
Figure 1.
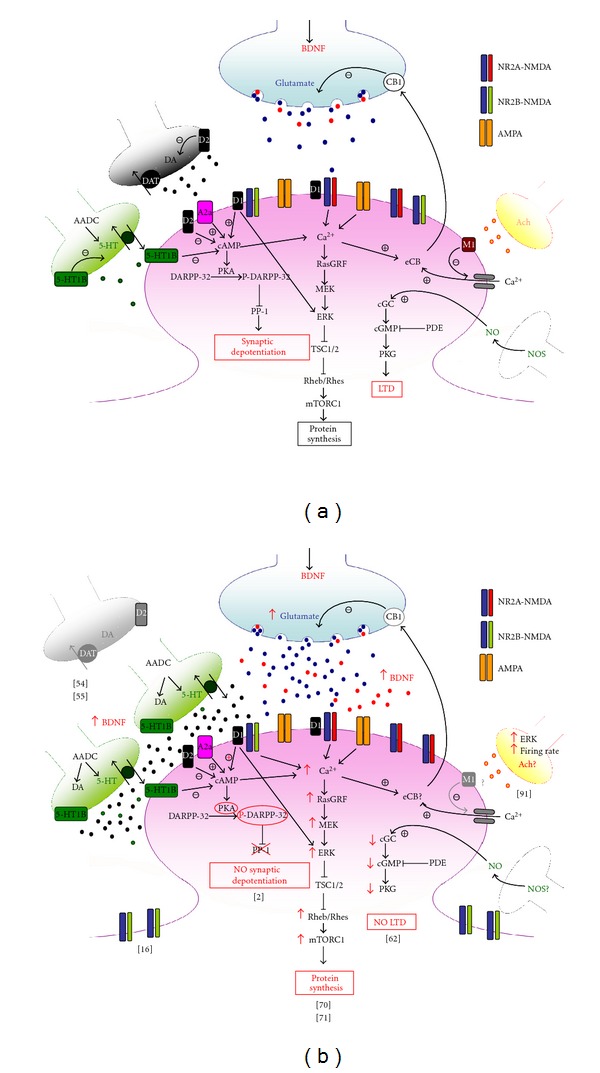
(a) In control condition, dopamine (DA) transmission is regulated by feedback control of release from nigrostriatal terminals (black) through D2 autoreceptors and uptake processes. DA binds striatal postsynaptic D1 receptors inducing the formation of cAMP, which in turn favours the activation of PKA, able to phosphorylate and activate DA- and cAMP-regulated phosphoprotein of 32 KDa (DARPP-32) and extracellular signal-regulated kinase (ERK). Once phosphorylated, DARPP-32 is able to inhibit protein phosphatase 1 (PP-1). Glutamate (blue) and BDNF (red) are released from corticostriatal terminals into the striatum. Glutamate release is regulated by endocannabinoids (eCB) activated by increases in intracellular calcium (Ca2+) concentrations through adenosine A2a and muscarinic M1 receptors activation, among other mechanisms, and retrogradely released by postsynaptic striatal neurons. Once released, glutamate activates metabotropic as well as NMDA and AMPA ionotropic receptors, whose activity and surface expression at postsynaptic membrane is also regulated by D1 receptors. In serotoninergic afferents, 5-hydroxytryptophan is converted to serotonin (5-HT) (green) by Aromatic-L-Amino Acid Decarboxylase (AADC) and released into the striatum. Cholinergic and nitric oxide synthase (NOS)-positive interneurons cooperate to induction of corticostriatal LTP and LTD. (b) In dyskinetic state L-DOPA is converted to DA by AADC and released from serotoninergic terminals in unregulated manner. Higher levels of striatal BDNF may support morphological changes in serotoninergic neurons. Excess of DA abnormally stimulates D1 pathway with hyperphosphorylation of ERK and uncontrolled activation of PKA that results in hyperphosphorylation of DARPP32, which persistently blocks PP-1 causing loss of synaptic depotentiation. Abnormal D1 receptor stimulation is associated to increased intracellular Ca2+ levels and dysregulation of NMDAR subunit composition with reduction of NR2B-containing NMDAR at synaptic sites, leading to increase in NR2A/NR2B ratio that has been suggested to have a role in the loss of depotentiation. Hyperactivation of ERK through convergent altered signalling pathways brings to increased inhibition of tuberous sclerosis complex (TSC)1/2, and consequent disinhibition of Rheb/Rhes, leading to excessive increase of signalling of mTORC1 that, in turn, exerts its long term effects through changes in protein synthesis. After chronic L-DOPA, cholinergic interneurons show increased phospho-ERK immunoreactivity and higher firing rates with increased release of acetylcholine (Ach). Striatal cGMP signalling is decreased and corticostriatal LTD, which strictly relies on the nitric-oxide- (NO-) dependent activation of protein kinase G (PKG) is abolished in dyskinetic state.