

Abstract
A concise and versatile approach toward the preparation of the cyanthiwigin family of cyathane natural products is described. By leveraging a unique double asymmetric catalytic alkylation procedure it is possible to quickly establish two of the most critical stereocenters of the cyanthiwigin framework with high levels of selectivity and expediency. The synthetic route additionally employs both a tandem ring-closing cross-metathesis reaction, and an aldehyde-olefin radical cyclization process, in order to rapidly arrive at the tricyclic cyathane core of the cyanthiwigin molecules. From this unifying intermediate, the preparations of cyanthiwigins B, F, and G are attained swiftly and without the need for protecting groups.
Keywords: Total Synthesis, Asymmetric Catalysis, Natural Product, Diterpene, Palladium
Introduction
Structural Features and Synthetic Challenges
The cyanthiwigin diterpenoid natural products were originally isolated from the marine sponge Epipolasis reiswigi.[1] Ten years after initial characterization, additional, novel cyanthiwigin molecules were found in extracts of the Jamaican sea sponge Mermekioderma styx.[2] To date, over 30 different examples of cyanthiwigin diterpenoids have been isolated and characterized from these two sources. The vast majority of these compounds are structurally similar, and are unified through the presence of a highly conserved tricyclic carbon scaffold (Figure 1). The cyanthiwigin molecules belong to a larger class of diterpene natural products known as the cyathanes. In keeping with the vast majority of other cyathane compounds, the 20 carbon atoms of the cyanthiwigins are arranged into a fused [5-6-7] tricyclic core skeleton (1–30).[3] However, in contrast to the remainder of the cyathanes, the dual all-carbon quaternary stereocenters found in the cyanthiwigin molecules at C(6) and C(9) are arranged with a syn relative stereochemical relationship, rather than an anti configuration. Furthermore, these diterpene natural products boast two additional points of stereogenicity at ring fusion carbons C(4) and C(5). These structural features establish a total of four contiguous stereocenters across the innermost bonds of the carbon scaffold. Indeed, the central ring of the cyanthiwigin [5-6-7] carbocyclic core imparts formidable challenge to the synthetic preparation of any of these natural products, due to the steric crowding surrounding and nested location of these critical stereocenters.
Figure 1.

The cyanthiwigin diterpenoid molecules
Structural differentiation among the members of the cyanthiwigin family is primarily manifest in variable oxidation of the peripheral carbocyclic skeleton. Oxidative diversity in the cyanthiwigins provides sparingly oxidized structures such as cyanthiwigin G (7), as well as more heavily oxidized examples as can be found in cyanthiwigin O (15). For many of the less-oxidized cyanthiwigin compounds (e.g., cyanthiwigin F (6)) preparative approaches toward these molecules can prove difficult owing to their sparse functionality. These minimally elaborated structures possess very few reactive handles upon which to leverage retrosynthetic planning. Because of this, synthetic routes toward the construction of less-oxygenated cyanthiwigins typically involve the installation and subsequent removal of superfluous moieties, an often cumbersome and inefficient method due to the introduction of nonessential functionality.
Biological Activity
The cyanthiwigin natural products boast a wide range of biological activities. The larger class of cyathanes in general possess a diverse set of bioactive properties, including antimicrobial and antineoplastic activity, as well as κ-opioid receptor agonism.[4] Most notably, some members of the cyathane natural products possess the capacity to stimulate the synthesis of nerve growth factor (NGF), a quality that implicates their potential application as therapeutic agents for neurodegenerative diseases and spinal injuries.[5] In addition, cyanthiwigin C has shown cytotoxic activity against both A549 human lung cancer cells (IC50 = 4.0 mg/mL) and P-388 human leukemia cells (IC50 = 11.2 mg/mL).[6] Cyanthiwigin F (6) has displayed cytotoxic activity against human primary tumor cells (IC50 = 3.1 mg/mL).[2] Unfortunately, exhaustive investigation of the entire family of cyanthiwigin molecules has been impeded by a lack of sufficient material with which to perform the required biological assays. As such, synthetic preparation of these natural products has become an appealing goal.
Previous Synthetic Efforts
Though the larger class of cyathane diterpenoid natural products has inspired numerous total syntheses to date,[3] the specific cyanthiwigin molecules have remained comparatively less explored. The first preparation of any cyanthiwigin natural product was the total synthesis of cyanthiwigin U (21) reported by Phillips and coworkers in 2005.[7] This elegant synthetic work boasted a ring opening, ring closing cross metathesis reaction to efficiently construct the completed tricyclic cyathane core. After the completion of cyanthiwigin U (21), Phillips was able to extend his preparative route in order to arrive at cyanthiwigin W (23) and cyanthiwigin Z (26).[8] In 2006, Reddy and coworkers were the first to disclose the preparation of the natural product cyanthiwigin AC (2 9 ), wherein they employed a unique deconjugative spiro-bis-alkylation strategy.[9]
Retrosynthetic Analysis
Because the members of the cyanthiwigin family of molecules differ from one another primarily in terms of oxygenation, we hypothesized that a synthetic route capable of rapidly preparing the carbocyclic core would provide simultaneous access to many of these marine natural products. Thus, our approach to the cyathane molecule cyanthiwigin F (6) was developed with a specific focus upon quickly constructing the tricyclic cyathane skeleton. In keeping with this goal, our initial retrosynthetic maneuver envisioned disconnection of either the five-membered A-ring or the seven-membered C-ring to lead back to either bicycle 31 or diketone 32, respectively (Scheme 1). In either case, further simplification was anticipated via retrosynthetic opening of the remaining peripheral ring, an operation that would be addressed in the forward sense via ring-closing metathesis. If the seven-membered C-ring were to be closed first, this strategy would lead to tetraolefin 33. Initial closure of the five-membered A-ring would invoke triolefin precursor 34. Regardless of the route employed, we expected that either intermediate 33 or 34 would be accessible via enantioenriched diketone 35.
Scheme 1.

Retrosynthetic analysis of cyanthiwigin F
In order to target diketone 35, we envisioned the use of enantioselective alkylation technology that had been previously developed in our lab.[10] At the outset of our synthetic efforts toward the cyanthiwigin natural products, this stereoselective methodology had already proven quite reliable for the formation of α-quaternary cyclohexanone products. We predicted that by implementing this reaction on an appropriately designed substrate, that it would be possible to forge two carbon-carbon bonds with enantiocontrol, thus providing rapid access to the critical diketone 35 in a single synthetic procedure. This double stereoselective decarboxylative alkylation reaction was anticipated to employ bis(β-ketoester) 36 as the crucial substrate, and in a forward sense, was expected to set both of the necessary all-carbon quaternary stereocenters of the natural product. Fortuitously, compounds similar to bis(β-ketoester) 36 have been known in the literature for nearly a century. As such we were confident that this material could be prepared from diallyl succinate (37) via an initial Claisen condensation and a subsequent Dieckmann cyclization.[11]
Results and Discussion
Forward Synthetic Efforts
The following sections describe the various reactions, routes, and experiments explored in order to synthetically prepare the cyanthiwigin marine diterpene compounds.[12]
Double Asymmetric Catalytic Alkylation
Studies toward the total synthesis of cyanthiwigin F commenced with the Fischer esterification of succinic acid (38) with allyl alcohol to afford diallylsucciniate (37, Scheme 2A). Exposure of diallyl succinate to a solution of allyl alkoxide in refluxing toluene initiated the desired Claisen condensation, a transformation immediately followed by subsequent Dieckmann cyclization to generate cyclohexadione product 39 exclusively as its bis-enol tautomer.[11] Thereafter, double methylation of bis-ester 39 under standard conditions provided access to bis(β-ketoester) 36 as a 1 : 1 mixture of racemic and meso diastereomers. Combination and optimization of the steps in this reaction sequence eventually facilitated the direct preparation of bis(β-ketoester) 36 from diallyl succinate (37). In the event, addition of diallyl succinate (37) to a suspension of sodium hydride in THF at room temperature, followed by subsequent quenching with methyl iodide, allowed for the generation of bis(β-ketoester) 36 under lower temperature conditions in a single step (Scheme 2B).[13] While the yield of the one step procedure is nominally lower than that of the two step process, the ease of operation and facile scalability of the more direct route outweigh these minimal losses. Interestingly, the diastereomers of bis(β-ketoester) 36 were found to be separable, with each possessing distinct physical properties. While the more polar of the two diastereomers of bis(β-ketoester) 36 was always observed to be a viscous oil, the less polar diastereomer was isolated as a fluffy white solid. With cyclohexadione 36 in hand, we were poised to address the double stereoselective decarboxylative alkylation reaction.
Scheme 2.
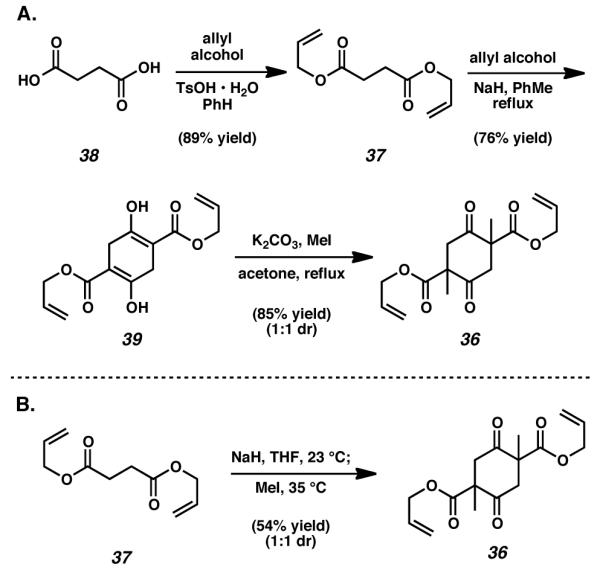
Preparation of the bis(β-ketoester) substrate for double allylation
Previous to these efforts, our group had developed a powerful suite of enantioselective decarboxylative catalytic alkylation reactions. Using this technology, it is possible to access enantioenriched cyclohexanone products bearing all-carbon quaternary stereocenters at the ketone α-position, starting from substrates containing an allyl enol carbonate, silyl enol ether, or β-ketoester moiety.[10] Having attained bis(β-ketoester) 36, we anticipated that exposure of this material to the conditions of our palladium-catalyzed alkylation would result in the formation of two independent C–C bonds, thus forging the all-carbon quaternary stereocenters corresponding to positions C(6) and C(9) of the cyanthiwigin core.
While stereoselective transformations to set more than one stereocenter have been reported in the literature prior to our efforts, the implementation of a double catalytic, stereoselective, C–C bond-forming reaction in the context of complex total synthesis has gone relatively unexplored.[14] Nevertheless, these types of transformations are both efficient and direct, as they set multiple stereocenters with a single catalytic species, and are therefore increasingly desirable for the rapid synthetic preparation of complex natural products.
Despite the potential benefits of double asymmetric transformations, it should be emphasized that the starting material (36) for our envisioned reaction was attained as a 1 : 1 blend of racemic and meso diastereomers. Exposing such a stereoisomeric mixture to an enantiopure catalyst is typically ill-advised, as the presence of pre-existing stereocenters in the substrate could interfere with inherent catalyst selectivity and afford reduced quantities of the desired product.[15] Indeed, the potential development of mismatched catalyst-substrate interactions, which would deleteriously impact yield and selectivity of the reaction, was a major concern. We were additionally mindful of the possibility for the reaction to proceed via an undesired kinetic resolution.[16]
Subjecting a diastereomeric mixture of bis(β-ketoesters) to the conditions of the stereoselective decarboxylative alkylation was a maneuver with many prospective outcomes. In the event of diastereomeric interference between the substrate and catalyst, the potential existed for incomplete allylation or impeded decarboxylation. Even in the case of complete transformation of bis(β-ketoester) 3 6 into the desired diketone (3 5 ), the stereochemical course of the reaction was difficult to predict. The diastereomeric mixture containing three stereoisomers of starting material could possibly traverse any of 16 distinct stereodefined pathways to give any of three potential product stereoisomers (Scheme 3).
Scheme 3.

Pathways of the double catalytic enantioselective alkylation
In order for the reaction to afford the desired diketone product with acceptable yield and selectivity, the catalyst employed had to meet several stringent requirements. First, initial decarboxylation of each stereoisomer of starting material ((R,R)-, meso-(R,S)-, or (S,S)-36) to give either intermediate ketone enolate ((R)- or (S)-40) would need to proceed at roughly equivalent rates regardless of configuration. If a large disparity in the rate of decarboxylation existed between different stereoisomers of starting bis(β-ketoester) 36, undesired kinetic resolution would influence the downstream stereoselective bond formation. The same requirement would be necessary for the subsequent decarboxylation of intermediates 41 to yield the transient enolates (R)- and (S)-42, and as such, all stereoisomers of 41 would need to react at equal rates. The second requirement envisioned was that all bond-forming reactions would occur under complete catalyst stereocontrol. This would dictate that any pre-existing or intermediately formed stereocenters present in any isomer of bis(β-ketoester) 36 or intermediate 41 should have no impact upon the selectivity of the allylation event. Third, the catalyst control in these situations would be required to be highly selective, so as to preferentially guide all of the possible ketone enolate stereoisomers ((S)- and (R)-40, (S)- and (R)-42) toward the single desired, enantioenriched product (3 5 ) .
Despite initial uncertainty concerning the course of this reaction, we reasoned that the stereoablative nature of the stereoselective decarboxylative alkylation methodology would minimize any undesired mismatched interactions.[17] This fact, combined with the relatively distant relationship between the two reactive centers in 36, gave us confidence in the success of our double alkylation approach. Therefore, we subjected a 1 : 1 diastereomeric mixture of racemic and meso-36 to a solution of Pd(dmdba)2 palladium(0) and (S)-tert-butyl phosphinooxazoline (t-BuPHOX, 43) in diethyl ether (Scheme 4).[18] To our delight, this reaction proceeded smoothly at 25 °C to give the desired, enantioenriched diketone (R,R)-35 in 64% yield and 99% ee, along with 14% of the meso diastereomer (4.4 : 1 diastereomeric ratio). This critical reaction established both of the all-carbon quaternary stereocenters necessary for completion of the cyanthiwigin molecules at an early stage of the synthesis. Creating these nested and difficult points of chirality well in advance was important to the versatility and flexibility of our route.
Scheme 4.
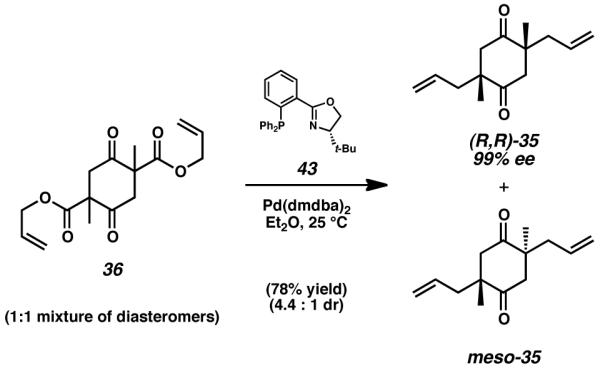
Double asymmetric decarboxylative catalytic allylation of 36
When considering the excellent enantioselectivity observed in the double alkylation reaction, the modest diastereoselectivity attained from this transformation was initially perplexing. Investigation of literature pertinent to double stereoselective transformations eventually revealed that the high levels of ee observed in diketone product (R,R)-35 come at the expense of a reduced diastereomeric ratio.[19],[20] When only a single undesired allylation event occurs while traversing the possible mechanistic pathways detailed in Scheme 3, an additional molecule of the undesired meso diastereomer is produced (meso-(R,S)-35) regardless of which alkylation occurs against preference. While this unwanted material negatively impacts the diastereomeric ratio of the isolated diketone, it nevertheless has no influence upon the ee of the desired product. In order to adversely affect the ee of cyclohexadione (R,R)-35, an individual molecule of bis(β-ketoester) (R,R)-36, meso-(R,S)-36, or (S,S)-36 must undergo two disfavored allylation events in sequence to afford product (S,S)-35. Because the likelihood of two allylation errors impacting a single substrate is very low in the presence of reasonable catalyst control, the total yield of the unwanted (S,S)-35 stereoisomer is negligible, and thus the product ee is excellent. However, due to the much higher likelihood of a single alkylation event yielding the undesired configuration, the amount of diketone meso-(R,S)-35 afforded through this reaction is larger than anticipated. In effect, generation of the meso diastereomer serves as a buffer against accumulation of undesired stereoisomer (S,S)-35, allowing this reaction to sacrifice some small measure of diastereomeric ratio in favor of exceptionally high levels of enantioselectivity. This phenomenon (sometimes referred to as the “Horeau Principle”), was first observed and rationalized by Langenbeck in 1936, and was later elaborated into more thorough mathematical representations by Horeau, Kagan, and Rautenstrauch.[19],[20]
Diketone Desymmetrization and Elaboration
With the successful generation of enantioenriched diketone (R,R)-35 attained, our efforts were thereafter focused on elaboration of this material via construction of the peripheral A- and C-rings of the cyathane tricycle.
At this juncture of the synthesis, advancement of diketone 35 required differentiation between the functional groups of this C2 symmetric substrate.[21] Initial desymmetrizing efforts were attempted via careful addition of various Grignard reagents to cyclohexadione 35 in the hopes of executing a single nucleophilic addition to either ketone moiety. Disappointingly, these experiments ultimately proved unsuccessful (Scheme 5). The steric encumbrance imposed by the α-quaternary stereocenters of diketone 35 very likely impede approach of any incoming nucleophile toward either of the carbonyl carbons, and thus renders this cyclohexadione intransigent to 1,2-addition conditions. An alternative desymmetrization strategy investigated involved the attempted mono-functionalization of the pendent allyl side chains of substrate 35. Regrettably, this approach also proved ineffective. In cases where dihydroxylation or epoxidation conditions were employed, only low yields were ever obtained, and in every case the sparing material isolated was a mixture of mono- and di-functionalized products.
Scheme 5.
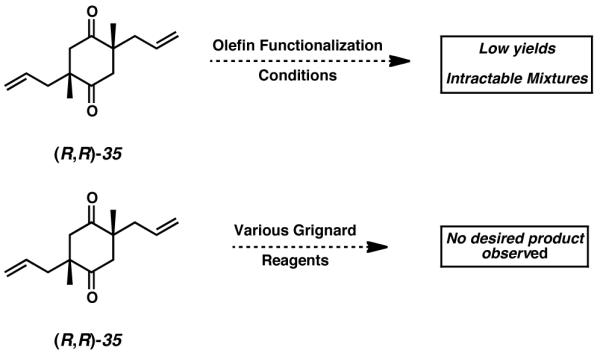
Attempts toward diketone functionalization
Greater selectivity and reactivity was ultimately achieved when diketone 35 was subjected to the conditions of enol triflate formation (Scheme 6A). Slow addition of diketone 35 to a solution of potassium bis(trimethylsilyl)amide in tetrahydrofuran allowed generation of a monoanionic ketone enolate. This intermediate was thereafter trapped via exposure to a solution of phenyl bis(trifluoromethane)sulfonimide to afford cyclohexanone 44 in reasonable yield. With this newly desymmetrized material in hand, we attempted to leverage the installed triflate to introduce functionality that would enable construction of a bicyclic structure.
Scheme 6.
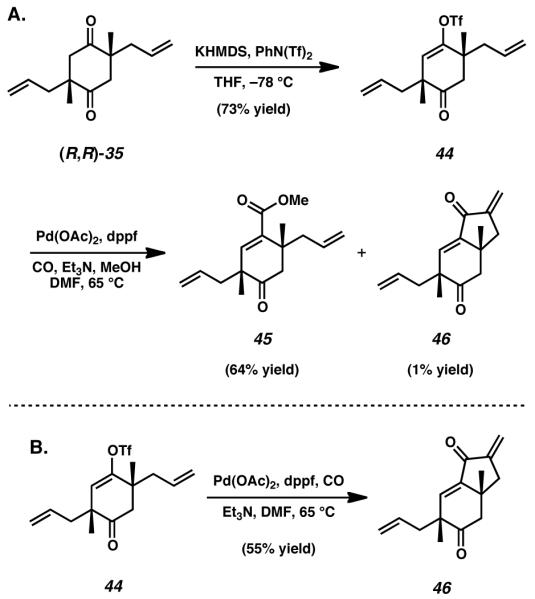
(A) Triflate formation and carboxylative attempts toward diketone elaboration, (B) An unanticipated carbonylative Heck reaction
By submitting enol triflate 44 to the conditions of a palladium-catalyzed carboxylation reaction in the presence of methanol, conjugated ester 45 was obtained as the major product. Unfortunately, purification of this material proved difficult, and isolates of enoate 45 were regularly contaminated with trace amounts of a strongly UV-active impurity. Thorough and careful investigation of this contaminant eventually revealed its identity as dienone 46, an unexpected yet intriguing byproduct of this carbonylative methodology. We suspected bicycle 46 to be the result of an intramolecular Heck-type reaction, wherein initial oxidative addition of palladium into the enol triflate bond was followed by subsequent carbon monoxide insertion and eventual olefin insertion into the pendant arm of 44. This hypothesis was later strengthened when the methoxycarbonylation reaction was repeated in the absence of the nucleophilic cosolvent (Scheme 6B). Without methanol, these conditions afforded exclusively the carbonylative Heck product 46.[22]
Interestingly, the structure of dienone product 46 suggests that oxidative addition and carbon monoxide incorporation occur before olefin complexation and insertion. While this phenomenon is known, typically in these reactions carbonylation occurs as the final step of the transformation, taking place only after olefin insertion.[22] This observed reversal in expected reactivity is likely due to the difficulty of olefin insertion directly following oxidative addition, a process that would require cyclobutane formation. The relatively greater ease of cyclopentanone formation provides a reasonable explanation for pre-emptive carbon monoxide incorporation. Regrettably, further elaboration of either enoate 45 or dienone 46 proved unproductive in our hands, and so alternative methods of functionalizing triflate 44 were sought.
In order to better gauge the reactivity of enol triflate 44, we executed a number of cross-coupling reactions to test the viability of sp2-sp, s p2-sp2, and s p2-sp3 hybridized carbon-carbon bond construction. Attempts at Sonogashira coupling of ketone 44 and trimethylsilyl acetylene smoothly provided access to enyne 47 in high yield (Scheme 7).[23] Similarly, copper-accelerated Stille reaction of triflate 44 with enol stannane 48 proved to be very effective.[24] After acidic workup of this sp2 -sp2 coupling reaction, enone 49 was obtained in reasonable yield.
Scheme 7.
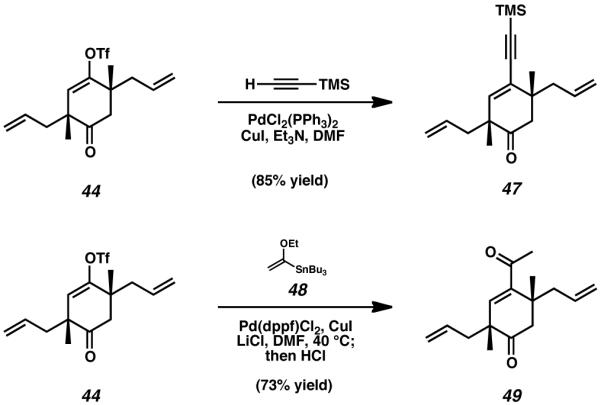
Palladium-catalyzed cross-coupling reactions of triflate 44.
Encouraged by the successes of the preliminary Stille coupling, we repeated this reaction while employing the more complicated enol stannane 50 in place of coupling partner 48 (Scheme 8).[25] This transformation progressed easily to generate cyclohexanone 51. However, purification and manipulation of this material was difficult, due to the rapid decomposition of the enol ether upon contact with silica gel or prolonged exposure to atmospheric conditions, both of which resulted in hydrolysis to reveal the latent ketone moiety. In order to circumvent issues of instability experienced with this intermediate, enol ether 51 was immediately exposed to the Grubbs/Hoveyda generation II catalyst (52) in order to execute ring-closing metathesis, and this process was subsequently followed by acidic workup. This synthetic procedure successfully closed the seven-membered C-ring of the cyathane tricycle, ultimately generating bicyclic ketone 53. While our efforts were bolstered by the successful formation of a [6,7]-bicyclic intermediate, this material nevertheless provided us with unanticipated difficulty. Formation of bicycle 53 was always accompanied by an undesired shift of the anticipated C(12)–C(13) olefin into conjugation with the newly revealed C(10) ketone.[26] If enone 53 were to be used to pursue the synthesis of the cyanthiwigin molecules, this new development would necessitate isomerization of the C(11)–C(12) olefin back into the C(12)–C(13) position, a task we regarded to be nontrivial. Furthermore, while the oxygenation present at C(10) was the result of functionality necessary for the stability of stannane 50, this newly formed ketone was superfluous to the structure of the completed natural product. Thus, removal of this moiety would be required at some later stage if enone 53 were to serve as a viable synthetic intermediate. Because of these difficulties, we opted not to pursue the further elaboration of dienone 53. Instead, we chose to investigate alternative cross coupling-conditions for triflate 44.
Scheme 8.
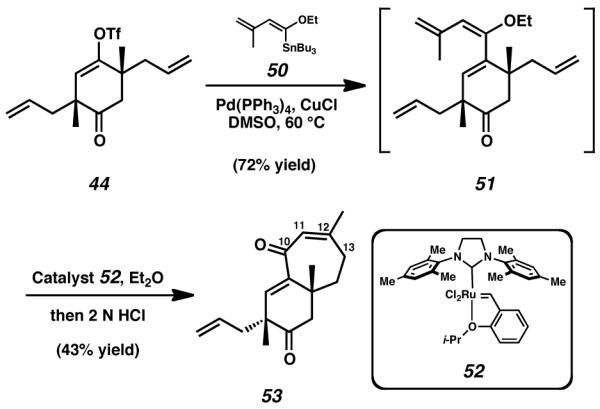
Formation, ring-closing metathesis, and acidic hydrolysis of the enol ether species 51
A reinvestigation of triflate 44 revealed the viability of a direct sp2 -sp3 bond formation via a Negishi cross-coupling procedure. Zinc dust was first treated with 1,2-dibromoethane and trimethylsilyl chloride, and to this activated metal was added alkyl iodide 54. After generation of the alkyl zinc species, a THF solution containing a palladium(0) catalyst and triflate 44 was introduced to the mixture, thus initiating Negishi cross coupling of the two fragments (Scheme 9).[27] After appropriate workup and purification, tetraolefin 33 was isolated as the sole product of this reaction. We were excited to find that ring-closing metathesis of cyclohexanone 33 to form the seven-membered C-ring furnished bicyclic product 31, a structure containing two of the three rings of the cyathane skeleton. Notably, we found that this ring-forming process was both faster and higher-yielding when modified Grubbs–Hoveyda catalyst 55 was employed in place of the Grubbs/Hoveyda second-generation catalyst 52.[28]
Scheme 9.
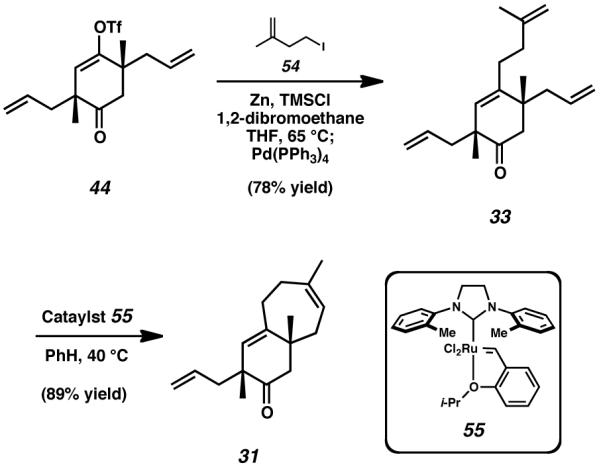
Negishi cross-coupling for the formation of an sp2-sp3 carbon-carbon bond
We anticipated that further advancement of this material toward the cyanthiwigin natural products would undoubtedly require selective functionalization of the remaining allyl side chain in the presence of the C(12)–C(13) olefin. For this reason, we turned our attention toward the possibility of using metathesis reactivity for the elaboration of the remaining terminal olefin. Cross-metathesis of vinyl boronate species 56 with newly attained bicycle 31 proved fruitful, and upon exposure to an oxidative workup involving aqueous sodium perborate, this process generated aldehyde 57 as the major product (Scheme 10A).[29] With the success of the ring-closing and cross-metathesis processes confirmed as independent reactions, we hypothesized that both transformations might be accomplished concurrently via the use of the same catalyst.
Scheme 10.
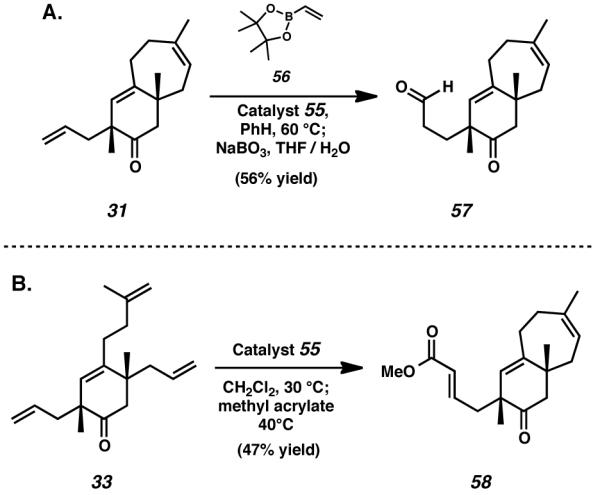
Functionalization of the C(2)–C(3) olefin via cross-metathesis
In the event, the addition of methyl acrylate to the conditions used for the ring-closing metathesis of tetraolefin 33 executed both formation of the seven-membered cyathane C-ring and functionalization of the terminal allyl moiety (Scheme 10B). By employing this reaction, monocyclic starting material 33 was smoothly transformed into bicyclic enoate 58 in a single synthetic operation. Though ester 58 was not amenable to further desired transformation, the technique elucidated by its formation nevertheless aided considerably in our synthetic efforts. By subjecting tetraolefin 33 to the conditions of concurrent ring-closing and cross-metathesis in the presence of vinyl boronate 56, and executing a subsequent oxidative workup with sodium perborate, it was possible to rapidly prepare bicyclic aldehyde 57 (Scheme 11).
Scheme 11.
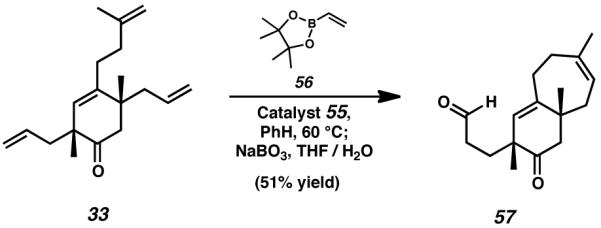
Ring-closing and cross-metathesis reaction to generate the bicyclic aldehyde speices 57
Tricycle Formation via Radical Cyclization
At this stage in the synthesis, the synthetic challenges remaining to be addressed included the finalization of the tricyclic cyathane core via installation of the five-membered A-ring, as well as the establishment of both the C(4) and C(5) stereocenters. We hypothesized that all of these pending goals might be accomplished in tandem through the use of a radical cyclization reaction. It was envisioned that the formation of a high-energy acyl radical species via hydrogen atom abstraction from aldehyde 57 would encourage thermodynamically-controlled carbon-carbon bond formation with the C(4)–C(5) olefin. To test this hypothesis, we turned our attention toward methods for the reliable production of acyl radical intermediates from aldehydes.[30]
Preliminary attempts to generate an acyl radical for the purpose of intramolecular cyclization were unfortunately unsuccessful. Subjecting bicyclic aldehyde 57 to tributylstannane or triphenylstannane did not yield the desired product. Both radical propagators were employed in combination with either azobisisobutyronitrile (AIBN) or 2,2′-azobis(4-methoxy-2,4-dimethyl valeronitrile) (V-70) initiators, yet in each case only unreacted substrate 57 or nonspecific decomposition were observed (Scheme 12).[31],[32]
Scheme 12.
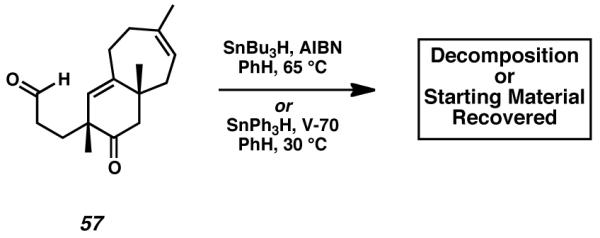
Initial attempts at radical cyclization for A-ring formation
In light of the failure of stannane reagents to furnish the targeted tricyclic product, we sought alternative conditions for the reliable formation of acyl radical species. After a thorough investigation of the literature, we became familiarized with aldehyde-olefin cyclization methodology developed by Tomioka et al. in 2005.[33] By implementing a bulky thiol radical propagator and an appropriate initiator, Tomioka was able to achieve the cyclization of aldehyde functional groups onto olefins to form cyclic ketone products. By employing this reaction, it was demonstrated that in the presence of tert-dodecanethiol and AIBN, linear aldehyde 59 could be cyclized onto an isolated olefin moiety to be smoothly converted into cyclopentanone 60 (Scheme 13). Similarly, tert-dodecanethiol and 1,1′-azobis(cyclohexane-1-carbonitrile) (V-40 initiator) enabled the conversion of aldehyde 61 into cyclohexanone 62.
Scheme 13.
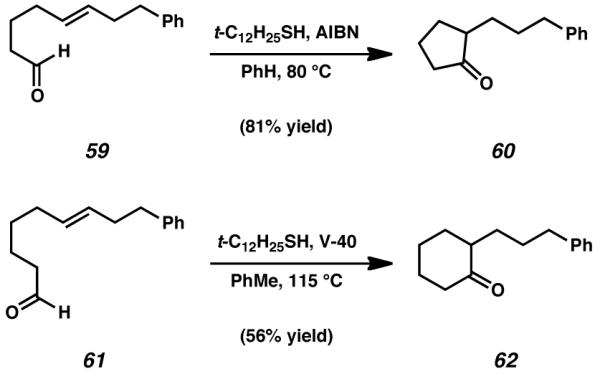
Radical cyclization conditions developed by Tomioka and coworkers[33]
Because our work required the cyclization of an aldehyde onto an unconjugated olefin, we attempted to subject bicyclic aldehyde 57 to Tomioka’s methodology with the intention of forming the required cyathane A-ring. Fortunately, employing tert-dodecanethiol and V-40 initiator in the radical cyclization of 57 forged the desired cyclopentanone and provided access to fully formed cyathane tricycle 63 (Scheme 14A). Minor optimization of this reaction was possible under lower temperature conditions, wherein using tert-butylthiol as propagator and AIBN as initiator afforded slightly increased yield of the desired product (Scheme 14B). Regardless of the exact reagents used, both reactions afforded 63 as a single diastereomer.
Scheme 14.
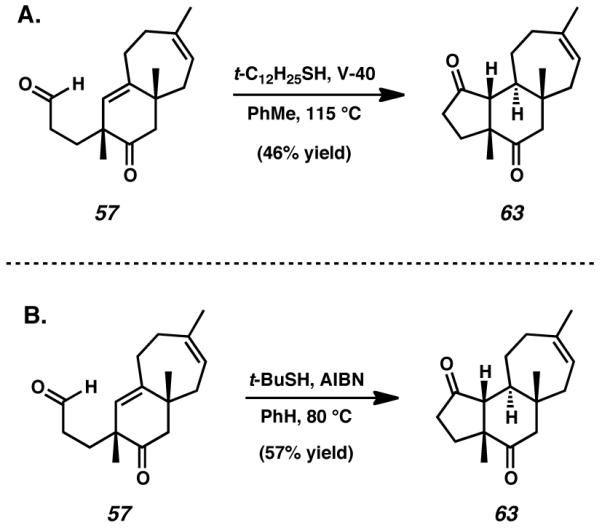
Application of Tomioka’s methodology to A-ring formation
It is possible that the diastereoselectivity observed in this cyclopentanone-forming reaction is a consequence of thermodynamic control. After initial hydrogen atom abstraction from aldehyde 57, acyl radical species 64 is formed (Scheme 15). Due to steric and conformational constraints, this radical species has limited access to the C(4)–C(5) olefin. Indeed, acyl radical approach and carbon-carbon bond formation may only occur from the bottom face of the bicyclic system as drawn in Scheme 15. Construction of this bond in line with such a trajectory establishes the desired stereochemistry at C(4), and additionally generates a rapidly equilibrating tertiary radical at C(5) (65). Further hydrogen atom abstraction from tert-butyl thiol to quench the radical found in intermediate 65 proceeds under thermodynamic control, and this affords the more stable trans-oriented [6,7] ring fusion in preference to the cis-fused alternative. Ultimately, these factors ensure that tricycle 63 is furnished as the sole stereoisomeric product of the reaction. The formation of the five-membered A-ring to construct tricyclic diketone 63 marked the completion of the cyathane core skeleton.
Scheme 15.
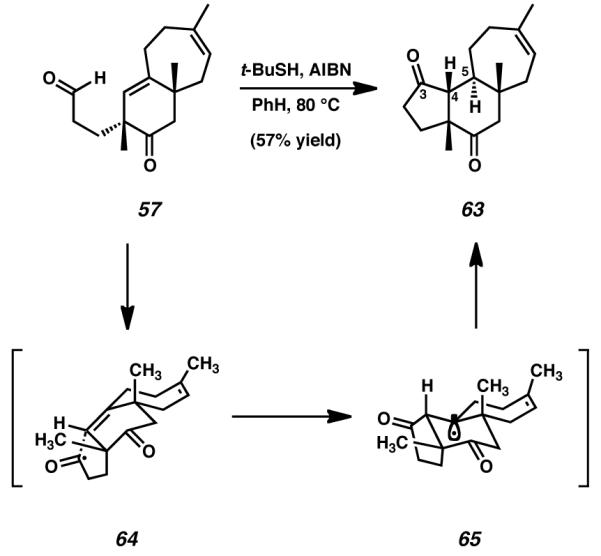
Mechanistic hypothesis and stereochemical rationale for the radical cyclization of bicyclic aldehyde 57
Attaining this material was of considerable significance to our synthetic efforts, not only because of its proximity to cyanthiwigin F (6), but also because we envisioned that this tricyclic diketone 63 could serve as a platform from which to access other cyanthiwigin natural products. Fortunately, solid crystals of this critical intermediate were amenable to X-ray analysis (Figure 2). The data collected from X-ray crystallography on diketone 63 revealed not only the stereochemistry set by the radical cyclization reaction, but also confirmed the relative stereochemistry established by the initial double alkylation reaction as well.
Figure 2.
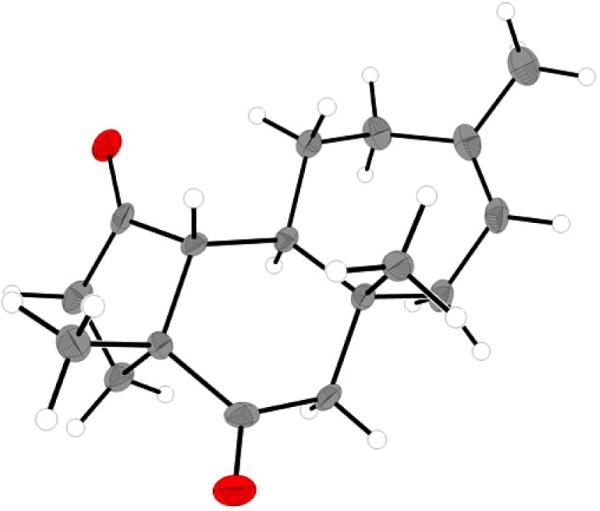
X-ray crystal structure of tricycle 63
Completion of the Cyanthiwigin Natural Products
With tricyclic diketone 63 in hand, the final challenges remaining in the total synthesis of cyanthiwigin F were the installation of the C(3) isopropyl group and introduction of the C(2)–C(3) olefin. In order to address these requirements, we envisioned harnessing the reactivity of the newly installed C(3) ketone to establish a vinyl triflate suitable for transition metal-catalyzed cross-coupling reactions. In the event, selective deprotonation of diketone 63 with KHMDS and trapping with N - phenyl bis(trifluoromethane)sulfonimide produced vinyl triflate 66 in reasonable yield (Table 1). Having achieved the synthesis of tricycle 66, we predicted that isopropyl group installation could be accomplished via a Negishi cross-coupling process similar to the one used previously.[27] Regrettably, when these coupling conditions were employed with triflate 66 and 2-iodopropane, the only isolated material was reductive deoxygenation product 67 (Table 1, entry 1).
Table 1.
Transition metal cross-coupling reactions toward preparation of cyanthiwigin
| Entry | i-PrX | Metal Catalyst |
Temp (° C) |
Yield 6 (%) |
Yield 67 (%) |
Yield 66 (%) |
|---|---|---|---|---|---|---|
| 1 | i-PrZnI | Pd(PPh3)4 | 65 °C | 0 | ND | 0 |
| 2 | i-PrMgCl | CuI | −10 °C | 0 | 65 | 0 |
| 3 | i-PrLi | CuCN | −20 °C | 0 | 0 | 73 |
| 4 | i-PrMgCl | CuBr•DMS | −20 °C | 17 | 7 | 57 |
| 5 |
i-PrMgCl + CuCN |
- | −78 → 0 °C |
0 | 0 | 99 |
| 6 |
i-PrMgCl + CuBr•DMS |
- | −78 → −20 °C |
11 | 10 | 0 |
| 7 |
i-PrLi + CuCN |
- | −78 → −20 °C |
8 | 3 | 27 |
| 8 |
i-PrLi + CuCN |
- | −20 °C | 0 | 0 | 99 |
| 9 | i-PrMgCl | Ni(dppp)Cl2 | 35 °C | 0 | 0 | 99 |
| 10 | i-PrMgCl | Pd(PPh3)4 | −78 → 23 °C |
0 | 37 | 19 |
| 11 | i-PrMgCl | Pd(dppf)Cl2 | 35 °C | 0 | ND | 0 |
| 12 |
i-PrMgCl + CuCN |
Pd(dppf)Cl2 | –78 → 0 °C |
41 | 23 | 0 |
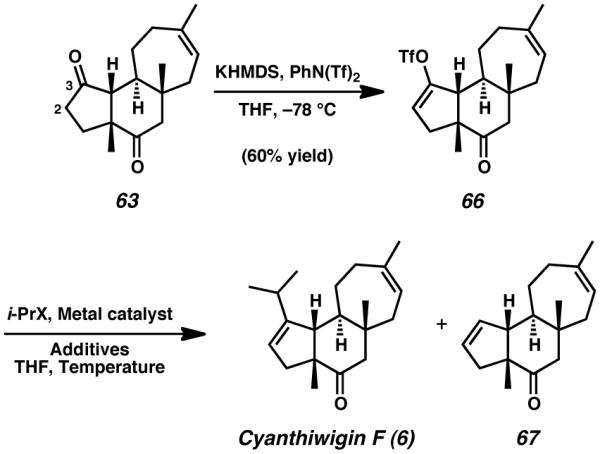
Because palladium-catalyzed cross-coupling of 66 and 2-iodopropane was met with difficulty, alternative copper-catalyzed methods to install the required three-carbon fragment were investigated. Preliminary experiments involved direct addition of isopropyl magnesium chloride to suspensions of triflate 66 and catalytic amounts of either copper iodide or copper cyanide. Disappointingly, these reactions were similarly ineffective, affording either the reduced tricycle 67, recovered triflate 66, and only sparing yields of cyanthwigin F 6 (Entries 2–4).[34] Use of copper bromide dimethyl sulfide complex for this direct-addition technique did yield small quantities of the natural product, but these sparing amounts of the desired compound were contaminated with larger amounts of the reduction product 67. To rectify this issue, we endeavored to approach the coupling via the use of stoichiometric quantities of pre-generated isopropyl-cuprate reagents, using a variety of different copper sources (Entries 5–8). In all cases, we observed either low reactivity or a preference for reductive deoxygenation.
Additional experiments into isopropyl cross-coupling involved employing a number of nickel and palladium catalysts in a series of Kumada-type reactions (Entries 9–11),[35] but the results overwhelmingly favored reduced product 67 in those instances where reactivity was observed. Finally, we discovered that introduction of a pre-generated, higher-order isopropyl cyanocuprate to a solution of triflate 66 and dichloro(1,1′-bis(diphenylphosphino)ferrocene)palladium(II) gave a combined 65% yield of the natural product (6) and tricycle 67, in a 1.8 : 1 mixture favoring cyanthiwigin F (Entry 12).[36],[37]
In addition to being instrumental in the total synthesis of cyanthiwigin F, it was our hope that the completed cyathane core represented by tricycle 63 would prove useful in the synthesis of other diterpenes of this natural product family. Starting from diketone 63, deprotonation at the α-position of the C(3) ketone and trapping of the incipient enolate with allyl chloroformate provided access to enol carbonate 68. Thereafter, treatment of this material with a catalytic quantity of palladium(0) in acetonitrile provided enone 69 in high yield (Scheme 16).[38] Unsaturated ketone 69 afforded an excellent opportunity for direct introduction of the C(3) isopropyl group via 1,2-addition, and so was exposed to isopropyl lithium under Luche-type activation conditions. Cerium-mediated alkyl lithium reactivity proceeded with exclusive addition to C(3), generating tertiary alcohol 70 as a mixture of inconsequential diastereomers.
Scheme 16.
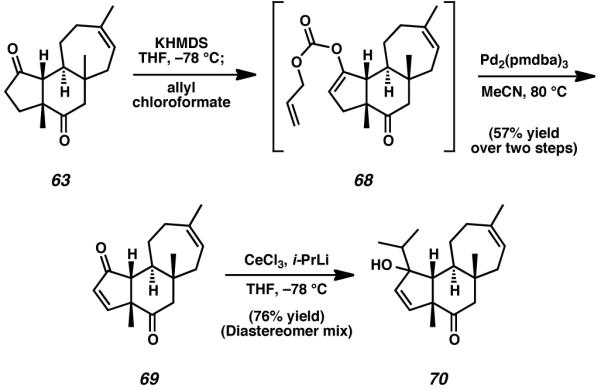
Advancement of tricycle 63 toward additional members of the cyanthiwigin nautral products
The mixture of alcohols, 70, was then subjected to PCC in dichloromethane, conditions anticipated to execute allylic oxidation with concomitant oxygen transposition. In the event, 70 was smoothly transformed into natural product 2, thus completing the total synthesis of cyanthiwigin B (Scheme 17).[39] We further envisioned that cyanthiwigin B (2) might be additionally advanced toward other members of this natural product family via selective carbonyl reduction at C(8).[40] Conditions reported to selectively reduce ketones in the presence of enones unfortunately provided exclusively over-reduction of both carbonyl moieties, but this difficulty was mitigated by immediate and selective reoxidation of the resulting material with manganese(IV) dioxide. This allylic oxidation process generated enone 71 as the sole product of reaction. Notably, tricycle 71 was found to be structurally identical to cyanthiwigin E, with the single exception of the configuration of the stereogenic alcohol present at C(8), which was determined to be epimeric to that found in the natural product. Nevertheless, 8-epi-cyanthiwigin E (71) served as an invaluable intermediate in the preparation of another cyanthiwigin compound. Treatment of enone 71 with Martin’s sulfurane in deuterated chloroform successfully eliminated the C(8) secondary alcohol to install the C(7)–C(8) olefin, thus finalizing the total synthesis of cyanthiwigin G (7).[41],[42]
Scheme 17.
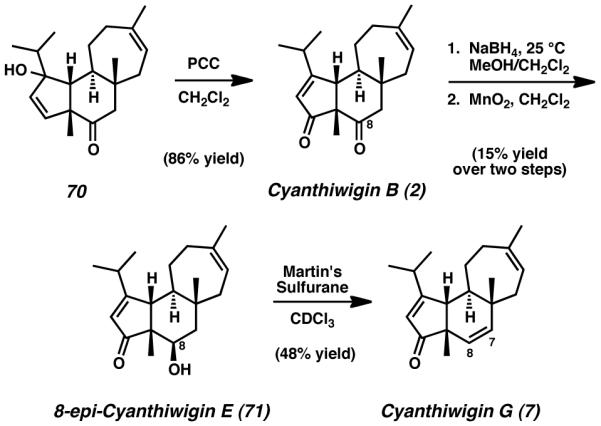
Completion of cyanthiwigings B and G
Conclusion
In summary, we have developed an efficient, versatile, and enantioselective route to the cyanthiwigin natural products. Our approach toward these molecules involves a rapid synthesis of the central six-membered B-ring, with a specific focus on early installation of both the C(6) and C(9) all-carbon quaternary stereocenters. The use of a double asymmetric decarboxylative catalytic alkylation reaction not only enables access to the critical enantioenriched cyclohexadione 35, but this methodology has additionally proven tolerant of a diastereomeric mixture of racemic and meso starting materials in the same catalytic transformation. Because of the ease with which stereoisomeric mixtures of precursor bis(β-ketoester) 36 can be prepared, this stereoablative approach expedites the early phases of our synthesis considerably. Our strategy also involves an efficient, single operation ring-closing and cross-metathesis reaction to generate a bicyclic aldehyde from a monocyclic tetraolefin. Combined with a powerful radical cyclization, these techniques furnish ready and rapid access to a versatile tricyclic intermediate representing the completed cyathane core (63). By leveraging this core compound as a branching point toward marine natural products, our group was able to expediently prepare multiple cyanthiwigin molecules. In particular, the total synthesis of cyanthiwigin F (6) was accomplished in nine total steps, seven of which form carbon-carbon bonds. Of the seven carbon-carbon bond-forming transformations in our synthetic sequence, four construct more than one carbon-carbon bond simultaneously. Additionally, the synthesis is highly efficient in terms of its use of redox reactions, as only minimal oxidative or reductive processes are employed. The flexibility and modularity of our synthetic route later accommodated further extrapolation of tricyclic intermediate 63 toward additional members of the cyanthiwigin family, thus facilitating the preparation of cyanthiwigins B (2) and G (7).
Experimental Section
Synthesis of diketone 35
A flame dried round bottom flask cooled under argon was charged with bis(3,5-dimethoxydibenzylideneacetone)palladium(0) (Pd(dmdba)2, 0.268 g, 0.330 mmol, 0.05 equiv) and (S)-t-BuPHOX (43) (0.140 g, 0.362 mmol, 0.055 equiv). The flask was purged under vacuum briefly, and then backfilled with argon. The solids were dissolved in Et2O (500 mL), and the resulting solution was stirred at 25 °C for 30 min. After precomplexation, neat 36 (2.00 g, 6.59 mmol, 1.00 equiv) was added to the reaction. The solution was stirred vigorously at 25 °C for 10 h (Note: continual stirring is necessary due to the apparent low solubility of Pd(dmdba)2 in Et2O.), after which time the solvent was removed in vacuo. The crude oil was purified over silica gel using 3% ethyl acetate in hexanes as eluent to afford 35 as a colorless oil (1.07 g, 78% yield, 4.4 : 1 dr, 99% ee): Rf = 0.7 (15:85 ethyl acetate/hexane); 1H NMR (300 MHz, CDCl3) δ 5.68 (dddd, J = 18.3, 10.2, 6.9, 6.9 Hz, 2H), 5.17–5.09 (comp. m, 3H), 5.07–5.04 (m, 1H), 2.82 (d, J = 14.7 Hz, 2H), 2.38 (d, J = 15 Hz, 2H), 2.34 (app ddt, J = 13.2, 6.9, 1.0 Hz, 2H), 2.09 (app ddt, J = 13.5, 7.8, 0.9 Hz, 2H), 1.10 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 212.8, 132.4, 120.0, 49.4, 48.4, 43.8, 24.3; IR (Neat film, NaCl) 3078, 2978, 1712, 1640, 1458, 1378, 1252, 1129, 1101, 998, 921 cm−1; HRMS (EI) m/z calc’d for C14H20O2 [M]+: 220.1463, found 220.1466; [α]25D −163.1 (c 0.52, CH2Cl2). Chiral GC assay (GTA column): 100 °C isothermal method over 90 min. Retention times: 67.7 min (Major enantiomer, C2 diastereomer, 81.7%), 74.1 min (Minor enantiomer, C2 diastereomer, 0.6%), 77.4 min (meso diastereomer, 17.6%). Achiral GC assay (DB-Wax column): 100 °C isotherm over 2.0 min, ramp 5 °C/min to 190 °C, then 190 °C isotherm for 10.0 min. Retention times: 18.5 min (C2 diastereomer, 81.0%), 18.7 min (meso diastereomer, 19.0%).
Supplementary Material
Acknowledgements
This publication is based on work supported by Award No. KUS-11-006-02, made by King Abdullah University of Science and Technology (KAUST). The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, Abbott, Boehringer Ingelheim, Merck, and Bristol-Myers Squibb, GlaxoSmithKline, Johnson and Johnson, Amgen, Merck Research Laboratories, Pfizer, Novartis, Roche, Abbott Laboratories, Boehringer-Ingelheim, AstraZeneca, and Caltech for financial support. We also wish to thank Dr. M. W. Day and Mr. L. M. Henling for X-ray crystallographic expertise, Dr. Andrew Harned, Dr. David White, Daniel Caspi, and J. T. Mohr for helpful discussions, and Professor Mark T. Hamann for authentic samples and spectra of Cyanthiwigins B, F, and G. Ruthenium olefin metathesis catalysts were generously donated by Materia.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- [1].Green D, Goldberg I, Stein Z, Ilan M, Kashman Y. Nat. Prod. Lett. 1992;1:193–199. [Google Scholar]
- [2].a) Peng J, Walsh K, Weedman V, Bergthold JD, Lynch J, Lieu KL, Braude IA, Kelly M, Hamann MT. Tetrahedron. 2002;58:7809–7819. [Google Scholar]; b) Peng J, Avery MA, Hamann MT. Org. Lett. 2003;5:4575–4578. doi: 10.1021/ol035592f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Peng J, Kasanah N, Stanley CE, Chadwick J, Fronczek M, Hamann MT. J. Nat. Prod. 2006;69:727–730. doi: 10.1021/np050197e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Wright DL, Whitehead CR. Org. Prep. Proced. Int. 2000;32:309–330. [Google Scholar]; b) Enquist JA, Jr., M B. Nat. Prod. Rep. 2009;26:661–680. doi: 10.1039/b811227b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Saito T, Aoki F, Hirai H, Inagaki T, Matsunaga Y, Sakakibara T, Sakemi S, Suzuki Y, Watanabe S, Suga O, Sujaku T, Smogowicz AA, Truesdell SJ, Wong JW, Nagahisa A, Kojima Y, Kojima N. J. Antibiot. (Tokyo) 2003;51:983–990. doi: 10.7164/antibiotics.51.983. [DOI] [PubMed] [Google Scholar]
- [5].a) Obara Y, Nakahata N, Kita T, Takaya Y, Kobayashi H, Hosoi S, Kiuchi S, Ohta T, Oshima Y, Ohizumi Y. Eur. J. Pharmacol. 1999;370:79–84. doi: 10.1016/s0014-2999(99)00077-1. [DOI] [PubMed] [Google Scholar]; b) Obara Y, Kobayashi H, Ohta T, Ohizumi Y, Nakahata N. Mol. Pharmacol. 2001;59:1287–1297. doi: 10.1124/mol.59.5.1287. [DOI] [PubMed] [Google Scholar]
- [6].Sennett SH, Pomponi SA, Wright AE. J. Nat. Prod. 1992;55:1421–1429. doi: 10.1021/np50088a006. [DOI] [PubMed] [Google Scholar]
- [7].Pfeiffer MWB, Phillips AJ. J. Am. Chem. Soc. 2005;127:5334–5335. doi: 10.1021/ja0509836. [DOI] [PubMed] [Google Scholar]
- [8].Pfeiffer MWB, Phillips AJ. Tetrahedron Lett. 2008;49:6860–6861. doi: 10.1016/j.tetlet.2008.09.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Reddy TJ, Bordeau G, Trimble L. Org. Lett. 2006;8:5585–5588. doi: 10.1021/ol062304h. [DOI] [PubMed] [Google Scholar]
- [10].a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. 2005;117:7084–7087. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2004;44:6924–6927. [Google Scholar]
- [11].Ebert H. Liebigs Ann. Chem. 1885;229:45–88. [Google Scholar]
- [12].Enquist JA, Jr., Stoltz BM. Nature. 2008;453:1228–1230. doi: 10.1038/nature07046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Conditions for this optimized procedure were adapted from a Dieckmann reaction previously published by our group in Organic Syntheses. See: Mohr JT, Krout MR, Stoltz BM. Org. Synth. 2009;86:194–211. doi: 10.15227/orgsyn.086.0194.
- [14].a) Masamune S, Choy W, Peterson J, Sita L. Angew. Chem. 1985;97:1–31. [Google Scholar]; Angew. Chem. Int. Ed. 1985;24:1–30. [Google Scholar]; b) Rieck H, Helmchen G. Angew. Chem. 1995;107:2881–2883. [Google Scholar]; Angew. Chem., Int. Ed. 1995;34:2687–2689. [Google Scholar]; c) Lebsack AD, Link JT, Overmann LE, Stearns BA. J. Am. Chem. Soc. 2002;124:9008–9009. doi: 10.1021/ja0267425. [DOI] [PubMed] [Google Scholar]; d) Kolodiazhnyi OI. Tetrahedron. 2003;59:5953–6018. [Google Scholar]; e) Welter C, Dahnz A, Brunner B, Streiff S, Dübon P, Helmchen G. Org. Lett. 2005;7:1239–1242. doi: 10.1021/ol047351t. [DOI] [PubMed] [Google Scholar]
- [15].Kagan H. Tetrahedron. 2001;57:2449–2468. [Google Scholar]
- [16].Eliel EL, Wilen SH. Stereochemistry of Organic Compounds. John Wiley & Sons, Inc.; New York: 1994. pp. 965–971. [Google Scholar]
- [17].Mohr JT, Ebner DC, Stoltz BM. Org. Biomol. Chem. 2007;5:3571–3576. doi: 10.1039/b711159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Helmchen G, Pfaltz A. Acc. Chem. Res. 2000;33:336–345. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]; b) Tani K, Behenna DC, McFadden RM, Stoltz BM. Org. Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- [19].Langenbeck W, Triem G. Z. Phys. Chem. A. 1936;117:401–409. [Google Scholar]
- [20].a) Vigneron JP, Dhaenens M, Horeau A. Tetrahedron. 1973;29:1055–1059. [Google Scholar]; b) Rautentrauch V. Bull. Soc. Chim. Fr. 1994;131:515–524. [Google Scholar]; c) Baba SE, Sartor K, Poulin J, Kagan H. Bull. Soc. Chim. Fr. 1994;131:525–533. [Google Scholar]
- [21].Poss CS, Schreiber SL. Acc. Chem. Res. 1994;27:9–17. [Google Scholar]
- [22].a) Negishi EI, Ma S, Amanfu J, Copéret C, Miller JA, Tour JM. J. Am. Chem. Soc. 1996;118:5919–5913. [Google Scholar]; b) Satoh T, Itaya T, Okuro K, Miura M, Nomura M. J. Org. Chem. 1996;60:7267–7271. [Google Scholar]; c) Matsuura T, Overmann LE, Poon DJ. J. Am. Chem. Soc. 1998;120:6500–6503. [Google Scholar]; d) Iwasawa N, Satoh H. J. Am. Chem. Soc. 1999;121:7951–7952. [Google Scholar]; e) Hayashi T, Tang J, Kato K. Org. Lett. 1999;1:1487–1489. [Google Scholar]; f) Artman GD, III, Weinreb SM. Org. Lett. 2003;5:1523–1526. doi: 10.1021/ol034314d. [DOI] [PubMed] [Google Scholar]; g) Wu X, Nilsson P, Larhed M. J. Org. Chem. 2005;70:346–349. doi: 10.1021/jo048375g. [DOI] [PubMed] [Google Scholar]; h) Wu XF, Neumann H, Beller M. Angew. Chem. 2010;122:5412–5426. [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:5284–5288. doi: 10.1002/anie.201002155. [DOI] [PubMed] [Google Scholar]; i) Bloome KS, Alexanian EJ. J. Am. Chem. Soc. 2010;132:12823–12825. doi: 10.1021/ja1053913. [DOI] [PubMed] [Google Scholar]
- [23].a) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975;16:4467–4470. [Google Scholar]; b) Eckhardt M, Brückner R, Suffert J. Tetrahedron Lett. 1995;36:5167–5170. [Google Scholar]
- [24].Han X, Stoltz BM, Corey EJ. J. Am. Chem. Soc. 1999;121:7600–7605. [Google Scholar]
- [25].Tivola PB, Deagostino A, Prandi C, Venturello P. J. Chem. Soc., Perkin Trans. 1. 2001:437–441. doi: 10.1039/b104449b. [DOI] [PubMed] [Google Scholar]
- [26].All attempts to directly isolate the bicyclic enol ether resulting from ring-closing metathesis of 51 only provided enone 53, presumably as the result of decomposition.
- [27].a) King AO, Okukado N, Negishi EI. J. Chem. Soc., Chem. Commun. 1977:683–684. [Google Scholar]; b) Taishi T, Takechi S, Mori S. Tetrahedron Lett. 1998;39:4347–4350. [Google Scholar]
- [28].Stewart IC, Ung T, Pletnev AA, Berlin J, Grubbs RH, Schrodi Y. Org. Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]
- [29].For development and synthetic application of the vinyl boronate cross-metathesis, See: Morrill C, Funk TW, Grubbs RH. Tetrahedron Lett. 2004;45:7733–7736. Morrill C, Grubbs RH. J. Org. Chem. 2003;68:6031–6034. doi: 10.1021/jo0345345. Njardarson JT, Biswas K, Danishefsky SJ. Chem. Commun. 2002:2759–2761. doi: 10.1039/b209941a.
- [30].a) Kharasch WH, Urry WH, Kuderna BM. J. Org. Chem. 1949;14:248–253. doi: 10.1021/jo01154a009. [DOI] [PubMed] [Google Scholar]; b) Starker LN, Cosulich DB, Smith JM., Jr. J. Am. Chem. Soc. 1955;77:2534–2536. [Google Scholar]; c) Stetter H, Kuhlmann H. Tetrahedron Lett. 1974;15:4505–4508. [Google Scholar]
-
[31].Faced with the difficulty of generating an appropriate acyl radical species, we postulated that formation of a ketyl radical would be more facile, as well as more reactive. See:
Yeh MCP, Wang FC, Tu JJ, Chang SC, Chou CC, Liao JW. Organometallics. 1998;17:5656–5662.
Molander GA, McKie JA. J. Org. Chem. 1992;57:3132–3139.
Molander GA, Kenny C. J. Org. Chem. 1991;56:1439–1445.
Toward this end, bicyclic aldehyde 57 was treated with a solution of samarium(II) iodide in the presence of hexamethylphosphoramide (HMPA), with the expectation that these conditions would afford an appropriate ketyl radical. Unfortunately, rather than undergo the radical-olefin cyclization process we envisaged, the ketyl radical instead engaged in an undesired intramolecular Pinacol-type coupling to yield [5-6-7]tricyclic system 72.
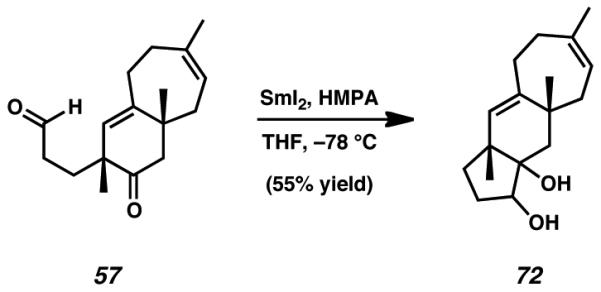 Diol 72. To a flame dried vial was added a THF solution of aldehyde 57 (20 mg, 0.077 mmol, 1.0 equiv, in 500 μL solvent). To this solution was added HMPA (25 μL), and the vial was cooled to −78 °C. Once this temperature had been reached, SmI2 (921 μL, 0.1 M in THF, 0.092 mmol, 1.2 equiv) was added dropwise to the reaction. After complete addition of a single portion of SmI2, the reaction had not reached completion. A second portion of SmI2 (921 μL, 0.1 M in THF, 0.092 mmol, 1.2 equiv) was added, and the reaction was allowed to reach 0 °C over 30 min. After this time had elapsed, the reaction was poured into a solution of brine (20 mL) that contained citric acid (770 mg). The phases were separated, and the aqueous layer was extracted with ethyl acetate (4 × 30 mL). Combined organic layers were washed with brine (40 mL), dried over MgSO4, filtered, and concentrated in vacuo. The resulting crude material was purified over silica using 10% → 12% → 15% ethyl acetate in hexanes as eluent. This afforded diol 72 as a colorless oil (11 mg, 55% yield). Selected characterization data is as follows: 1H NMR (500 MHz, CDCl3) δ 5.33 (app t, J = 6.8 Hz, 1H), 5.13 (s, 1H), 3.83 (app dt, J = 6.8, 4.3 Hz, 1H), 2.38 (d, J = 4.4 Hz, 1H), 2.35-2.33 (m, 1H), 2.31 (s, 1H), 2.24-2.16 (m, 1H), 2.12 (ddd, J = 14.5, 6.8, 0.8 Hz, 1H), 2.03 (app dt, J = 14.7, 5.2 Hz, 2H), 1.95 (dddd, J = 14.0, 8.9, 6.8, 5.3 Hz, 1H), 1.88 (dd, J = 14.5, 6.8 Hz, 1H), 1.74 (ddd, J = 12.7, 9.1, 5.3 Hz, 1H), 1.66 (s, 3H), 1.65-1.58 (m, 1H), 1.62 (d, J = 4.8 Hz, 1H), 1.47 (d, J = 14.1 Hz, 1H), 1.44 (ddd, J = 12.7, 8.9, 7.2 Hz, 1H), 1.15 (s, 3H), 1.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 142.9, 139.3, 131.5, 121.3, 79.5, 79.0, 46.4, 44.6, 40.1, 39.7, 38.4, 35.9, 30.5, 29.8, 27.7, 25.7, 23.7.
Diol 72. To a flame dried vial was added a THF solution of aldehyde 57 (20 mg, 0.077 mmol, 1.0 equiv, in 500 μL solvent). To this solution was added HMPA (25 μL), and the vial was cooled to −78 °C. Once this temperature had been reached, SmI2 (921 μL, 0.1 M in THF, 0.092 mmol, 1.2 equiv) was added dropwise to the reaction. After complete addition of a single portion of SmI2, the reaction had not reached completion. A second portion of SmI2 (921 μL, 0.1 M in THF, 0.092 mmol, 1.2 equiv) was added, and the reaction was allowed to reach 0 °C over 30 min. After this time had elapsed, the reaction was poured into a solution of brine (20 mL) that contained citric acid (770 mg). The phases were separated, and the aqueous layer was extracted with ethyl acetate (4 × 30 mL). Combined organic layers were washed with brine (40 mL), dried over MgSO4, filtered, and concentrated in vacuo. The resulting crude material was purified over silica using 10% → 12% → 15% ethyl acetate in hexanes as eluent. This afforded diol 72 as a colorless oil (11 mg, 55% yield). Selected characterization data is as follows: 1H NMR (500 MHz, CDCl3) δ 5.33 (app t, J = 6.8 Hz, 1H), 5.13 (s, 1H), 3.83 (app dt, J = 6.8, 4.3 Hz, 1H), 2.38 (d, J = 4.4 Hz, 1H), 2.35-2.33 (m, 1H), 2.31 (s, 1H), 2.24-2.16 (m, 1H), 2.12 (ddd, J = 14.5, 6.8, 0.8 Hz, 1H), 2.03 (app dt, J = 14.7, 5.2 Hz, 2H), 1.95 (dddd, J = 14.0, 8.9, 6.8, 5.3 Hz, 1H), 1.88 (dd, J = 14.5, 6.8 Hz, 1H), 1.74 (ddd, J = 12.7, 9.1, 5.3 Hz, 1H), 1.66 (s, 3H), 1.65-1.58 (m, 1H), 1.62 (d, J = 4.8 Hz, 1H), 1.47 (d, J = 14.1 Hz, 1H), 1.44 (ddd, J = 12.7, 8.9, 7.2 Hz, 1H), 1.15 (s, 3H), 1.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 142.9, 139.3, 131.5, 121.3, 79.5, 79.0, 46.4, 44.6, 40.1, 39.7, 38.4, 35.9, 30.5, 29.8, 27.7, 25.7, 23.7.
- [32].Namy JL, Souppe J, Kagan HB. Tetrahedron Lett. 1983;24:765–766. [Google Scholar]
- [33].Yoshikai K, Hayama T, Nishimura K, Yamada K, Tomioka K. J. Org. Chem. 2005;70:681–683. doi: 10.1021/jo048275a. [DOI] [PubMed] [Google Scholar]
- [34].Rawat DS, Gibbs RA. Org. Lett. 2002;4:3027–3030. doi: 10.1021/ol026176i. [DOI] [PubMed] [Google Scholar]
- [35].Tamao K, Sumitani K, Kiso Y, Zembayashi M, Fujioka H, Kodama SI, Nakajima I, Minato A, Kumada M. Bull. Chem. Soc. Jpn. 1976;49:1958–1969. [Google Scholar]
- [36].Although, to our knowledge, there have been no reports of cuprate-mediated palladium cross-coupling reactions, there nevertheless do exist reports of copper-accelerated Negishi reactions. See: Weichert A, Bauer M, Wirsig P. Synlett. 1996:473–474.
- [37].Products 6 and 67 were ultimately separated via reverse phase HPLC purification.
- [38].a) Shimizu I, Tsuji J. J. Am. Chem. Soc. 1982;104:5844–5846. [Google Scholar]; b) Shimizu I, Minami I, Tsuji J. Tetrahedron Lett. 1983;24:1797–1800. [Google Scholar]; c) Minami I, Takahashi K, Shimizu I, Kimura T, Tsuji J. Tetrahedron. 1986;42:2971–2977. [Google Scholar]
- [39].The final two steps of our synthetic route toward cyanthiwigin B were adapted from Phillips’ synthesis of cyanthiwigin U. See: Pfeiffer MWB, Phillips AJ. J. Am. Chem. Soc. 2005;127:5334–5335. doi: 10.1021/ja0509836.
- [40].Ward DE, Rhee CK. Can. J. Chem. 1989;67:1206–1211. [Google Scholar]
- [41].a) Arhart RJ, Martin JC. J. Am. Chem. Soc. 1972;94:5003–5010. [Google Scholar]; b) Martin JC, Franz JA, Arhart RJ. J. Am. Chem. Soc. 1974;96:4604–4611. [Google Scholar]
- [42].An alternative approach toward cyanthiwigin G involving formation of an enol triflate species from cyanthiwigin B (2) via deprotonation at C(7) and trapping with N-phenyl bis(trifluoromethane)sulfonimide, failed to achieve the desired product. Under these conditions, only γ-deprotonation at C(18) was observed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.