated tumors (Fig. 4 G and H). Reduction in vascular leakiness correlates with increased vascular perfusion measured by delivery of FITC-conjugated lectin to tumor vessels (Fig. 4 I and J). Overall, 2 μg of TNFα-RGR treatment over 2 wk improves vessel maturity, reduces vascular leakiness, and, thus, enhances tumor perfusion.
Fig. 4.
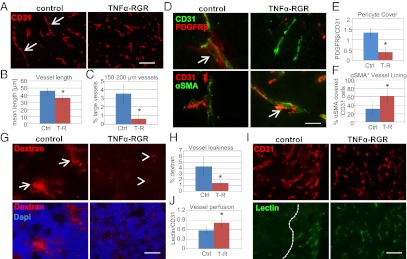
Tumor-targeted TNFα stabilizes vessels and enhances vascular functionality. (A) Representative pictures of CD31-positive vessels in control (untreated) RIP1-Tag5 tumors and after 2 wk of treatment with 2 μg of TNFα-RGR. Arrows point at large vessels. (Original magnification: 20×.) (Scale bar: 100 μm.) (B) Quantification of mean vessel length in control (Ctrl) and treatment groups (T-R, TNFα-RGR) (P = 0.01). (C) Quantification of percentage of large vessels (size: 150–200 μm) (P = 0.003). (D Upper) CD31+ vessels (green) and coverage with PDGFRβ+ pericytes (red). Arrow points at a pericyte-covered area in controls. (Lower) CD31+ vessels (red) and association of αSMA+ perivascular cells (green). Arrow points at close vascular lining in TNFα-RGR treated tumors. (Original magnification: 40×.) (Scale bar: 50 μm.) (E) Ratio of PDGFRβ-positive pericytes to CD31-positive endothelial cells (P = 0.003). (F) Percent αSMA+ covered endothelial cells (P = 0.02). (G) Vascular permeability assessed by injection of 70-kDa Texas-red labeled dextran followed by saline perfusion. (Upper) Dextran signals in tumors. Arrows point at areas of dextran extravasation. Arrowheads point at residual dextran associated with vessels. (Lower) Dextran/dapi double staining. (Original magnification: 20×.) (Scale bar: 100 μm.) (H) Quantification of percentage of dextran in tumors as readout for vascular leakiness (P = 0.02). (I) CD31-positive vessels (Upper) in relation to i.v. injected FITC-lectin (Lower). Dashed line indicates perfused and nonperfused tumor areas. (Original magnification: 20×.) (Scale bar: 100 μm.) (J) Ratio lectin-positive vessels to CD31-positive vessels (P = 0.03, n = 3–8 for all groups).
TNFα Effects on Macrophage Polarization.
Functional improvement of angiogenic vessels per se can enhance influx of immune effector cells into tumor parenchyma (8). In addition, TNFα is also a potent inducer of endothelial activation (23) which in turn facilitates leukocyte extravasation into tumors (21, 24). Indeed, staining with the activation marker VCAM reveals strong up-regulation in TNFα-RGR treated tumors (Fig. S4). Strikingly, VCAM signals are not confined to CD31-positive vessels, but comprise the whole stromal compartment, including fibroblasts and macrophages (Fig. S4). Low-dose, intratumoral TNFα treatment has no effect on the recruitment of myeloid cells into tumors (Fig. S5A). However, CD68+ tumor-resident macrophages change in phenotype (VCAM+; Fig. S5B) and preferentially cluster around vessels after treatment (Fig. S5C). We therefore hypothesized that macrophage polarization during TNFα therapy may play a modulatory role in vessel remodeling and antitumor immunity. To test this hypothesis, we isolated CD68+ macrophages from untreated and TNFα–RGR-treated tumors and analyzed their gene expression signature. Interestingly, under TNFα treatment macrophages specifically up-regulate immunostimulatory genes such as MCP-1, IL6, iNOS, and Mig, which is consistent with a switch to M1 macrophages (Fig. 5A). Moreover, these tumor-resident macrophages are Tie2-negative and neither proangiogenic (VEGFlow, PLGFlow) nor immunosuppressive (Ccl17low, IL10low), suggesting a skewing away from a tumor-promoting phenotype. Indeed, CD68+ macrophages isolated from untreated tumors suppress anti-Tag CD8+ T-cell proliferation in vitro, whereas macrophages from TNFα-RGR treated tumors alleviate suppression (Fig. 5B). Interestingly, angiopoetin 2 (Ang2), a Tie2 tyrosine kinase receptor ligand, is also up-regulated in treated macrophages. Ang2 has been shown to modulate TNFα-dependent, vascular VCAM expression and promote leukocyte adhesion to the activated endothelium (25). Ang2 alone does not induce expression of adhesion molecules on endothelial cells; however, it sensitizes HUVECs for VCAM induction under low-dose TNFα, which in itself is insufficient to induce vessel inflammation (Fig. S6). Macrophages isolated from untreated tumors secrete low amounts of TNFα and Ang2 and do not induce VCAM expression on endothelial cells (Fig. 5C). In contrast, HUVECs cocultured with Ang2+ macrophages from TNFα-RGR treated tumors express VCAM. This effect requires both Ang2 and TNFα and is abolished with corresponding blocking antibodies (Fig. 5C). These data strongly suggest that differential cytokine production of TNFα-polarized macrophages modulates both endothelial activation and antitumor T-cell responses.
Fig. 5.

Tumor macrophages are activated and reprogrammed to express immunostimulatory factors and angiogenic modulators. (A) Quantitative PCR analysis of isolated CD68+ macrophages from TNFα-RGR treated tumors, expressed as fold change relative to CD68+ from control (untreated) RIP1-Tag5 tumors (n = 3). (B Left) Quantitative analysis of TagTCR8 cell proliferation, unstimulated (-) or stimulated with Tag-specific peptide/IL2 (+) in the presence of macrophages (MØ) isolated from untreated controls (ctrl) or tumors after 2 wk of treatment with 2 μg of TNFα-RGR (T-R) (P = 0.01). (Right) Representative histograms showing percent proliferation of CFSE-labeled T cells from all groups. (C Left) HUVEC were incubated with macrophages isolated from untreated (MØ ctrl) or TNFα-RGR treated tumors (MØ T-R) in the presence of Ang2 receptor (Tie2) (Ang2 block) or TNFα blocking antibodies (TNFα block). Arrows delineate VCAM positive, cellular HUVEC staining. (Original magnification: 40×.) (Scale bar: 50 μm.) (C Right) Quantification of percent VCAM-positive cells in relation to DAPI-positive cells (P = 0.08).
Discussion
Our previous work has implicated IFNγ and TNFα in vascular remodeling and antitumor immunity, but their actual role in the tumor environment has been elusive (6). Here, we developed intratumoral targeting strategies and demonstrate distinct antitumor effector mechanisms for both cytokines.
IFNγ, when targeted to endothelial cells, predominantly induces vessel death. Destruction of the angiogenic vasculature in itself shows therapeutic efficacy reminiscent of antiangiogenic drugs. Interestingly, endogenous IFNγ induced during antitumor immune responses has also been shown to act on stroma with profound antivascular effects (12). Nevertheless, intratumoral IFNγ fails to elicit a potent immune response either because vessel destruction ultimately interferes with lymphocyte infiltration or because of counterregulatory mechanisms in the tumor environment (18, 26).
In contrast, intratumoral TNFα has dual effects by remodeling tumor stroma and enhancing adaptive immunity. As such, it shows survival benefits as a single agent, an effect that exclusively depends on CD8+ effector T cells. Activation of adoptive immunity has been observed in the context of intratumoral TNFα treatment in previous studies (27, 28). However, we expand on this observation and demonstrate that intratumoral TNFα therapy is a strong adjunct to immunotherapy. Vaccination against the model tumor antigen SV40 Large T antigen, which is ineffective once solid tumors have formed in RIP-Tag mice (5), becomes highly efficient when combined with TNFα-RGR. Similarly, adoptive transfers of activated antitumor effector cells with limited impact on overall survival (8) become highly effective in conjunction with TNFα-RGR.
TNFα has a long history as an anticancer agent. It is most potent when used in combination with chemotherapy either in local high-dose treatment regimens such as isolated limb perfusion (14) or specifically targeted into tumors in picogram quantities (15, 29). The rationale for using local TNFα therapy is based on increased endothelial permeability followed by damage of the tumor vasculature and necrosis. In patients undergoing isolated limb perfusion, high-dose TNFα causes endothelial activation and redistribution of junctional and cytoskeletal molecules (30, 31). This rearrangement is followed by suppression of αvβ3 integrin, progressive detachment of endothelial cells, and apoptosis (11). Vascular effects of low-dose TNFα treatment are less clear. Importantly, our study demonstrates that intratumoral low-dose TNFα treatment (2 μg over 2 wk) induces initial vessel stabilization. This effect is documented by induction of a more regular vascular network with small vessel calibers and mural stabilization. These vessels are less leaky and, thus, improve tumor perfusion. Interestingly, TNFα has been reported to reduce interstitial tumor pressure when injected systemically in tumor-bearing mice (32). Reduced interstitial pressure, in turn, may increase drug penetration (33) and is more likely a result of vascular stabilization than leakiness or destruction. Intriguingly, vascular remodeling under low-dose TNFα treatment as shown here is reminiscent of transient vascular normalization under VEGF blockade, which improves drug penetration and enhances chemotherapy (34).
Although targeted to endothelial cells, TNFα effects are not restricted to the vascular compartment. We show that tumor-resident macrophages cluster around vessels, display a distinct M1 phenotype, and are activated to secrete a variety of inflammatory and angiogenic modulators. Skewing of macrophages towards an M1 profile promotes antitumor immunity. Importantly, however, recent reports highlight another role of macrophages in vascular remodeling; for instance, polarization of macrophages away from a tumor-promoting M2 profile promotes vessel normalization (35, 36). Intriguingly, TNFα-treated tumor macrophages up-regulate Ang2, a context-dependent Tie2 agonist that has been shown to reduce vascular leakiness (37). Moreover, Fiedler et al. have demonstrated that autocrine Ang2 is a potent modulator of TNFα-induced vascular inflammation (25). Our data provide evidence for a potential paracrine effect of Ang2 to enhance TNFα-mediated vascular inflammation that can be exploited to increase leukocyte adhesion and antitumor immunity. Thus, our study suggests that macrophages play a modulatory role in mediating intratumoral TNFα effects by promoting vessel perfusion, activation, and antitumor immunity (Fig. S7).
Long-term TNFα treatment over weeks results in stromal destruction and resolution of T-cell infiltration (Fig. S8). Therefore, early stromal activation/remodeling provides the opportunity to combine intratumoral TNFα therapy with active immunotherapy. Once tumor vessels are destroyed, immunotherapy is less effective, most likely due to limited access of effector cells into the tumor tissue. Apparently, dose and scheduling of intratumoral TNFα are critical for the development of effective combination therapies. Our findings open insights into the anti-tumor role of TNFα and offer therapeutic opportunities. Targeting low-dose TNFα into solid tumors with resultant vessel stabilization can be exploited to “precondition” the tumor microenvironment for active immunotherapy. This adjuvant effect to immunotherapy has so far been unexplored.
Materials and Methods
A detailed description of methods is provided in SI Materials and Methods. In summary, TNFα and IFNγ fusion proteins were produced by recombinant technology and purified by using His-tag/ Ni-NTA beads. Transgenic RIP1-Tag5 mice were treated at 22–23 wk (long term) or at 27 wk (short term) to monitor survival or intratumoral effects by histology, respectively. Vascular morphology and functionality was analyzed by using a Nikon Ti-E microscope and quantified by using NIS software (version 3.0). IFNγ/IFNγ-RGR was used at 2 μg and 25 μg; TNFα/TNFα-RGR was used at 0.2 ng, 0.2 μg, and 2 μg. CD8 T cells were depleted by using antibodies (clone 53–6.7). Vaccination was performed with a mixture of purified Tag protein and CpG oligonucleotides in a prime (s.c.)/boost regimen (i.p. every third week). Adoptive transfers were performed by using TCR transgenic mice specific for the model tumor antigen Tag.
Supplementary Material
Acknowledgments
We thank H. Ee for critical reading of the manuscript. This work was supported by grants from the Medical Research Foundation, Royal Perth Hospital, the National Health and Medical Research Council, the Cancer Council Western Australia (grants and fellowship to R.G.), the University of Western Australia (to A.J.), and the Swedish Research Council (fellowship to A.J.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118296109/-/DCSupplemental.
References
- 1.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson D, Ganss R. Tumor growth or regression: Powered by inflammation. J Leukoc Biol. 2006;80:685–690. doi: 10.1189/jlb.1105646. [DOI] [PubMed] [Google Scholar]
- 3.Johansson M, Denardo DG, Coussens LM. Polarized immune responses differentially regulate cancer development. Immunol Rev. 2008;222:145–154. doi: 10.1111/j.1600-065X.2008.00600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganss R, Hanahan D. Tumor microenvironment can restrict the effectiveness of activated antitumor lymphocytes. Cancer Res. 1998;58:4673–4681. [PubMed] [Google Scholar]
- 5.Garbi N, Arnold B, Gordon S, Hämmerling GJ, Ganss R. CpG motifs as proinflammatory factors render autochthonous tumors permissive for infiltration and destruction. J Immunol. 2004;172:5861–5869. doi: 10.4049/jimmunol.172.10.5861. [DOI] [PubMed] [Google Scholar]
- 6.Ganss R, Ryschich E, Klar E, Arnold B, Hämmerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 2002;62:1462–1470. [PubMed] [Google Scholar]
- 7.Dirkx AE, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J. 2006;20:621–630. doi: 10.1096/fj.05-4493com. [DOI] [PubMed] [Google Scholar]
- 8.Hamzah J, et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature. 2008;453:410–414. doi: 10.1038/nature06868. [DOI] [PubMed] [Google Scholar]
- 9.Shrimali RK, et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. 2010;70:6171–6180. doi: 10.1158/0008-5472.CAN-10-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talmadge JE, Tribble HR, Pennington RW, Phillips H, Wiltrout RH. Immunomodulatory and immunotherapeutic properties of recombinant gamma-interferon and recombinant tumor necrosis factor in mice. Cancer Res. 1987;47:2563–2570. [PubMed] [Google Scholar]
- 11.Rüegg C, et al. Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat Med. 1998;4:408–414. doi: 10.1038/nm0498-408. [DOI] [PubMed] [Google Scholar]
- 12.Qin Z, Blankenstein T. CD4+ T cell—mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. 2000;12:677–686. doi: 10.1016/s1074-7613(00)80218-6. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, et al. Responsiveness of stromal fibroblasts to IFN-gamma blocks tumor growth via angiostasis. J Immunol. 2009;183:6413–6421. doi: 10.4049/jimmunol.0901073. [DOI] [PubMed] [Google Scholar]
- 14.Lejeune FJ, Liénard D, Matter M, Rüegg C. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun. 2006;6:6. [PubMed] [Google Scholar]
- 15.Curnis F, Sacchi A, Corti A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J Clin Invest. 2002;110:475–482. doi: 10.1172/JCI15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung M, Dimtchev A, Velena A, Dritschilo A. Combining radiation therapy with interstitial radiation-inducible TNF-α expression for locoregional cancer treatment. Cancer Gene Ther. 2011;18:189–195. doi: 10.1038/cgt.2010.69. [DOI] [PubMed] [Google Scholar]
- 17.Santoro A, et al. Activity and safety of NGR-hTNF, a selective vascular-targeting agent, in previously treated patients with advanced hepatocellular carcinoma. Br J Cancer. 2010;103:837–844. doi: 10.1038/sj.bjc.6605858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curnis F, et al. Targeted delivery of IFNgamma to tumor vessels uncouples antitumor from counterregulatory mechanisms. Cancer Res. 2005;65:2906–2913. doi: 10.1158/0008-5472.CAN-04-4282. [DOI] [PubMed] [Google Scholar]
- 19.Ebbinghaus C, et al. Engineered vascular-targeting antibody-interferon-gamma fusion protein for cancer therapy. Int J Cancer. 2005;116:304–313. doi: 10.1002/ijc.20952. [DOI] [PubMed] [Google Scholar]
- 20.Joyce JA, et al. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4:393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 21.Hamzah J, et al. Targeted liposomal delivery of TLR9 ligands activates spontaneous antitumor immunity in an autochthonous cancer model. J Immunol. 2009;183:1091–1098. doi: 10.4049/jimmunol.0900736. [DOI] [PubMed] [Google Scholar]
- 22.van Laarhoven HW, et al. Effects of the tumor vasculature targeting agent NGR-TNF on the tumor microenvironment in murine lymphomas. Invest New Drugs. 2006;24:27–36. doi: 10.1007/s10637-005-4540-2. [DOI] [PubMed] [Google Scholar]
- 23.Briscoe DM, Cotran RS, Pober JS. Effects of tumor necrosis factor, lipopolysaccharide, and IL-4 on the expression of vascular cell adhesion molecule-1 in vivo. Correlation with CD3+ T cell infiltration. J Immunol. 1992;149:2954–2960. [PubMed] [Google Scholar]
- 24.Hamzah J, et al. Vascular targeting of anti-CD40 antibodies and IL-2 into autochthonous tumors enhances immunotherapy in mice. J Clin Invest. 2008;118:1691–1699. doi: 10.1172/JCI33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiedler U, et al. Angiopoietin-2 sensitizes endothelial cells to TNF-alpha and has a crucial role in the induction of inflammation. Nat Med. 2006;12:235–239. doi: 10.1038/nm1351. [DOI] [PubMed] [Google Scholar]
- 26.Gasparri AM, et al. Critical role of indoleamine 2,3-dioxygenase in tumor resistance to repeated treatments with targeted IFNgamma. Mol Cancer Ther. 2008;7:3859–3866. doi: 10.1158/1535-7163.MCT-08-0538. [DOI] [PubMed] [Google Scholar]
- 27.Curnis F, et al. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13) Nat Biotechnol. 2000;18:1185–1190. doi: 10.1038/81183. [DOI] [PubMed] [Google Scholar]
- 28.Balza E, et al. Targeted delivery of tumor necrosis factor-alpha to tumor vessels induces a therapeutic T cell-mediated immune response that protects the host against syngeneic tumors of different histologic origin. Clin Cancer Res. 2006;12:2575–2582. doi: 10.1158/1078-0432.CCR-05-2448. [DOI] [PubMed] [Google Scholar]
- 29.Borsi L, et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood. 2003;102:4384–4392. doi: 10.1182/blood-2003-04-1039. [DOI] [PubMed] [Google Scholar]
- 30.Renard N, et al. Early endothelium activation and polymorphonuclear cell invasion precede specific necrosis of human melanoma and sarcoma treated by intravascular high-dose tumour necrosis factor alpha (rTNF alpha) Int J Cancer. 1994;57:656–663. doi: 10.1002/ijc.2910570508. [DOI] [PubMed] [Google Scholar]
- 31.Petrache I, Birukova A, Ramirez SI, Garcia JG, Verin AD. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol. 2003;28:574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 32.Kristensen CA, Nozue M, Boucher Y, Jain RK. Reduction of interstitial fluid pressure after TNF-alpha treatment of three human melanoma xenografts. Br J Cancer. 1996;74:533–536. doi: 10.1038/bjc.1996.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seynhaeve AL, et al. Tumor necrosis factor alpha mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res. 2007;67:9455–9462. doi: 10.1158/0008-5472.CAN-07-1599. [DOI] [PubMed] [Google Scholar]
- 34.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 35.Stockmann C, et al. Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature. 2008;456:814–818. doi: 10.1038/nature07445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rolny C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Daly C, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci USA. 2006;103:15491–15496. doi: 10.1073/pnas.0607538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.