

Abstract
αkap, a muscle specific anchoring protein encoded within the Camk2a gene, is thought to play a role in targeting multiple calcium/calmodulin kinase II isoforms to specific subcellular locations. Here we demonstrate a novel function of αkap in stabilizing nicotinic acetylcholine receptors (AChRs). Knockdown of αkap expression with shRNA significantly enhanced the degradation of AChR α-subunits (AChRα), leading to fewer and smaller AChR clusters on the surface of differentiated C2C12 myotubes. Mutagenesis and biochemical studies in HEK293T cells revealed that αkap promoted AChRα stability by a ubiquitin-dependent mechanism. In the absence of αkap, AChRα was heavily ubiquitinated, and the number of AChRα was increased by proteasome inhibitors. However, in the presence of αkap, AChRα was less ubiquitinated and proteasome inhibitors had almost no effect on AChRα accumulation. The major sites of AChRα ubiquitination reside within the large intracellular loop and mutations of critical lysine residues in this loop to arginine increased AChRα stability in the absence of αkap. These results provide an unexpected mechanism by which αkap controls receptor trafficking onto the surface of muscle cells and thus the maintenance of postsynaptic receptor density and synaptic function.
Introduction
The clustering of nicotinic acetylcholine receptors (AChRs) at the postsynaptic membrane is a hallmark of the neuromuscular junction (NMJ) (Sanes and Lichtman, 2001). The initial steps involved in the formation of receptor clusters have been studied extensively, and several key components of this multistep process have been identified. Based on mouse genetic studies, MuSK (muscle-specific tyrosine kinase), LRP4 (low-density lipoprotein receptor-related protein 4), rapsyn, and Dok7 (dedicator of cytokinesis family member 7) all have been shown to be required for the initial formation of receptor clusters (Gautam et al., 1995; DeChiara et al., 1996; Sanes and Lichtman, 2001; Okada et al., 2006; Weatherbee et al., 2006; Kim et al., 2008; Zhang et al., 2008; Wu et al., 2010). Components of the dystrophin glycoprotein complex have also been shown to be involved in clustering and in the maintenance of postsynaptic receptor density. For example, mice deficient in α-dystrobrevin or α-syntrophin exhibit dramatic reductions in AChR number and density and an abnormal pattern of receptor clustering (Adams et al., 2000; Grady et al., 2000; Martinez-Pena y Valenzuela et al., 2011).
The maintenance of a high density of AChR in the postsynaptic membrane may also depend on the mechanisms that regulate their progression through the secretory pathway from the endoplasmic reticulum (ER) to the cell surface. However, the molecular machinery that controls the processing of AChR within the secretory pathway remains poorly defined. In an effort to understand the machinery involved in AChR trafficking, we used a multidisciplinary approach that led us to identify a novel function for αkap, a protein implicated previously in localizing multiple isoforms of calcium/calmodulin-dependent protein kinase II (CaMKII). αkap is a muscle-specific protein encoded within the Camk2a gene that contains a putative transmembrane domain and an association domain but lacks the catalytic domain that is responsible for the kinase activity of CaMKII subunit α (Bayer et al., 1996, 1998, 2002). In skeletal and cardiac muscles, it is thought that αkap is involved in targeting CaMKII isoforms to the sarcoplasmic reticulum in which they regulate calcium homeostasis (Campbell and MacLennan, 1982; Chu et al., 1990; Wang and Best, 1992; Xu et al., 1993; Hain et al., 1995; Pawson and Scott, 1997; Bayer et al., 1998; Nori et al., 2003). In mammalian muscle cells, at least three isoforms of CaMKII (βm, γ, and δ) have been found to be expressed (Damiani et al., 1995; Bayer et al., 1998) and concentrated at the NMJs (Martinez-Pena y Valenzuela et al., 2010). In the present study, we investigated the role of αkap in muscle cells by knocking down its expression with short-hairpin RNA (shRNA) and discovered that αkap has a novel function, regulating the degradation of AChR protein.
Materials and Methods
Plasmid constructs.
Plasmids containing mouse AChR subunit cDNAs driven by the CMV promoter were a generous gift from Dr. William Green (University of Chicago, Chicago, IL), and Lrp4 cDNA was a generous gift from Dr. Stephan Kröger (University of Munich, Munich, Germany). Throughout the paper, the respective subunits are referred to as AChRα, AChRβ, AChRδ, and AChRε. The HA–αkap construct was a generous gift from Dr. Ulrich Bayer (University of Colorado, Denver, CO). GFP–αkap was generated using the primers 5′-GGAATTCTATGCTG CTCTTTCTCACGCTGTGG and 5′-GGGTACCATGCGGCAGGACGGAGGGCGCCCCAGATCTGT and cloned into the pEGFP–C3 vector, in which alanine was mutated to lysine at position 207 (A207K) to prevent the dimerization of GFP (monomeric-GFP–C3).
A plasmid encoding mouse protein disulfide isomerase associated protein 3 (pdiA3) cDNA was purchased from Origen (MC200134) and subcloned into the mCherry–C1 vector (Clontech) using the following primers: 5′-GGGAATTCTATGCGCTTCAGCTGCCTAGC and 5′-GGGGTACCTTAGAGGTCCTCTTGTGCC.
Mutations of lysine residues (K359, K347, K388, and K393) to arginine within the intracellular loop of AChRα were generated using the QuickChange XL Site-Directed Mutagenesis Kit (Stratagene). The following primers were used for mutagenesis: 359 forward, 5′-CTATGTCTTCTGTAAAAATCCTTCTCTCTTGTTTATCTCTGGATGGTC; 359 reverse, 5′-GACCATCCAGAGATAAACAAGAGAGAAGGATTTTTACAGAAGACATAG; 374 forward, 5′-GATATATCTGACATCTCTGGGAGACCGGGTCCTCCACCTATGGGC; 374 reverse, 5′-GCCCATAGGTGGAGGACCCGGTCTCCCAGAGATGTCAGATATATC; 388 forward, 5′-GGCTTTCACTCTCCGCTGATCAGACACCCTGAGGTGAAAAGCGCC; 388 reverse, 5′-GGCGCTTTTCACCTCAGGGTGTCTGATCAGCGGAGAGTGAAAGCC; 393 forward, 5′-CACCCTGAGGTGAGGAGCGCCATCGAGGGG; and 393 reverse, 5′-CCCCTCGATGGCGCTCCTCACCTCAGGGTG.
Two shRNA constructs against murine αkap were generated by cloning the annealed target sequences (nucleotide starting position at either 137 or 337) into BglII/HindIII sites of the pSUPER vector or pSUPER.Neo.GFP according to the recommendations of the manufacturer. The target sequence of the shRNA to αkap at position 137 is 5′-GTTCAGTTAATGGAATCTTC and at position 337 is 5′-GGCCTGGACTTTCATCGATTC. A scrambled shRNA (as negative control) was also generated: 5′-GGTTATCTATTAGCGATTCTA. All shRNA constructs were verified by DNA sequencing.
Cell culture and transfection.
The C2C12 muscle cell line and the HEK293T human embryonic kidney cell line were purchased from the American Type Culture Collection. HEK293T cells were grown at 37°C in 5% CO2 and maintained in DMEM containing 10% (v/v) fetal bovine serum (FBS) (Invitrogen) and 1% penicillin/streptomycin. C2C12 myoblasts were maintained in DMEM supplemented with 20% FBS and switched to DMEM with 5% horse serum (Invitrogen) when cell density reached confluency of ∼90%. For AChR staining, C2C12 cells were grown on dishes coated with laminin (to promote the formation of large receptor clusters).
All transfections were performed using Lipofectamine 2000 (Invitrogen) as per the recommendation of the manufacturer with minor modifications. C2C12 transfection was performed in serum-free medium for 8 h before switching cells to the differentiation medium. HEK293T cells were either harvested for immunoblotting or fixed for confocal imaging 40–48 h after transfection.
Pharmacological agents.
The proteasome inhibitor MG132 was purchased from Enzo Life Sciences and was used at 5 μm. Leupeptin, lactacystin, and chloroquine were purchased from Sigma-Aldrich and used at 20, 10, and 25 μg/ml, respectively.
Acetylcholine chloride (Sigma) was made as a 1 m stock solution in distilled water and kept frozen. On the day of recording, it was diluted to the concentration of choice in extracellular recording solution.
RT-PCR.
Total RNA was isolated from cultured myotubes using the RNAeasy Mini Kit (Qiagen). cDNA was synthesized using Superscript First Strand System and oligo-dT primers (Invitrogen). PCR was performed using the following primers: AChRα, 5′-ACGGCGACTTTGCCATTGTC and 5′-CAGGCGCTGCATGACGAAGT; GAPDH, 5′-TGAAGGTCGGTGTGAACGGATTTGGC and 5′-CATGTAGGCCATGAGGTCCACCAC.
RT-PCR products were analyzed from samples collected in the exponential phase of amplification (35 cycles for AChR and 30 cycles for GAPDH).
Immunofluorescence and confocal microscopy.
To assess AChR clustering, C2C12 myoblasts grown on laminin-coated dishes were transfected with 4 μg of pSuper.GFP expressing scrambled shRNA or shRNA(αkap). Eight hours after transfection, myoblasts were transferred into differentiation medium. Four to five days later, myotubes were bathed with α-bungarotoxin (α-BTX) conjugated to Alexa Fluor-594 (BTX–Alexa594) (Invitrogen) at 4°C for 1 h, washed in PBS, and then fixed in 2% paraformaldehyde (PFA). Cells were washed with PBS, mounted in DAPI Prolong Gold anti-fade medium (Invitrogen), and imaged with a Leica SP5 confocal microscope. The z-stacks were then collapsed, and the contrast was adjusted with Photoshop (Adobe Systems).
To compare the proportion of the myotube surface covered by AChR clusters under control conditions and when αkap was knocked down, we identified fields in dishes transfected with pSuper.GFP–shRNA(αkap) in which there were both green fluorescent (transfected) and untransfected control myotubes and then used functions in NIH ImageJ (version 1.45i) to quantify the extent of clustering. We first applied a binary threshold to the entire red image (BTX–Alexa594), so that all regions with intensity below the threshold (background autofluorescence and fluorescence attributable to nonclustered receptors) were assigned to white, and the higher-intensity regions of clustered receptors were assigned to black. We then outlined at least one fluorescent and one nonfluorescent myotube on the green image (EGFP) and overlaid these regions of interest onto the thresholded images of AChR. We then applied the histogram function to each region of interest to get the number of pixels that were black or white. Finally, we calculated the proportion of each myotube covered by clustered receptors as (number of black pixels)/(total number of pixels in the region of interest).
To assess the localization of AChRα within HEK293T cells, cells were cotransfected with plasmids expressing GFP–αkap, AChRα, and pdiA3–mCherry. Forty to forty-eight hours later, cells were washed with PBS, fixed in 4% PFA for 10 min at room temperature, bathed in a blocking solution (10% BSA) for 15 min, and permeabilized with a solution containing 0.2% Triton X-100 and 2% BSA in PBS for 15 min. Cells were incubated overnight with primary monoclonal anti-AChRα antibody MAB35 (The Developmental Studies Hybridoma Bank) diluted at 1:1000. Alexa Fluor-647-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch) was added to cells for 1 h. Coverslips were mounted onto glass slides using DAPI-Prolong Gold antifade medium (Invitrogen) and imaged at the appropriate wavelengths with a Leica SP5 confocal microscope.
Immunoprecipitation, pull down, and Western blotting.
Transfected and untransfected C2C12 myotubes or HEK293T cells were lysed in buffer containing 50 mm HEPES, 300 mm NaCl, 2 mm EDTA, 0.5% NP-40, 0.8% CHAPS, 1 mm PMSF, 2 mm activated sodium orthovanadate, 5 mm NaF, and protease inhibitors. Lysates were incubated for 1 h at 4°C and cleared by centrifugation. Each lysate was assayed for anti-tubulin reactivity to assess the amount of protein present, and then aliquots of each lysate were typically used for several different procedures.
To selectively pull down surface AChR, living myotubes (untransfected or transfected with or without αkap shRNA) were incubated with BTX–biotin for 1 h at 4°C. Cells were then lysed, and lysates were incubated with NeutrAvidin beads (Thermo Fisher Scientific) overnight. To selectively pull down intracellular AChR, living myotubes were bathed with unlabeled BTX to saturate all surface receptors for 1 h at 4°C. Cells were then lysed, and lysates were incubated with BTX–biotin, followed by NeutrAvidin beads overnight. To pull down total AChR, cells were lysed with no pretreatment, and then lysates were incubated with BTX–biotin, followed by NeutrAvidin beads overnight. For all three conditions, the beads were collected by centrifugation and washed in PBS four times. The bound proteins were released by boiling in reducing 2× lithium dodecyl sulfate (LDS) buffer (Invitrogen).
To pull down CaMKIIs, muscle cell lysates were incubated overnight with calmodulin beads (Sigma) in the presence of 2 mm CaCl2. The beads were collected and washed four times with buffer containing 0.5% NP-40. Because αkap is known to bind to multiple CaMKII isoforms, this procedure was expected to pull down αkap, although it lacks a calmodulin binding domain.
The ProFound Mammalian HA-tag IP/Co-IP Kit (Thermo Fisher Scientific) was used to examine αkap and AChRα interaction in HEK293T cells following the instructions of the manufacturer in the presence of 300 mm NaCl in the M-PER Mammalian Protein extraction reagent. The immunoprecipitate was then probed with anti-HA (Covance) or anti-αkap (Santa Cruz Biotechnology) and anti-AChRα (MAB210; Covance) antibodies.
To assay AChR ubiquitination, HEK293T cells were cotransfected with αkap, HA-tagged ubiquitin (HA–Ub), and either wild-type or mutated AChRα constructs (see above). The cells were harvested and lysed in denaturing buffer [50 mm Tris HCl, 150 mm NaCl, 2 mm EDTA, 5 mm N-ethylmaleimide (Sigma), 2 mm activated sodium orthovanadate, 2% SDS, and protease inhibitors], boiled for 10 min, and briefly sonicated. The lysates were diluted to 0.1% SDS using buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1 mm PMSF, 2 mm activated sodium orthovanadate, 5 mm NaF, and protease inhibitors). Precleared protein lysates (20 μg) were incubated with 2 μg of anti-AChRα (MAB153; Santa Cruz Biotechnology) for 1 h at 4°C, followed by additional incubation with 20 μl of Protein A/G Plus-agarose beads (Santa Cruz Biotechnology) overnight at 4°C. Beads were collected by centrifugation, extensively washed with lysis buffer without SDS, resuspended in LDS reducing sample buffer, and boiled for 5 min. Proteins were separated by SDS-PAGE and transferred to PVDF membranes, which were then incubated in 10 mm Tris, pH 7.4, 1 mm PMSF, 10 mm β-mercaptoethanol, and 6 m guanidine for 1 h at 4°C, followed by extensive washing in PBS. Membranes were then incubated with primary antibodies diluted in blocking buffer (2% fat-free milk in PBS) overnight at 4°C. After extensive washing, the membranes were then incubated with secondary HRP-conjugated antibodies and developed using SuperSignal West Femto Maximum Sensitivity Substrate or West Pico (Thermo Fisher Scientific). Mouse anti-ubiquitin (Santa Cruz Biotechnology) and mouse anti-HA (clone A1; Covance) antibodies were diluted at 1:1000, mouse anti-αkap (clone A1; Santa Cruz Biotechnology) was diluted at 1:200, rabbit anti-CaMKII (M176; Santa Cruz Biotechnology) was diluted at 1:500, rabbit anti-GFP and rat anti-AChRα MAB210 (Covance) were diluted at 1:5000, mouse anti-tubulin (Developmental Studies Hybridoma Bank) was diluted 1:10,000, rabbit anti-Lrp4 was diluted 1:10,000, anti-LAMP1 was diluted 1:1000, and anti-P2X2 was diluted 1:5000. All horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch) were diluted at 1:10,000.
The intensity of bands on Western blots was quantified by scanning the film at 8 bits with white assigned a value of 0 and black a value of 255. We then conducted the analysis using NIH ImageJ (version 1.45). We first verified that there was no saturation in the darkest regions of the blot and then selected a region of the film with no signal, which was used to calculate a background level that was subtracted from all pixel values so that the subtracted baseline was assigned a value of 0. We then used the “magic wand” function to select the pixels of each band and the measure function to determine the integrated intensity of the selected region. This value is linearly proportional to the amount of reaction product imaged on the film but not necessarily to the amount of protein present. For all data presented as histograms, results from several blots were averaged, with the number indicated in the figure legend. The error bars represent the SD.
Electrophysiology.
HEK293T cells were cotransfected with either all four wild-type AChR subunits (α, β, δ, and ε) or the 4R mutant of α and wild-type β, δ, and ε. Each transfection mix also included a plasmid encoding EGFP.
Whole-cell recordings from green fluorescent cells were made ∼48 h after transfection, using a Dagan 3900 patch clamp and Clampex 9.0 software (Molecular Devices). All recordings were made at room temperature with the holding potential set to −50 mV. The perfusion solution was switched from extracellular solution alone to extracellular solution plus acetylcholine with a BPS8 solution switcher (ALA). The cell was perfused by solutions applied from a narrow-bore pipette placed ∼1 mm from the cell. The 10–90% exchange time of this system was ∼25 ms.
The extracellular recording solution contained the following (in mm): 135 NaCl, 1.5 KCl, 1 CaCl2, 1 MgCl2, and 15 HEPES, pH 7.5. The intracellular solution contained the following (in mm): 140 KCl, 1 MgCl2, 10 EGTA, and 10 HEPES, pH 7.5.
Results
Acetylcholine receptors form complexes with αkap
Previous studies have shown that αkap is able to directly interact with many CaMKII isoforms (Bayer et al., 1996, 2002), and because CaMKII has been shown to control the density of AChR at the mature NMJ (Martinez-Pena y Valenzuela et al., 2010), we asked whether αkap could be involved in this process. To test this idea, lysates from cultured C2C12 myotubes were incubated with BTX–biotin to label AChR, and then AChR–BTX–biotin complexes were isolated with NeutrAvidin-coated beads and subjected to Western blot analysis with anti-AChRα and anti-αkap antibodies. αkap was pulled down with AChRα (Fig. 1A), indicating that AChRs are present in the same complex as αkap. Similarly, when calmodulin-coated beads were used, we also pulled down AChRα with αkap (Fig. 1B). It should be noted that neither AChR nor αkap have calmodulin binding sites, indicating that the pull down by calmodulin beads is indirect, presumably because of the known ability of αkap to bind to multiple calmodulin-binding CaMKII isoforms. Thus, proteins besides AChRα and αkap are present in the material pulled down from cells that endogenously express both proteins. The interaction of αkap and AChRα was also observed when HEK293T cells (which lack endogenous αkap and AChR subunits) were cotransfected with AChRα and HA-tagged αkap constructs (Fig. 1D). AChRα was not pulled down by anti-HA-coupled agarose beads when it was expressed alone, but both αkap and AChRα were present in the lysate that was pulled down by anti-HA when cells were cotransfected. This result suggests that either αkap directly interacts with AChRα or other components of the complex that link these two proteins are expressed in both muscle and HEK293T cells.
Figure 1.

αkap forms complexes with AChR in skeletal muscle cells. A, Lysates from cultured C2C12 myotubes were incubated with (+ BTX) or without (− BTX) BTX–biotin, and then biotin containing complexes were isolated with NeutrAvidin beads. Pull-down proteins (PD) were probed with anti-αkap or anti-AChRα antibody. B, Myotube lysates were incubated with calmodulin beads in the absence of calcium (− Ca) or in the presence 2 mm calcium (+ Ca) to pull down calcium/calmodulin binding proteins. Blots were probed with anti-αkap or anti-AChRα antibody. C, Lysates from human embryonic kidney (HEK293T) cells transfected with AChRα and αkap–HA (+ αkap) or with AChRα and empty plasmid (− αkap) were immunoprecipitated with anti-HA-coupled agarose beads, and blots were probed with anti-αkap or anti-AChRα antibodies. D, Aliquots from the lysates used for the pull downs in C probed for tubulin. For all four panels, at least three independent experiments were performed.
Knockdown of αkap decreased AChR protein levels
Having demonstrated that αkap is present in a complex with AChRα, we next tested whether changing the expression of αkap had an effect on the clustering of AChR on the muscle cell surface. To do this, we used αkap-specific shRNA to knock down the expression of αkap in C2C12 muscle cells. We designed two different shRNAs (shRNA137 and shRNA337), and as an initial test of the efficiency of the knock down, we transfected C2C12 myoblasts with GFP–αkap alone or cotransfected with GFP–αkap and either shRNA137 or shRNA337. When myotubes were viewed 4–5 d later, there were many green fluorescent fibers in the dishes transfected with GFP–αkap alone but very few fluorescent fibers in the dishes cotransfected with GFP–αkap and shRNAs (data not shown), indicating that both shRNAs were effective in knocking down the expression of exogenous αkap–GFP. Western blot analysis of equal amount of proteins with anti-αkap antibody showed that transfection with either shRNA drastically reduced the endogenous expression of αkap protein compared with untransfected myotubes or myotubes transfected with a scrambled shRNA (Fig. 2A). In contrast, neither of the shRNAs had an effect on the expression level of three CaMKII isoforms (βm, γ, and δ) that have several domains similar to domains of αkap (Fig. 2A). We performed all remaining experiments using shRNA337, which will be referred to as shRNA(αkap).
Figure 2.
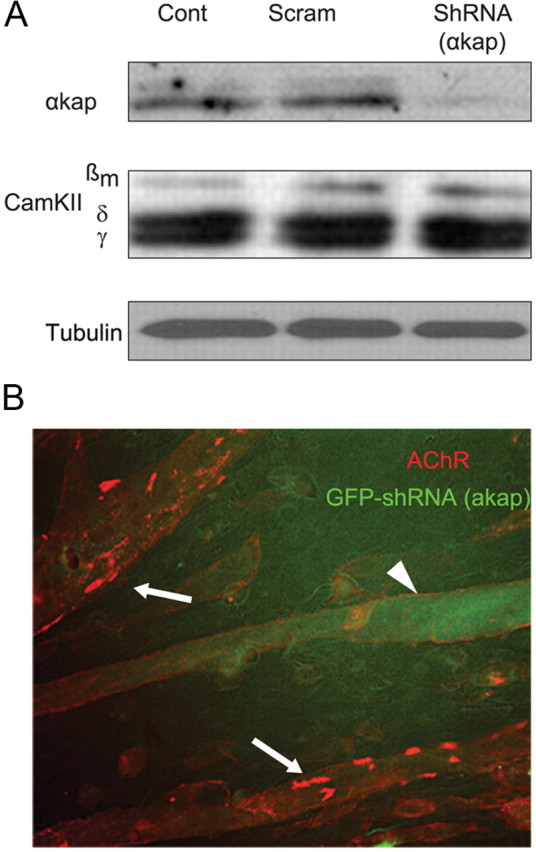
Knockdown of αkap significantly reduced the formation of AChR clusters on the surface of muscle cells. C2C12 myoblasts were transfected with either shRNA(αkap) or scrambled shRNA, and then 4–5 d after their differentiation, myotubes were either harvested for protein extraction and immunoblotting or bathed with BTX–Alexa594 to label surface AChR. A, Western blots of equal amount of proteins from untransfected myotubes (Cont), myotubes transfected with shRNA(αkap) (a plasmid expressing shRNA specific to αkap), or a plasmid containing a scrambled version of the shRNA sequence (Scram). Blots were probed with anti-αkap or anti-CaMKII (M176) antibody. M176 recognizes multiple CaMKII isoforms, including three that are expressed in skeletal muscle (βm, δ, and γ). Tubulin was used as loading control. B, Untransfected myotubes (arrows) and a myotube transfected with GFP–shRNA(αkap) viewed in the same dish to allow comparison of the number of AChR clusters when αkap is knocked down. Very few clustered receptors were present on transfected (green fluorescent, arrowhead) myotubes.
Next we examined the effect of the knockdown of αkap on AChR clusters on the surface of muscle cells. C2C12 myoblasts were cotransfected with plasmids encoding EGFP and either shRNA(αkap) or its scrambled version and grown on laminin-coated dishes. The AChR clusters (identified by their labeling with fluorescent BTX) were fewer and smaller on myotubes transfected with shRNA (identified by their labeling with EGFP) compared with either untransfected myotubes in the same dish (Fig. 2B) or to myotubes in separate dishes transfected with the scrambled shRNA alone (data not shown). The average percentage of the surface covered by clustered receptors in control myotubes was 6.8 ± 3.3%, but this fell to 0.7 ± 0.2% in shRNA(αkap) knockdown myotubes (±SEM, n = 10 fibers each).
The decrease in the number of clusters of AChR on the surface of myotubes transfected with shRNA could have been produced by several mechanisms. The morphology of cultured myotubes cotransfected with EGFP and shRNA(αkap) plasmids was indistinguishable from untransfected myotubes, indicating that αkap knockdown had no effect on the differentiation of myoblasts into myotubes. Because AChR forms a complex with several other postsynaptic proteins, another possibility was that depletion of αkap might lead to a decrease in other components of the complex that are involved in receptor clustering. However, Western blot results showed that shRNA(αkap) produced a dramatic decrease in AChRα but no change in the level of the AChR clustering protein rapsyn (Fig. 3A). We next tested whether the decrease in the amount of AChRα was attributable to a decrease in AChR mRNAs. However, when RT-PCR products specific to AChRα were analyzed (Fig. 3B), we found that there was no difference in the amount of AChRα mRNAs between untransfected and αkap-depleted myotubes. If αkap is involved specifically in clustering AChR on the surface of myotubes, the amount of AChR on the plasma membrane should be the same in myotubes transfected with shRNA(αkap) and scrambled shRNA and in untransfected myotubes. However, in myotubes transfected with shRNA(αkap), the amount of surface AChRα was dramatically reduced compared with scrambled or untreated myotubes (Fig. 3C,D). We also found that fewer receptors were detected in the internal pool of myotubes transfected with shRNA(αkap) compared with myotubes transfected with scrambled shRNA or to control myotubes (Fig. 3E,F). Thus, both the intracellular and the surface pools of AChRα were depleted by similar amounts.
Figure 3.

Knockdown of αkap reduced AChR protein levels. A, Equal amounts of proteins from lysates of myotubes transfected with shRNA(αkap) or scrambled shRNA (Scram) or untransfected myotubes (Cont) were probed with four antibodies to assess the effect of the shRNA on αkap, AChRα, rapsyn, and tubulin. B, RT-PCR products of AChRα from cultured myotubes transfected with shRNA(αkap), scrambled shRNA, and untransfected myotubes. The PCR reaction conditions did not allow saturation to be reached for any of the samples, so band intensity is proportional to the amount of mRNA in the original sample. C, Western blots of surface AChRα. Living myotubes were treated with BTX–biotin at 4°C to saturate all surface receptors before cell lysis. Biotin-labeled surface AChRs were precipitated with NeutrAvidin beads. AChRα subunits were detected with MAB210. D, Quantification of three blots as in C. E, Western blots of intracellular AChRα. Surface receptors were saturated with unlabeled BTX, and then muscle lysates were saturated with BTX–biotin to selectively label intracellular AChR. F, Quantification of three blots as in E.
αkap controls AChR degradation
Having shown that knockdown of αkap has no effect on mRNA levels for AChRα but drastically reduced AChRα protein levels, we hypothesized that αkap might control the stability of AChR. As an initial test, muscle cells were treated with MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal), a widely used proteasome inhibitor that can prevent degradation of many ER-associated degradation (ERAD) substrates (Rock et al., 1994) or with leupeptin, a lysosome inhibitor. We found that, in untransfected myotubes or myotubes transfected with the scrambled shRNA, both MG132 and leupeptin increased the amount of AChRα protein to ∼160% of the untreated control (Fig. 4), indicating that both inhibitors were able to prevent AChRα degradation. However, the two inhibitors had quite different effects on C2C12 myotubes that had been transfected with shRNA(αkap). The effect of MG132 on cells transfected with shRNA(αkap) was quite similar to the effect of MG132 on untransfected cells (Fig. 4A,B), whereas the effect of leupeptin plus shRNA(αkap) was quite similar to shRNA(αkap) alone (Fig. 4C,D). Thus, in the presence of shRNA(αkap), MG132 produced an average increase of 578 ± 16% in AChRα, whereas leupeptin produced no change (95 ± 8%). A second pair of inhibitors showed similar trends. In the presence of shRNA(αkap), the proteasome inhibitor lactacystin produced a significant increase in AChRα (average of 333 ± 62%, p < 0.05, n = 4), but the lysosomal inhibitor chloroquine produced no significant increase in AChRα (average of 161 ± 47%, p > 0.7). These results suggest that αkap protects AChR from degradation by the proteasome but not from degradation within lysosomes.
Figure 4.

A proteasome inhibitor prevented AChR degradation in cells transfected with shRNA(αkap). A, Myotubes transfected with shRNA(αkap), scrambled shRNA (Scram), or untransfected myotubes (Cont) were treated with the proteasome inhibitor MG132 (at 5 μm), and lysates were immunoblotted with MAB210 antibody against AChRα. Tubulin was used as a loading control. B, Quantification of three independent experiments as in A. 100% was the average intensity of the samples from untransfected myotubes in the absence of MG132. C, Myotubes transfected with plasmids encoding shRNA(αkap) or the scrambled shRNA were treated with leupeptin (20 μg/ml). D, Quantification of three independent experiments as in C. 100% was the average intensity of the samples from myotubes transfected with the scrambled shRNA in the absence of leupeptin.
To better understand the mechanism by which αkap controls the amount of AChR in myotubes, we used heterologous expression in HEK293T cells, which lack endogenous αkap and AChRα. First we tested whether αkap promotes the stability of AChRα in HEK293T cells, as it does in cultured myotubes. Lysates from cells transfected with EGFP and AChRα with or without αkap were immunoblotted with anti-AChRα antibody, and AChRα levels were compared. The amount of AChRα was nearly threefold higher in cells cotransfected with αkap compared with cells transfected with only AChRα, indicating that αkap promotes the stability of AChR in HEK293T cells (Fig. 5A,C). Treatment of HEK293T cells expressing AChRα and a control plasmid with MG132 also increased AChRα levels by approximately threefold compared with untreated control cells (Fig. 5A–C). However, the combined effect of αkap and MG132 on AChRα accumulation was no larger than either treatment alone, indicating that αkap is sufficient to maximally protect AChRα from degradation by the proteasome (Fig. 5C).
Figure 5.
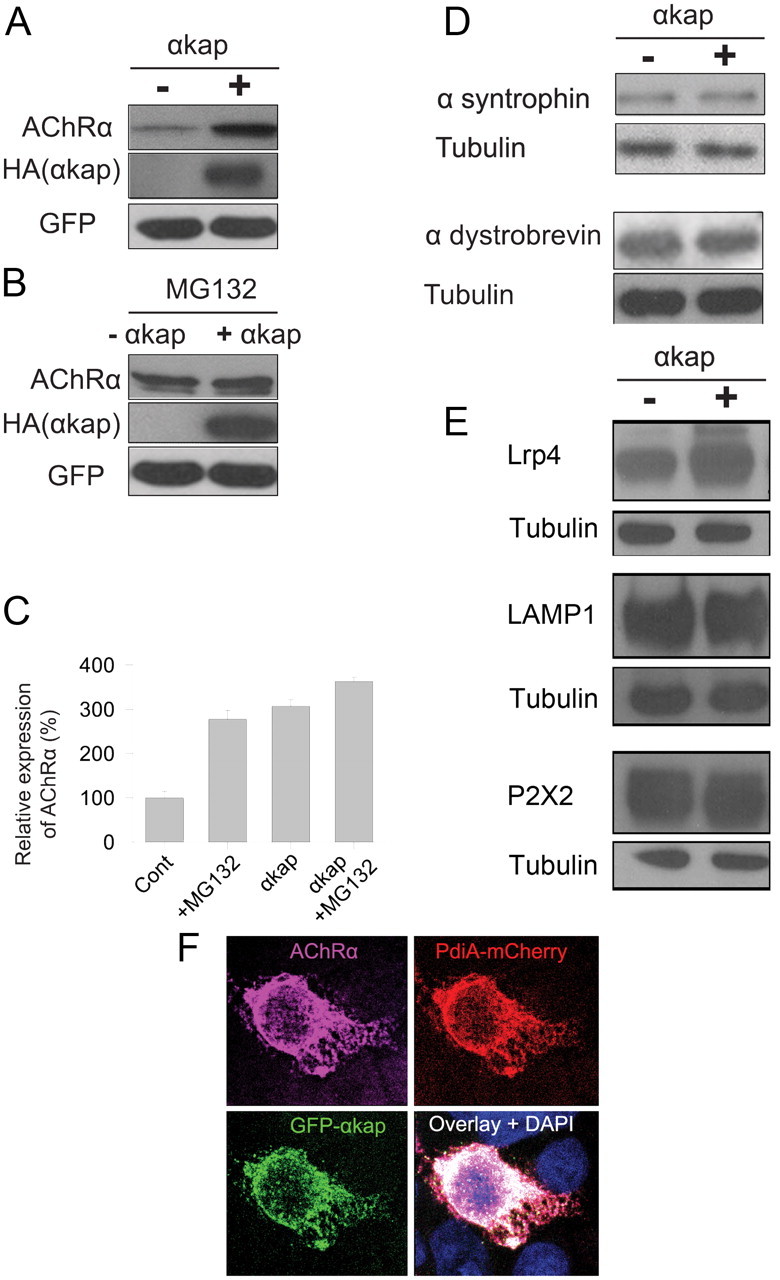
αkap increased AChRα accumulation in transfected HEK cells. A, Western blots of homogenates from HEK293T cells cotransfected with AChRα, HA-αkap and pEGFP-C3(+) constructs or with AChRα, pEGFP-C3 constructs, and an empty version of the same vector into which αkap–HA was inserted (−). GFP was used as a control for the transfection efficiency and for the loading of total proteins. Anti-HA antibody was used to confirm the expression of αkap. B, Experiment identical to A, except that MG132 was present. C, Quantification of two independent replicates of experiments as in A and B. D, Western blots of lysates from HEK293T cells transfected with α-syntrophin or α-dystrobrevin with or without αkap. E, Lysates from HEK293T cells transfected with Lrp4 or P2X2 with or without αkap were probed with antibodies against Lrp4, P2X2, and LAMP1. F, Confocal images of an HEK293T cell coexpressing GFP–αkap, AChRα, and pdiA–mCherry (a marker of the ER). The nucleus was labeled with DAPI. Cells were fixed, permeabilized, and stained with anti-AChR antibody (MAB35), followed by anti-rat Alexa Fluor-647-labeled secondary antibody to visualize AChR. pdiA and αkap were visualized by the fused fluorescent proteins.
Next, we tested whether αkap is involved in controlling the stability of other AChR-associated components expressed in HEK293T cells. In contrast to AChRα, the amount of α-syntrophin or α-dystrobrevin (two components of the postsynaptic dystrophin glycoprotein complex that tethers AChR) was similar in the presence or absence of αkap (Fig. 5D). However, like AChRα, the amount of Lrp4, a transmembrane protein that transmits a signal required for clustering of AChRs, was significantly higher in cells cotransfected with αkap (190 ± 10% of the level in cells transfected with Lrp4 only, n = 4; Fig. 5E). To explore the possibility that αkap might be acting as a general regulator of the stability of transmembrane proteins, we tested two additional transmembrane proteins expressed in HEK293T cells. Neither rat P2X2 (a plasma membrane protein) nor LAMP1 (a lysosomal membrane protein) was sensitive to the presence of αkap (Fig. 5E). Thus, αkap is not a general regulator of transmembrane proteins.
Previous studies have shown that, in several cell types, αkap is primarily localized to the ER compartment (Nori et al., 2003; Singh et al., 2009) and that, when AChRα is expressed without other AChR subunits, it remains mostly trapped in the ER (Christianson and Green, 2004). We therefore examined whether AChRα colocalizes with αkap in the ER of HEK293T cells. Confocal microscopy analysis of cells cotransfected with monomeric GFP–αkap, AChRα, and the ER marker mCherry–pdiA3 showed that they all reside in the ER under these conditions (Fig. 5E).
Because MG132 does not promote any additional AChR accumulation when αkap is present, we hypothesized that αkap might prevent ubiquitination of AChRα and thus degradation. As a first test of this hypothesis, we cotransfected HEK293T cells with AChRα and HA–Ub (Fang and Kerppola, 2004) in the presence or absence of αkap and estimated the amount of AChRα in each sample by Western blots (Fig. 6A). We then used this information to equalize the amount of AChRα (Fig. 6B), which was then immunoprecipitated with AChRα antibody and probed on an immunoblot with anti-ubiquitin antibody. The blots showed a smear, representing the addition of several ubiquitin moieties to the various lysine residues within AChRα (Fig. 6C). By quantifying the intensity of the staining in the smear, we found that, when AChR was expressed alone, the level of AChRα ubiquitination was substantially higher (210 ± 10%) than when it was coexpressed with αkap.
Figure 6.

αkap inhibits ubiquitination of AChRα. Two independent replicates are shown in all three panels. A, The level of expression of AChRα in lysates from HEK293T cells cotransfected with AChRα, HA–Ub, and either a plasmid encoding αkap (+) or empty vector (−) as a control. Tubulin staining (data not shown) indicated that equal amounts of protein were loaded in the two lanes. B, C, Lysates with equal amounts of proteins were immunoprecipitated with anti-AChRα antibody (MAB153) and probed with AChRα antibody MAB210 (B) or with anti-ubiquitin antibody (C). To allow the amount of ubiquitination to be easily compared between experimental conditions, before the immunoprecipitation step the concentration of total proteins in each sample was adjusted so that similar amounts of AChRα would be precipitated (see B) although the amounts of AChR in the original samples were quite different (see A). The material recognized by anti-ubiquitin appears as a smear because the extent of polyubiquitination varies widely.
There are four lysines (K359, K374, K388, and K393) within the major intracellular loop of AChRα that are predicted to be ubiquitinated (Radivojac et al., 2010). If the absence of αkap promotes ubiquitination of lysine residues on AChRα leading to AChR degradation, changing lysine to arginine residues (so ubiquitin cannot be conjugated) should lead to less degradation of AChRα. To directly test this idea, we used site-directed mutagenesis to make four single mutant constructs (K359R, K374R, K388R, or K393R, 1R), a construct that contained three mutations (K359R/K388R/K393R, 3R), and a construct that had all four mutations (K359R/K374R/K388R/K393R, 4R). When coexpressed with HA–Ub in HEK293T cells, the 3R and 4R AChRα mutants exhibited a large decrease in ubiquitination compared with the wild-type AChRα or any of the four 1R mutants (Fig. 7A; data for only one of the 1R mutants is shown). The 4R mutant AChRα was capable of assembling into a functional AChR complex when coexpressed with β, δ, and ε subunits (Fig. 7B). However, we decided not to pursue an electrophysiological approach to assess expression levels, because of the highly variable extent of transfection and because the rapid desensitization to maximal concentrations of ACh made the amplitude of the peak currents dependent on the position of the drug application pipette and the rate of flow out of it, as well as on the number of receptors present. When a biochemical approach was used to assess expression levels, the reduced levels of ubiquitination in the 3R and 4R mutants correlated with a significant increase in the stability of AChRα mutants compared with wild-type (Fig. 7C,D). These results demonstrate that lysines 359, 374, 388, and 393 represent major sites for ubiquitination and that ubiquitinating several of these lysines promotes degradation. The presence of αkap did not increase the amount of 3R or 4R mutants compared with wild-type AChRα (Fig. 7E,F), suggesting that the effect of αkap on AChRα stability is mediated by controlling AChRα ubiquitination.
Figure 7.

αkap had no effect on the stability of AChRα when lysines essential for ubiquitination were mutated. HEK293T cells were cotransfected with plasmids encoding the indicated AChRα variants and HA–Ub, and 48 h later protein lysates were collected. The anti-AChRα antibody MAB153 was used to selectively immunoprecipitate proteins from each sample. A, The extent of ubiquitination of AChRα detected with an anti-HA antibody. In this experiment, the amount of material loaded onto each lane was adjusted so that comparable amounts of AChRα were present in all samples. The top gel shows the amount of AChRα detected with anti-AChRα (MAB210), and the bottom gel shows the anti-HA immunoreactivity. The control lane (Cont) was from cells transfected with HA–Ub only. WT, Wild type. The 1R lane is the K359R mutant, the 3R lane is the K359R/K388R/K393R mutant, and the 4R lane is the K359R/K374R/K388R/K393R mutant. The lower band present in all lanes of the anti-HA blot (arrow) is the 25 kDa light chain of the antibody used for immunoprecipitation and served as a loading control. The smear present in some lanes (brackets, ∼70–250 kDa) is polyubiquinated AChRα and so is absent in the control because no AChR were present. Polyubiquitination of AChRα was greatly decreased in the 3R and 4R mutants. B, Whole-cell recordings from HEK293T cells cotransfected with EGFP, wild-type AChR β, δ, and ε subunits, and either the wild-type or the 4R mutant AChRα. The holding potential was −50 mV. ACh was applied for the duration indicated by the bar above each trace. C, Western blots of homogenates from HEK293T cells cotransfected with EGFP and wild-type AChRα or the ubiquitination-deficient 3R or 4R mutant. In these experiments, the same amount of material (as determined from anti-GFP staining) was loaded onto each lane. No αkap was present (−αkap). D, Quantification of three independent experiments similar to C. In this panel as well as F, 100% represents the intensity of signal produced when cells were transfected with wild-type AChRα in the absence of αkap. E, Experiment similar to C, except that the cells were also cotransfected with a plasmid encoding αkap (+ αkap). F, Quantification of three experiments similar to E.
Discussion
In this work, we provide evidence for a novel function of αkap in controlling AChR stability. We demonstrated the following: (1) loss of αkap drastically reduced AChR levels, leading to fewer receptor clusters on the surface of muscle cells; (2) two proteasome inhibitors prevented degradation of AChRα in the absence of αkap; (3) proteasome inhibition does not enhance AChRα accumulation any further in the presence of αkap; (4) the absence of αkap increased ubiquitination of AChRα; (5) major sites for lysine ubiquitination reside within the intracellular domain of AChRα; and finally, (6) mutations of AChRα lysine residues required for ubiquitination slow the degradation of AChRα.
Previous studies have shown that, in heterologous cells transfected with all receptor subunits, unassembled AChR subunits are rapidly degraded in the ER and that assembly into a pentameric complex is essential for protein stability (Claudio et al., 1989; Blount and Merlie, 1990). This degradation is thought to occur in the ERAD pathway in which ubiquitination and the proteasome are involved, because unassembled subunits were accumulated in cells treated with proteasome inhibitors (Christianson and Green, 2004). In the present study, we demonstrated that overexpression of αkap substantially increased the amount of unassembled AChRα in the ER of HEK cells, even when the proteasome was functioning normally. In skeletal and cardiac muscles, αkap is highly expressed and localized to the sarcoplasmic reticulum (Nori et al., 2003; Singh et al., 2009). It was established previously that αkap associates with multiple CaMKII isoforms and anchors them to the sarcoplasmic reticulum through its hydrophobic domain (Campbell and MacLennan, 1982; Damiani et al., 1995; Bayer et al., 1998). This has the functional consequence of modulating Ca2+ release from the sarcoplasmic reticulum and so regulating excitation/contraction coupling in muscle cells (Bers, 2002a,b). Our work indicates that αkap has a second important function as a regulator of AChR stability in muscle cells. The action of αkap on protein stability was not unique to AChR, because at least one additional muscle protein (Lrp4) was also αkap sensitive. However, two other transmembrane proteins (P2X2 and LAMP1) and several intracellular synaptic proteins (rapsyn, α-dystrobrevin, and α-syntrophin) were αkap insensitive. Thus, it is possible that modulation of αkap activity in vivo might have relatively selective actions on synaptic function.
The present experiments show that αkap interacts with AChRα and promotes their stability and that the effect of αkap can be mimicked by elimination of ubiquitin conjugating sites from AChRα. How could αkap be linked to AChR stability? One possible molecular mechanism would be an indirect effect, with αkap acting as a chaperone to promote the forward trafficking of the AChR to the Golgi so that the receptors are not in the ER long enough to be modified. A second possibility is that αkap more directly regulates the state of ubiquitination of AChRα in the ER. Our results favor the latter possibility, because in HEK cells expressing AChRα alone, there is no movement to the Golgi and yet αkap still is effective. As far as how αkap might act on AChR within the ER, one possibility is that αkap may act as a deubiquitinase. This seems unlikely because αkap does not share any similarities with known deubiquitinases. A related possibility is that αkap may control AChR deubiquitination through an association with deubiquitinase(s). It has been reported that the ubiquitin-specific protease 19 (USP19) localizes in the ER and is involved in processing ERAD substrates (Hassink et al., 2009). Interestingly, Usp19 also interacts with CaMKII (Sowa et al., 2009), and, because αkap forms complexes with CaMKII, it is possible that αkap may function as a scaffold for Usp19. Other mechanisms by which αkap could decrease ubiquitination of AChR would be by inhibiting the E3 ubiquitin ligase(s) that conjugate ubiquitin to lysines on the receptors or by masking the lysines of AChRα so they cannot be ubiquitinated.
Recent studies have shown that the ubiquitin–proteasome system is involved in regulating synaptic protein turnover and synaptic efficacy at a variety of synapses (Ehlers, 2003; Speese et al., 2003; Christianson and Green, 2004; Mabb and Ehlers, 2010). Ubiquitination has been shown to play a key regulatory mechanism for many cell-surface proteins, in addition to its classical function of targeting cytosolic proteins for degradation by the proteasome. For example, ubiquitination of AMPA receptors, NMDA receptors, and PSD scaffold proteins has emerged as the major mechanism for controlling the accelerated turnover and decreased amounts of receptor proteins and thus synaptic plasticity (Juo and Kaplan, 2004; Mabb and Ehlers, 2010). At the fly NMJ, a balance between ubiquitination and deubiquitination plays a key role in shaping the morphology of the synapse. Loss of function of the USP highwire or its ortholog in C. elegans (rpm-1) produces profound defects in synaptic transmission (Schaefer et al., 2000; Wan et al., 2000). At developing mammalian NMJ, ubiquitination of the receptor tyrosine kinase MuSK leads to a reduction in postsynaptic MuSK expression levels and consequently defective synapses (Lu et al., 2007). Our results indicate that an additional mechanism by which ubiquitination regulates receptor trafficking in an αkap-dependent manner is critical for maintaining the steady state of the postsynaptic receptor density and thus synaptic strength.
Footnotes
This work was supported by National Institutes of Health Grant NS-047332. The anti-AChR (mab35) antibodies were obtained from the Developmental Studies Hybridoma Bank. We thank Dr. Stephan Kröger and his group for providing antibodies and Lrp4 construct and members of our laboratory for their comments on this manuscript.
The authors declares no competing financial interests.
References
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, Froehner SC. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, Löhler J, Harbers K. An alternative, nonkinase product of the brain-specifically expressed Ca2+/calmodulin-dependent kinase II alpha isoform gene in skeletal muscle. Mol Cell Biol. 1996;16:29–36. doi: 10.1128/mcb.16.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, Harbers K, Schulman H. alphaKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J. 1998;17:5598–5605. doi: 10.1093/emboj/17.19.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Schulman H. Alternative splicing modulates the frequency-dependent response of CaMKII to Ca2+ oscillations. EMBO J. 2002;21:3590–3597. doi: 10.1093/emboj/cdf360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002a;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bers DM. Sarcoplasmic reticulum Ca release in intact ventricular myocytes. Front Biosci. 2002b;7:d1697–d1711. doi: 10.2741/A873. [DOI] [PubMed] [Google Scholar]
- Blount P, Merlie JP. Mutational analysis of muscle nicotinic acetylcholine receptor subunit assembly. J Cell Biol. 1990;111:2613–2622. doi: 10.1083/jcb.111.6.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KP, MacLennan DH. A calmodulin-dependent protein kinase system from skeletal muscle sarcoplasmic reticulum. Phosphorylation of a 60,000-dalton protein. J Biol Chem. 1982;257:1238–1246. [PubMed] [Google Scholar]
- Christianson JC, Green WN. Regulation of nicotinic receptor expression by the ubiquitin-proteasome system. EMBO J. 2004;23:4156–4165. doi: 10.1038/sj.emboj.7600436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A, Sumbilla C, Inesi G, Jay SD, Campbell KP. Specific association of calmodulin-dependent protein kinase and related substrates with the junctional sarcoplasmic reticulum of skeletal muscle. Biochemistry. 1990;29:5899–5905. doi: 10.1021/bi00477a003. [DOI] [PubMed] [Google Scholar]
- Claudio T, Paulson HL, Green WN, Ross AF, Hartman DS, Hayden D. Fibroblasts transfected with Torpedo acetylcholine receptor beta-, gamma-, and delta-subunit cDNAs express functional receptors when infected with a retroviral alpha recombinant. J Cell Biol. 1989;108:2277–2290. doi: 10.1083/jcb.108.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiani E, Picello E, Saggin L, Margreth A. Identification of triadin and of histidine-rich Ca2+-binding protein as substrates of 60 kDa calmodulin-dependent protein kinase in junctional terminal cisternae of sarcoplasmic reticulum of rabbit fast muscle. Biochem Biophys Res Commun. 1995;209:457–465. doi: 10.1006/bbrc.1995.1524. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Fang D, Kerppola TK. Ubiquitin-mediated fluorescence complementation reveals that Jun ubiquitinated by Itch/AIP4 is localized to lysosomes. Proc Natl Acad Sci U S A. 2004;101:14782–14787. doi: 10.1073/pnas.0404445101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- Hassink GC, Zhao B, Sompallae R, Altun M, Gastaldello S, Zinin NV, Masucci MG, Lindsten K. The ER-resident ubiquitin-specific protease 19 participates in the UPR and rescues ERAD substrates. EMBO Rep. 2009;10:755–761. doi: 10.1038/embor.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juo P, Kaplan JM. The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Curr Biol. 2004;14:2057–2062. doi: 10.1016/j.cub.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Je HS, Young P, Gross J, Lu B, Feng G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. J Cell Biol. 2007;177:1077–1089. doi: 10.1083/jcb.200610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y, Valenzuela I, Mouslim C, Akaaboune M. Calcium/calmodulin kinase II-dependent acetylcholine receptor cycling at the mammalian neuromuscular junction in vivo. J Neurosci. 2010;30:12455–12465. doi: 10.1523/JNEUROSCI.3309-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y, Valenzuela I, Mouslim C, Pires-Oliveira M, Adams ME, Froehner SC, Akaaboune M. Nicotinic acetylcholine receptor stability at the NMJ deficient in α-syntrophin in vivo. J Neurosci. 2011;31:15586–15596. doi: 10.1523/JNEUROSCI.4038-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nori A, Lin PJ, Cassetti A, Villa A, Bayer KU, Volpe P. Targeting of alpha-kinase-anchoring protein (alpha KAP) to sarcoplasmic reticulum and nuclei of skeletal muscle. Biochem J. 2003;370:873–880. doi: 10.1042/BJ20021624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada K, Inoue A, Okada M, Murata Y, Kakuta S, Jigami T, Kubo S, Shiraishi H, Eguchi K, Motomura M, Akiyama T, Iwakura Y, Higuchi O, Yamanashi Y. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- Radivojac P, Vacic V, Haynes C, Cocklin RR, Mohan A, Heyen JW, Goebl MG, Iakoucheva LM. Identification, analysis, and prediction of protein ubiquitination sites. Proteins. 2010;78:365–380. doi: 10.1002/prot.22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, Hadwiger GD, Nonet ML. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- Singh P, Salih M, Tuana BS. Alpha-kinase anchoring protein alphaKAP interacts with SERCA2A to spatially position Ca2+/calmodulin-dependent protein kinase II and modulate phospholamban phosphorylation. J Biol Chem. 2009;284:28212–28221. doi: 10.1074/jbc.M109.044990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol. 2003;13:899–910. doi: 10.1016/s0960-9822(03)00338-5. [DOI] [PubMed] [Google Scholar]
- Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Wang J, Best PM. Inactivation of the sarcoplasmic reticulum calcium channel by protein kinase. Nature. 1992;359:739–741. doi: 10.1038/359739a0. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133:4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu A, Hawkins C, Narayanan N. Phosphorylation and activation of the Ca2+-pumping ATPase of cardiac sarcoplasmic reticulum by Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 1993;268:8394–8397. [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]