

Abstract
Almost all theoretical and experimental studies of the mechanisms underlying learning and memory focus on synaptic efficacy and make the implicit assumption that changes in synaptic efficacy are both necessary and sufficient to account for learning and memory. However, network dynamics depends on the complex interaction between intrinsic membrane properties and synaptic strengths and time courses. Furthermore, neuronal activity itself modifies not only synaptic efficacy but also the intrinsic membrane properties of neurons. This paper presents examples demonstrating that neurons with complex temporal dynamics can provide short-term “memory” mechanisms that rely solely on intrinsic neuronal properties. Additionally, we discuss the potential role that activity may play in long-term modification of intrinsic neuronal properties. While not replacing synaptic plasticity as a powerful learning mechanism, these examples suggest that memory in networks results from an ongoing interplay between changes in synaptic efficacy and intrinsic membrane properties.
Keywords: neuronal oscillators, plateau neurons, neuromodulation, dynamic clamp
The behavior of rhythmic networks depends on the complex interaction between the dynamics of the individual neurons (their intrinsic properties) and the strengths and time courses of the synapses among them (1, 2, 3). Years of research on networks as diverse as central pattern generators in invertebrates and vertebrates (3), the thalamus (4, 5, 6), and the cerebellum (7) have clearly shown that complex neuronal characteristics, such as oscillatory and plateau properties, play crucial roles in shaping neural network output. Recent work has expanded the brain regions that show neuronal oscillations to the cortex (8), suggesting that complex intrinsic properties are likely to shape the computational dynamics of many, if not all, brain regions.
It is commonly assumed that short and long-lasting changes in synaptic efficacy are both necessary and sufficient to account for the stable changes in network dynamics that we call “memory”. The remarkable success of early neural network models, in which only synaptic strengths were modified but memories were stored, showed that changes in network output could result solely from changes in synaptic strength (9). An attractive feature of synaptic modification is that it can be restricted to a subset of the synaptic connections made by a neuron, or made onto a neuron. And last, but not least, the experimental work on the Aplysia gill withdrawal reflex and hippocampal slices, in which long-lasting changes in synaptic strength could be produced, and the mechanisms underlying those changes studied (10, 11) provided strong impetus to look primarily at synaptic plasticity as the mechanism underlying memory in intact animals. However, in this paper, we show that intrinsic neuronal currents can also play a role in memory phenomena in at least two different ways. First, a variety of “short-term” memory mechanisms can result from slowly activating and inactivating membrane conductances that make the response of a cell depend on its recent history of activity. Second, activity can modify, on a longer time scale, the intrinsic properties of neurons, resulting in long-lasting experience-dependent changes. These “intrinsic memory phenomena” should be viewed as powerful additions to the compendium of mechanisms that can be called into play to allow experience to modify neural circuits.
Neurons Have a Variety of Intrinsic Membrane Properties
Neurons can display a variety of activity patterns that depend on the number and type of voltage channels in their membranes (Fig. 1). Some neurons are silent unless excited; others are spontaneously active. Some neurons display intrinsic oscillatory properties involving periodic bursts of action potentials. Modifications in the number of each channel type present in the membrane can change a variety of neuronal properties, including firing frequency and threshold, rate of spike repolarization, degree of postinhibitory rebound, burst amplitude, and burst period. Moreover, changes in channel density can move neurons from one kind of activity pattern (such as tonic firing) to another (such as bursting).
Figure 1.
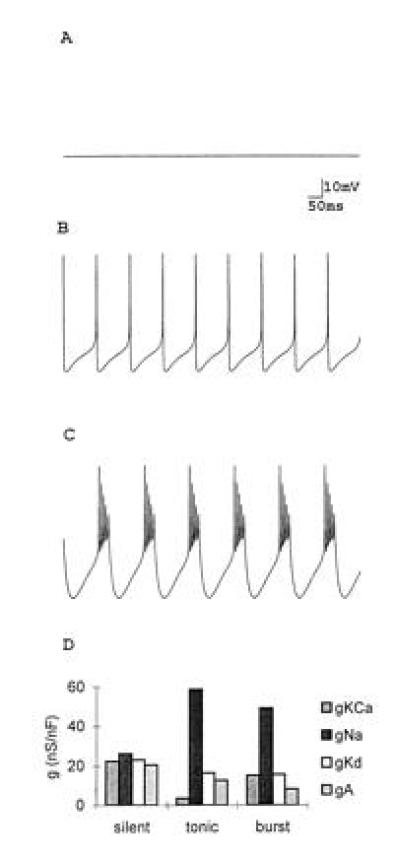
Different patterns of activity in a model neuron. Computer simulations using a model modified from that in ref. 12. (A) Silent neuron. (B) Tonic activity. (C) Oscillatory activity. (D) Plots of four of the seven currents in the model showing the relative balance of conductances giving rise to the patterns of activity shown in A, B, and C (Z.L., unpublished data). Taken from ref. 12.
Bistable Neurons
Sustained neuronal activity in response to a brief stimulus has been proposed to underlie some short-term memory tasks (see other papers in this colloquium). For many years, the assumption was made that such sustained activity resulted from reverberating activity through excitatory feedback loops. However, many neurons display plateau properties (whose expression can be controlled by neuromodulatory substances), which provide an alternative mechanism for the production of sustained neuronal excitability in response to a brief stimulus (13, 14, 15).
Plateau neurons have two states, a hyperpolarized state which is usually subthreshold or silent, and a more depolarized state in which the cell fires action potentials (13). The essential feature of these neurons is that, like a flip-flop in an electronic circuit, the neurons can be switched from one stable state to the other by a transitory synaptic event. The example shown in Fig. 2 is of a neuron that does not display plateau properties in control saline but does in the presence of a neuropeptide (15). In control saline, the neuron only fires during the depolarizing current pulse, but, in the presence of the peptide, a short depolarizing current pulse flips the neuron from its hyperpolarized state to its depolarized state. Note that the neuron continues to fire long after the current pulse has been terminated, but that a short hyperpolarizing pulse can switch the neuron back to its hyperpolarized state. Plateau neurons, like electronic flip-flops, store information about whether their last input was excitatory or inhibitory. A perfect bistable element can, of course, store information indefinitely. The key issue to ask is how long can plateau neurons in the nervous system store information?
Figure 2.
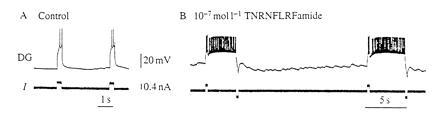
Plateau neuron (modified from ref. 15). Intracellular recording from the dorsal gastric (DG) neuron of the crab stomatogastric ganglion. (A) In control saline, a short pulse of depolarizing current elicits firing only during the current pulse. (B) In the presence of 10−7M TNRNFLRFamide, a short pulse of depolarizing current elicits firing that outlasts the current pulse and continues until it is terminated by a short hyperpolarizing pulse.
Most plateau neurons will not fire indefinitely but will spontaneously return to their hyperpolarized state after a period of time. However, this time can be quite long, hundreds of milliseconds in many cases, and some neurons sustain plateaus for many seconds or even minutes. One interesting possibility is that plateau properties that can be gated on and off by dopamine might play a role in working memory tasks in prefrontal cortex (X. J. Wang and M. Camperi, personal communication).
Another very interesting bistability is seen in theoretical and experimental work on the R15 neuron of Aplysia (16, 17, 18, 19). R15 is a prototypic bursting neuron, an extensive biophysical literature on its membrane currents and their modulation has been gathered (19), and a detailed model of this neuron and its modulation has been developed (16, 17). This model has the interesting feature that it can display different modes of activity and can be switched among them by transient synaptic inputs (16, 17). We illustrate a similar result in a model of a cultured stomatogastric ganglion neuron in Fig. 3A. The model was initially in a bursting mode, and a short depolarization switched it into a tonic firing mode. A subsequent depolarization flipped it back into the bursting mode.
Figure 3.
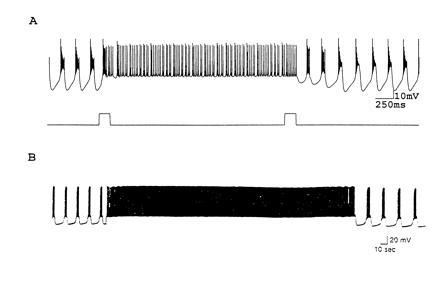
Mode switches between tonic firing and oscillatory action. (A) Model neuron initially in a bursting mode. A short depolarization flips the model into the tonic firing mode, where it remains until a second perturbing pulse, which switches it back into the bursting model (Z.L., unpublished data). (B) Intracellular recording from R15 in Aplysia. The neuron was initially in the bursting mode but was switched into the tonic firing mode by a short depolarizing current pulse (modified from ref. 18).
Mode switches such as these are found in only limited parameter regimes of oscillatory model neurons. Therefore the demonstration that these are seen in biological neurons under modulatory control is quite significant. Lechner et al. (18) showed that the R15 neuron of Aplysia exhibited this behavior (Fig. 3B). In this experiment the neuron was initially in a stable bursting mode. Then a short depolarizing pulse switched it into a tonic firing mode, where it stayed for several minutes before falling back into the bursting mode. The neuron could be switched back and forth between the bursting and tonically firing mode by brief pulses of current. In the theoretical work on R15 (16, 17), the mode switches were stable. However, in the experimental work, the mode switches appear to last from 1 min to several minutes, before reverting to initial conditions, and the ability of these neurons to display this mode switch behavior depends on the concentration of modulators, such as serotonin (18).
Memories or “Cellular Priming” from Slow Conductances
There are numerous voltage- and time-dependent conductances that have slow kinetics. These can provide the biophysical substrate for short-term memories. For example, a variety of voltage-dependent K+ conductances slowly inactivate and also slowly recover from inactivation. One such conductance is that of the Kv1.3 potassium channel (20, 21). Marom and Abbott (21) modeled this conductance, added it to a Hodgkin–Huxley model of a simple spiking neuron, and showed that its slow recovery from inactivation produced an enhanced response to a depolarizing current pulse subsequent to a long depolarization. Subsequently we (22) used the dynamic clamp (23, 24) to introduce the modeled Kv1.3 conductance into cultured stomatogastric ganglion neurons (Figs. 4 and 5).
Figure 4.
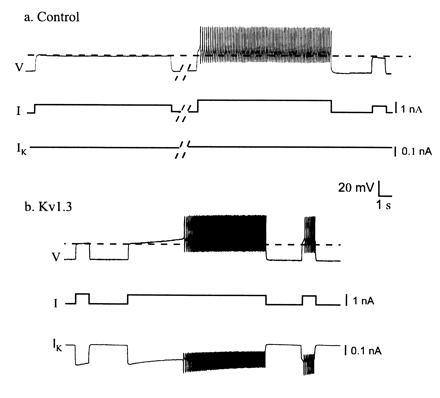
Addition of Kv1.3. to a tonically firing cultured stomatogastric ganglion neuron. (a) Control, in the absence of dynamic clamp-added Kv1.3. (b) With the addition of Kv1.3 (modified from ref. 22). V, intracellularly recorded membrane potential; I, injected current; IK, dynamic clamp-injected Kv1.3 current.
Figure 5.
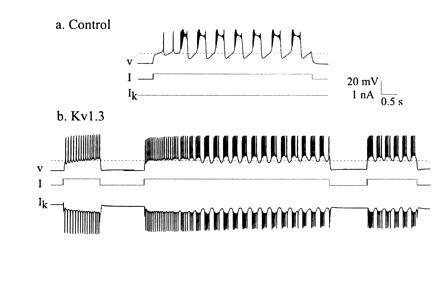
Addition of Kv1.3 to a bursting neuron. Locants and abbreviations are the same as in Fig. 4 (modified from ref. 22).
Fig. 4a illustrates the response of a neuron to two levels of depolarizing current. The first depolarization was subthreshold, while the second, slightly larger depolarization was suprathreshold. This neuron showed no appreciable spike frequency adaptation, and started firing very soon after the onset of the current pulse. Fig. 4b shows the same neuron after the Kv1.3 current was applied using the dynamic clamp. The neuron was first depolarized with a short subthreshold current pulse. Then the cell was given a long depolarizing current pulse. In response to this, as the Kv1.3 current slowly inactivated, and the cell depolarized, until after several seconds the cell started firing action potentials. When the current pulse was turned off, the neuron stopped firing. However, several seconds later, when a current pulse of the same amplitude as that given before the long depolarization was once again applied, the neuron fired. This memory effect occurs because the recovery from inactivation of Kv1.3 is very slow (22). Thus, a synaptic input that is subthreshold when given to a neuron that was relatively inactive is primed by a long period of depolarization to become suprathreshold. This priming effect decays away as the Kv1.3 conductance recovers from inactivation.
Fig. 5 shows the effect of the same Kv1.3 conductance applied with the dynamic clamp to a cultured stomatogastric ganglion neuron with different intrinsic membrane properties (22). In the absence of applied Kv1.3, this neuron behaved as an oscillator when depolarized (Fig. 5a). When the Kv1.3 current was applied, the additional K+ conductance provided by the Kv1.3 current caused the cell to fire tonically when depolarized but not to fire bursts of action potentials in response to a short depolarization. When a longer current pulse was applied, the neuron initially fired tonically, but then as Kv1.3 slowly inactivated, the cell moved back into its oscillatory regime. Again, because the recovery from inactivation of Kv1.3 is slow, a subsequent current pulse elicits bursts of action potentials, not tonic firing. Here, the Kv1.3 has produced a long-lasting “mode switch” in the response of a neuron to a depolarizing input. In summary, the response of a neuron with a conductance such as Kv1.3 to a given synaptic input will reflect the temporal pattern of past synaptic inputs it has received over a time scale of many seconds.
Oscillators as Postsynaptic Elements
When neurons have complex membrane properties, changes in synaptic strength can produce seemingly paradoxical changes in the effect of a synapse on its follower neuron. This is clearly shown when the postsynaptic neuron is an oscillator.
Fig. 6 shows a single compartment model of a neural oscillator (loosely adapted from that in ref. 12) and illustrates some results of changing the strength of a synaptic input to that oscillator. The top trace of Fig. 6 shows the behavior of the unperturbed oscillator. The middle trace shows the response of the model neuron to a simulated small inhibitory postsynaptic potential (IPSP) introduced at the time of the third spike in the second burst. This IPSP reset the phase of the oscillator, delaying the onset of the next and subsequent bursts. The bottom trace shows the effect of increasing the conductance of the simulated IPSP 5-fold. In this case the IPSP phase-advanced the oscillator when applied at the same phase as that shown in the previous example.
Figure 6.
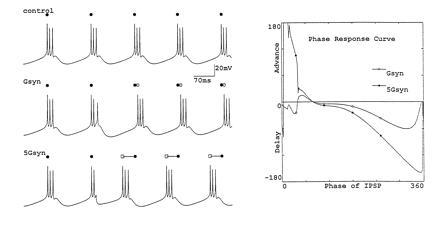
Effect of changing synaptic strength on the phase—response curve of an oscillator. (Left) Voltage traces of a model oscillator. Top trace shows control; filled circles indicate the first spike in each burst. Middle trace shows an IPSP simulated by a conductance (Gsyn) applied at the end of active phase of the second burst produced a modest phase delay (the time of the unperturbed oscillator is shown by the filled circle, and the first spike of the perturbed burst is shown in the open circle). The bottom trace shows an IPSP that was 5-fold larger (5Gsyn), applied at the same time as that in the middle trace. Note that now the oscillator is phase-advanced rather than phase-delayed. (Right) Full phase–response curve for the effect of the small and large IPSPs (Z.L., unpublished work).
Fig. 6 also shows the full phase–response curves for the effects of the large and small IPSPs on the oscillator. An interesting feature of this plot is that during much of the cycle, the effect of increasing the synaptic strength increases the phase shift produced by the IPSP. However, when the IPSP is applied during the active phase (when the neuron is firing), changing the synaptic strength changes the effect of the perturbation. Specifically, a small IPSP applied during this phase of the cycle produces a phase delay, but a large IPSP produces a phase advance.
In the past few years, the oscillatory properties in invertebrate and vertebrate nervous systems have been intensively studied by both theoretical and experimental methods (3, 12, 16, 17, 18, 19, 25, 26, 27, 28, 29, 30). Indeed, a variety of functions in sensory and motor systems have been ascribed to network oscillations, and there is tremendous interest in understanding how they arise. The fact that different synaptic strengths can produce opposite changes in an oscillator’s response to a synaptic input at a given phase of the oscillator illustrates the subtlety of controlling and modulating true oscillatory neurons.
Long-Term Control of Intrinsic Conductances: The Role of Activity
Experience is known to influence synaptic strength and connectivity in both development and learning. In this section, we will discuss both theoretical and experimental work that suggests that activity may also play a critical role in determining the intrinsic properties of neurons (12, 31, 32, 33, 34).
The initial motivation for the first model we developed (31) was to suggest mechanisms that could ensure that neuronal activity patterns were stably maintained despite the stochastic processes of channel turnover. Therefore, we designed a simple model in which neuronal activity, as reflected by the intracellular Ca2+ concentration, was used to slowly regulate the maximal conductances of each of the ionic currents expressed in the neuron (31). This is a biologically plausible mechanism, as there are an increasing number of reports suggesting that intracellular Ca2+ and activity can regulate membrane currents (e.g., refs. 35, 36, 37).
The first generation dynamically regulating model (31) was constructed from a model of the lateral pyloric (LP) neuron of the stomatogastric ganglion (38) and contained seven voltage- and time-dependent currents. We reasoned that if neuronal activity increased, this would lead to an increase in intracellular Ca2+, and therefore we used high levels of intracellular Ca2+ as a signal to decrease the maximal conductances of the inward currents (which tend to make neurons more excitable) and increase the maximal conductances of the outward currents (which tend to make neurons less excitable). Similarly, low intracellular Ca2+ concentrations in this model led to increases in the inward currents and decreases in the outward currents. Fig. 7 illustrates the essential behavior of the model in response to a perturbation. The model was initially in a bursting mode. When the extracellular K+ was increased, the cell first depolarized and fired tonically and then slowly recovered its bursting activity pattern, as the dynamically regulating process changes the balance of conductances to bring the cell back to its initial activity pattern.
Figure 7.
Effect of a perturbation on a dynamically regulating model neuron. The neuron was initially active in bursts. At the arrowhead, the extracellular K+ concentration in the simulation was increased. The neuron initially depolarized but over time regulated its conductances to return to its initial activity patterns. [Reprinted with permission from LeMasson, G., Marder, E. & Abbott, L. F. (1993) Science 259, 1915–1917] Copyright (1993). American Association for the Advancement of Science.
The dynamic regulating models have several interesting properties, in addition to their ability to maintain a homeostatic level of activity. First, if the dynamically regulating rule is implemented in an extended compartmental model of a neuron (33), a nonuniform distribution of conductances results. Second, if dynamically regulating neurons are coupled electrically, despite the symmetrical nature of the electrical coupling, the two neurons can “differentiate” and end up with different intrinsic properties, although they continue to fire together because of the coupling (ref. 31; Z.L., unpublished work).
Several lines of evidence suggest that the maintenance of the intrinsic properties of the neurons in the stomatogastric ganglion may depend on the normal rhythmic activity of the ganglion. The stomatogastric ganglion generates two important rhythmic motor patterns: the pyloric rhythm, with an intrinsic period of ≈1 sec, and the gastric mill rhythm, with a period of ≈5–10 sec. Therefore, most of the 25–30 neurons in the stomatogastric ganglion are normally rhythmically active. However, when stomatogastric ganglion neurons are isolated from the network, and their intrinsic properties are studied in the absence of modulatory and presynaptic inputs, most of them are either tonically active or silent but do not rhythmically burst (39, 40, 41, 42). Under normal conditions, it is the circuit dynamics and modulatory influences, which maintain circuit rhythmicity, that result in almost all of the neurons in the ganglion being rhythmically active, although they are not intrinsically rhythmically active.
Interestingly, when individual stomatogastric ganglion neurons are removed from the adult ganglion and placed in dissociated cell culture, after 3 days in culture, >70% of the neurons fire in bursts when depolarized (12). This suggests that when the neurons are removed from the synaptic and modulatory inputs that maintain their rhythmic activity in the intact circuit, they somehow “sense” their lack of rhythmic activity and actively modify the balance of their membrane conductances to ensure intrinsic rhythmicity. Indeed, Fig. 8 shows that as these cultured neurons change their activity patterns, they down-regulate the density of their outward currents while up-regulating the density of their inward currents.
Figure 8.
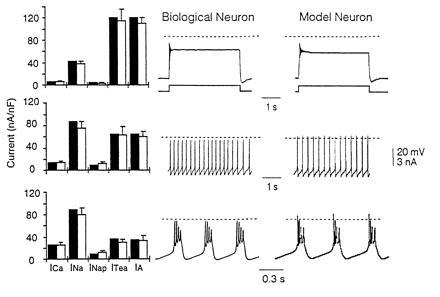
Changes in activity patterns and current densities of cultured stomatogastric ganglion neurons with time in culture. (Left) Current densities measured in voltage clamp on day 1 (Top), day 2 (Middle), and day 3 (Bottom). Measured currents are shown in the open bars, currents used in the model (Right) are shown in filled bars. (Middle) Recordings from the biological neuron are shown. The behavior of a model constructed using the current densities shown in the filled bars is shown (Right). Taken from ref. 12.
A simple prediction of our model would argue that if the cultured neurons were rhythmically driven, in a manner that mimics the rhythmic inhibitory drive that they receive in the intact stomatogastric ganglion, they might return to a tonic firing mode of activity, which more closely resembles their activity in the intact ganglion. The results of this experiment are shown in Fig. 9. In response to 1 hr of hyperpolarizing pulses, which resulted in strong postinhibitory rebound bursts, the cultured neurons lost their intrinsic bursting behavior and became tonically active. This process appears to depend on changes in intracellular Ca2+ (34). When this experiment was simulated in the dynamically regulating model (G. LeMasson and G.G.T., unpublished work; Z.L., unpublished work), identical results were obtained.
Figure 9.
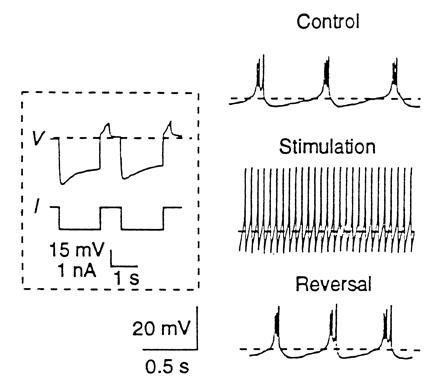
Activity alters the intrinsic properties of cultured stomatogastric ganglion neurons. (Inset) Hyperpolarizing current pulses trigger a depolarizing sag and a postinhibitory rebound burst. V, membrane potential; and I, current pulse. (Right) The top trace shows neuronal activity before stimulation; the middle trace shows neuronal activity after 1 hr of stimulation using the protocol in the Inset; and the bottom trace shows neuronal activity 1 hr after the stimulation was stopped. [Reprinted with permission from Turrigiano, G. G., Abbott, L. F. & Marder, E. (1994) Science 264, 974–977] Copyright (1994). American Association for the Advancement of Science.
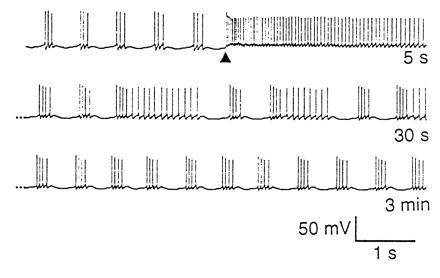
Another indication of ongoing processes that use activity to regulate the intrinsic properties of neurons is seen when the stomatogastric ganglion is placed into long-term organ culture. Ordinarily, robust pyloric and gastric mill rhythmicity depends on the presence of modulatory inputs to the stomatogastric ganglion. When these modulatory inputs are removed, the activity in the stomatogastric ganglion decreases dramatically, or sometimes ceases entirely. This is illustrated in the first two traces in Fig. 10. The top trace shows strong rhythmic activity from the stomatogastric ganglion of a crab with modulatory inputs from anterior ganglia left attached. At time (t) = 0, the nerve connecting the stomatogastric ganglion to the source of the modulatory inputs was cut, and the ganglion quickly lost its rhythmic activity patterns. Note, it remained non-rhythmic for almost 2 days, but after that time, rhythmic activity returned (t = 44 hr). (The J. Simmers laboratory has obtained similar data with no indication of synaptic strength changes; personal communication.) Because the modulatory inputs are no longer present, the simplest interpretation of these data is that rhythmic activity depends on modulatory inputs in the intact animal. However, if the modulatory input is no longer present and the network becomes arhythmic for an extended period of time, the neurons of the stomatogastric ganglion (like the cells in dissociated cell culture) may change their intrinsic properties to resume the same pattern of activity, but by a different set of cellular mechanisms.
Figure 10.
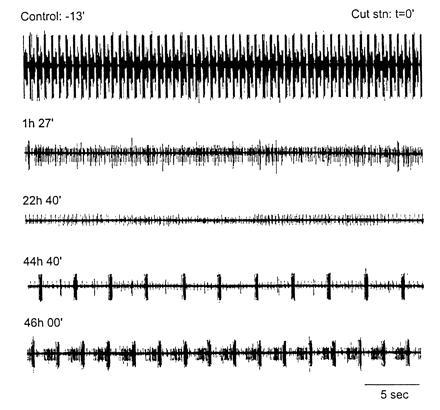
Resumption of rhythmic pyloric activity after 2 days in organ culture. Extracellular recordings from the main motor nerve of the stomatogastric ganglion. Under control conditions, there was strong rhythmic activity. The nerve bringing modulatory inputs into the stomatogastric ganglion was cut at t = 0. Recordings were taken at the various time points indicated. Note that rhythmic activity resumed at t = 44 hr, 40 min, and was strong at t = 46 hr (J.G., unpublished data).
Conclusions
The output of all neuronal circuits depends critically on the interaction between synaptic strength and intrinsic properties. In some cases, changes that occur during short- and long-term memory may result primarily from modifications in synaptic efficacy. However, there are mechanisms by which both short- and long-term changes in intrinsic neuronal properties can also modify circuit dynamics. These may occur independently of, or concurrently with, changes in synaptic efficacy. Therefore, the elucidation of how changes in circuit dynamics provide the substrate for the behavioral changes that we call learning and memory will require understanding not only the mechanisms that couple activity to synaptic strength but also the mechanisms that control intrinsic neuronal excitability.
Acknowledgments
This work was supported by a McKnight Investigators Award, National Institute of Mental Health Grant MH46742, the Alfred P. Sloan Foundation, and the W. M. Keck Foundation.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviation: IPSP, inhibitory postsynaptic potential.
References
- 1.Getting P A. Annu Rev Neurosci. 1989;12:185–204. doi: 10.1146/annurev.ne.12.030189.001153. [DOI] [PubMed] [Google Scholar]
- 2.Harris-Warrick R M, Marder E. Annu Rev Neurosci. 1991;14:39–57. doi: 10.1146/annurev.ne.14.030191.000351. [DOI] [PubMed] [Google Scholar]
- 3.Marder E, Calabrese R L. Physiol Rev. 1996;76:687–717. doi: 10.1152/physrev.1996.76.3.687. [DOI] [PubMed] [Google Scholar]
- 4.McCormick D A, Pape H-C. J Physiol (London) 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCormick D A, Pape H-C. J Physiol (London) 1990;431:319–342. doi: 10.1113/jphysiol.1990.sp018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huguenard J R, McCormick D A. J Neurophysiol. 1992;68:1373–1383. doi: 10.1152/jn.1992.68.4.1373. [DOI] [PubMed] [Google Scholar]
- 7.Llinás R R. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- 8.Steriade M, Nuñez A, Amica F. J Neurosci. 1993;13:3266–3283. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Churchland P S, Sejnowski T J. The Computational Brain. Cambridge, MA: MIT Press; 1992. [Google Scholar]
- 10.Hawkins R D, Kandel E R, Siegelbaum S A. Annu Rev Neurosci. 1993;16:625–665. doi: 10.1146/annurev.ne.16.030193.003205. [DOI] [PubMed] [Google Scholar]
- 11.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 12.Turrigiano G, LeMasson G, Marder E. J Neurosci. 1995;15:3640–3652. doi: 10.1523/JNEUROSCI.15-05-03640.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marder E. Curr Biol. 1991;1:326–327. doi: 10.1016/0960-9822(91)90101-2. [DOI] [PubMed] [Google Scholar]
- 14.Kiehn O. Trends Neurosci. 1991;14:68–73. doi: 10.1016/0166-2236(91)90023-n. [DOI] [PubMed] [Google Scholar]
- 15.Weimann J M, Marder E, Evans B, Calabrese R L. J Exp Biol. 1993;181:1–26. doi: 10.1242/jeb.181.1.1. [DOI] [PubMed] [Google Scholar]
- 16.Canavier C C, Baxter D A, Clark J W, Byrne J H. J Neurophysiol. 1993;66:2252–2257. doi: 10.1152/jn.1993.69.6.2252. [DOI] [PubMed] [Google Scholar]
- 17.Canavier C C, Baxter D A, Clark J W, Byrne J H. J Neurophysiol. 1994;72:872–882. doi: 10.1152/jn.1994.72.2.872. [DOI] [PubMed] [Google Scholar]
- 18.Lechner H A, Baxter D A, Clark J W, Byrne J H. J Neurophysiol. 1996;75:957–962. doi: 10.1152/jn.1996.75.2.957. [DOI] [PubMed] [Google Scholar]
- 19.Benson J A, Adams W B. In: Neuronal and Cellular Oscillators. Jacklet J W, editor. New York: Dekker; 1989. pp. 87–120. [Google Scholar]
- 20.Marom S, Levitan I B. Biophys J. 1994;67:579–589. doi: 10.1016/S0006-3495(94)80517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marom S, Abbott L F. Biophys J. 1994;67:515–520. doi: 10.1016/S0006-3495(94)80518-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turrigiano G G, Marder E, Abbott L F. J Neurophysiol. 1996;75:963–966. doi: 10.1152/jn.1996.75.2.963. [DOI] [PubMed] [Google Scholar]
- 23.Sharp A A, O’Neil M B, Abbott L F, Marder E. J Neurophysiol. 1993;69:992–995. doi: 10.1152/jn.1993.69.3.992. [DOI] [PubMed] [Google Scholar]
- 24.Sharp A A, O’Neil M B, Abbott L F, Marder E. Trends Neurosci. 1993;16:389–394. doi: 10.1016/0166-2236(93)90004-6. [DOI] [PubMed] [Google Scholar]
- 25.Gray C M. J Comput Neurosci. 1994;1:11–38. doi: 10.1007/BF00962716. [DOI] [PubMed] [Google Scholar]
- 26.Steriade M, McCormick D A, Sejnowski T J. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- 27.Marder E, Abbott L F. Curr Opin Neurobiol. 1996;5:832–840. doi: 10.1016/0959-4388(95)80113-8. [DOI] [PubMed] [Google Scholar]
- 28.Wang X J, Rinzel J. Neural Comput. 1992;4:84–97. [Google Scholar]
- 29.Traub R D. J Comput Neurosci. 1995;2:283–289. doi: 10.1007/BF00961440. [DOI] [PubMed] [Google Scholar]
- 30.Cohen A H, Ermentrout G B, Kiemel T, Kopell N, Mellen N, Sigvardt K A, Williams T L. Trends Neurosci. 1992;15:434–438. doi: 10.1016/0166-2236(92)90006-t. [DOI] [PubMed] [Google Scholar]
- 31.LeMasson G, Marder E, Abbott L F. Science. 1993;259:1915–1917. doi: 10.1126/science.8456317. [DOI] [PubMed] [Google Scholar]
- 32.Abbott L F, LeMasson G. Neural Comput. 1993;5:823–842. [Google Scholar]
- 33.Siegel M, Marder E, Abbott L F. Proc Natl Acad Sci USA. 1994;91:11308–11312. doi: 10.1073/pnas.91.24.11308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turrigiano G G, Abbott L F, Marder E. Science. 1994;264:974–977. doi: 10.1126/science.8178157. [DOI] [PubMed] [Google Scholar]
- 35.Linsdell P, Moody W J. J Neurosci. 1995;15:4507–4514. doi: 10.1523/JNEUROSCI.15-06-04507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong S J, Lnenicka G A. J Neurosci. 1995;15:3539–3547. doi: 10.1523/JNEUROSCI.15-05-03539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martínez J J, Onetti C G, García E, Hernández S. J Neurophysiol. 1991;66:1455–1461. doi: 10.1152/jn.1991.66.5.1455. [DOI] [PubMed] [Google Scholar]
- 38.Buchholtz F, Golowasch J, Epstein I R, Marder E. J Neurophysiol. 1992;67:332–340. doi: 10.1152/jn.1992.67.2.332. [DOI] [PubMed] [Google Scholar]
- 39.Miller J P, Selverston A I. J Neurophysiol. 1982;48:1378–1391. doi: 10.1152/jn.1982.48.6.1378. [DOI] [PubMed] [Google Scholar]
- 40.Marder E, Eisen J S. J Neurophysiol. 1984;51:1362–1373. doi: 10.1152/jn.1984.51.6.1362. [DOI] [PubMed] [Google Scholar]
- 41.Flamm R E, Harris-Warrick R M. J Neurophysiol. 1986;55:866–881. doi: 10.1152/jn.1986.55.5.866. [DOI] [PubMed] [Google Scholar]
- 42.Hooper S L, Marder E. J Neurosci. 1987;7:2097–2112. doi: 10.1523/JNEUROSCI.07-07-02097.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]