

Background: P14ARF is capable of triggering p53/Bax-independent mitochondrial apoptosis.
Results: Expression of p14ARF in p53/Bax-deficient cells causes the deprivation of anti-apoptotic Mcl-1 and Bcl-xL.
Conclusion: P53/Bax-independent apoptosis induced by forced expression of p14ARF is facilitated by de-repression of Bak.
Significance: Derepression of Bak following deprivation of Mcl-1 and/or Bcl-xL by p14ARF may be sufficient to induce apoptosis in the absence of BH3-only protein activation.
Keywords: Apoptosis, ARF, Bax, Bcl-2 Family Proteins, Mitochondrial Apoptosis, BH3-only Proteins, Bcl-xL, Mcl-1
Abstract
The p14ARF tumor suppressor plays a central role in regulating cell cycle arrest and apoptosis. We reported previously that p14ARF is capable of triggering apoptosis in a p53-independent manner. However, the mechanism remained unclear. Here we demonstrate that the p53-independent activation of the mitochondrial apoptosis pathway by p14ARF is primarily mediated by the pro-apoptotic Bax-homolog Bak. Expression of p14ARF exclusively triggers a N-terminal conformational switch of Bak, but not Bax, which allows for mitochondrial permeability shift, release of cytochrome c, activation of caspases, and subsequent fragmentation of genomic DNA. Although forced expression of Bak markedly sensitizes toward p14ARF-induced apoptosis, re-expression of Bax has no effect. Vice versa, knockdown of Bak by RNA interference attenuates p14ARF-induced apoptosis, whereas down-regulation of Bax has no effect. Bak activation coincides with a prominent, caspase-independent deprivation of the endogenous Bak inhibitors Mcl-1 and Bcl-xL. In turn, mitochondrial apoptosis is fully blocked by overexpression of either Mcl-1 or Bcl-xL. Taken together, these data indicate that in the absence of functional p53 and Bax, p14ARF triggers mitochondrial apoptosis signaling by activating Bak, which is facilitated by down-regulating anti-apoptotic Mcl-1 and Bcl-xL. Moreover, our data suggest that the simultaneous inhibition of two central endogenous Bak inhibitors, i.e. Mcl-1 and Bcl-xL, may be sufficient to activate mitochondrial apoptosis in the absence of BH3-only protein regulation.
Introduction
The p14ARF (p19ARF in the mouse) tumor suppressor was originally identified as the second product of the INK4A gene locus, which also encodes the cyclin-dependent kinase-inhibitor p16 (1–4). ARF and p16 share common exons 2 and 3. However, as transcription of ARF initiates at a separate first exon, i.e. exon 1β, and proceeds in an alternative reading frame, ARF and p16 are both structurally and functionally unrelated. ARF plays a key role in mediating stress signals elicited by cellular or viral oncogenes, DNA damage, or starvation (5–11). Hardly detectable under normal conditions, ARF is rapidly up-regulated by appropriate stimuli and counteracts Hdm-2 (Mdm-2), the natural antagonist of p53. Thereby, ARF indirectly launches a p53-driven response, which, depending on the cellular context, leads to cell cycle arrest, apoptosis, or senescence (12, 13). More recently, ARF has been implicated in the induction of autophagy as well (14, 15).
The Bcl-2 family of proteins plays a central role in regulating the intrinsic apoptosis signaling machinery (16, 17). Functionally, Bcl-2 proteins can be divided into pro- and anti-apoptotic family members. Depending on the presence or absence of specific Bcl-2 homology (BH)2 domains, the former group is subdivided into the multidomain proteins, including Bax and Bak, and the BH3-only subfamily, which shares homologies only in the BH3-domain (16, 18, 19). Bax and Bak facilitate the permeabilization of the outer mitochondrial membrane, presumably by forming pore-like structures, which allows for the release of cytochrome c and the subsequent activation of the caspase cascade. BH3-only proteins are essential initiators of apoptotic cell death and primarily act as up-stream regulators of Bax and Bak. Functionally, BH3-only proteins constitute a life/death-switch that integrates the diverse pro- and anti-apoptotic signals. Their apoptosis-promoting activity is held in check by anti-apoptotic Bcl-2 proteins such as Bcl-2, Bcl-xL, or Mcl-1.
We reported previously that in p53-proficient cells, apoptosis induction by ARF is preferentially executed via a Bax-mediated mitochondrial cell death pathway (20, 21). The pro-apoptotic BH3-only protein Puma is a critical mediator of ARF-induced apoptosis in this setting. Puma is rapidly up-regulated following expression of p14ARF in p53-proficient human cancer cells. Although loss of Puma blocks the activation of its downstream effector Bax and, thereby, almost completely abrogates ARF-induced mitochondrial cell death, the functional reconstitution of Puma in Puma-deficient cells fully resensitizes toward p14ARF-induced apoptosis (22). Whether Puma or other “direct activator” BH3-only proteins bring Bax into action directly (“direct activator model”) or rather indirectly, i.e. by sequestration of anti-apoptotic Bcl-2 proteins that keep Bax in an inactive state (“inhibitor/derepressor model”), remains controversial (18, 23, 24). Nonetheless, these data delineate that ARF, Hdm-2/p53, Puma, and Bax act in a sequential manner.
A number of reports, including some of our group, indicate that p14ARF is capable of inducing mitochondrial apoptosis in p53-deficient cancer cells as well (25–29). However, the signaling pathways involved remained unclear. Therefore, we investigated the role of pro- and anti-apoptotic Bcl-2 family proteins in regulating mitochondrial apoptosis signaling in response to the forced expression of the p14ARF tumor suppressor.
EXPERIMENTAL PROCEDURES
Cell Culture
DU145 prostate carcinoma cells and SAOS-2 osteosarcoma cells were obtained from Deutsche Sammlung für Mikroorganismen und Zellkulturen (Braunschweig, Germany) or the ATCC. Cells were grown in DMEM medium supplemented with 10% FCS, 10,000 units/liter penicillin, and 0.1 g/liter streptomycin (all from Invitrogen). DU145 cells stably expressing Bax were generated and cultured as described (20). The generation of DU145 Bax-GFP and DU145 Bak-GFP transfectants is described elsewhere (30).
Adenoviral Vectors and Infection
A recombinant, replication-deficient adenoviral vector expressing a human p14ARF cDNA (Ad-p14ARF) was established as described (25). An adenoviral vector expressing β-galactosidase (Ad-lacZ) was used as a control. Cells were infected with adenoviral vectors in DMEM/high glucose in the absence of FCS or antibiotics for 2 h at 37 °C.
Measurement of Apoptotic Cell Death
Apoptotic DNA fragmentation was determined on the single cell level by measuring the DNA content of individual cells as described (25). Data are given in percent hypoploidy, i.e. the percentage of cells with a sub-G1 DNA content, which reflects the percentage of apoptotic cells with fragmented genomic DNA.
Measurement of Mitochondrial Permeability Transition
Mitochondrial permeability transition was determined by using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanin iodide (JC-1; Molecular Probes, Leiden, The Netherlands) as described (31) and quantified by flow cytometric detection of cells with decreased red fluorescence, indicating mitochondria with a lowered membrane potential (ΔΨm). Data are given in % cells with low ΔΨm.
Quantitative Polymerase Chain Reaction
RNA extraction, reverse transcription, and quantitative PCR were performed according to standard procedures as described (32). Quantitative PCR was performed using the ABI Prism 7000 sequence detection system (Applied Biosystems, Weiterstadt, Germany) using Abelson murine leukemia viral oncogene homolog as an internal reference.
Antibodies
Mouse monoclonal antibodies against p14ARF (NeoMarkers, Fremont, CA), p53 (BD Biosciences), cytochrome c (BD Biosciences), or GFP (Santa Cruz Biotechnology, Santa Cruz, CA) were used at a dilution of 1:1,000. Antibodies against Bax (Trevigen, Gaithersburg, MD) or Bad (BD Biosciences) were mouse monoclonal antibodies used at a dilution of 1:10,000 or 1:500. Rabbit polyclonal antibodies against actin (Sigma-Aldrich, St. Louis, MO), Bcl-x (BD Biosciences), or Bak (Dako Cytomation, Glostrup, Denmark) were used at a dilution of 1:1000, 1:200, or 1:100. Anti-Bid is a rabbit polyclonal antibody from R&D Systems (Wiesbaden, Germany) used at a dilution of 1:100. Anti-Mcl-1, anti-Puma, and anti-Bim were rabbit polyclonal antibodies from Santa Cruz Biotechnology used at a dilution of 1:200. Antibodies against Noxa or Nbk were goat polyclonal antibodies from Santa Cruz Biotechnology and used at a dilution of 1:200. Secondary goat-anti-mouse IgG and goat-anti-rabbit IgG antisera coupled to HRP were from Southern Biotechnology Associates (Birmingham, AL) and used at a dilution of 1:10,000 in PBS-T (PBS supplemented with 0.05% Tween-20). A rabbit polyclonal antibody specifically detecting Bax protein that has undergone a N-terminal conformational change (BaxNT*) was from Upstate Biotechnology (Lake Placid, NY) and was used at a final concentration of 1 μg/ml. A monoclonal mouse antibody specifically detecting Bak protein that has undergone a N-terminal conformational change antibody (BakNT*) was from Calbiochem (Darmstadt, Germany).
SDS-PAGE and Western Blot Analysis
Cells were harvested, washed, and lysed as described (25). Equal amounts of protein (25 μg/lane) were separated by SDS-PAGE. Immunoblotting and visualization of the proteins were performed using enhanced chemiluminescence (25). Equal loading was confirmed by reprobing with an antibody against actin.
Detection of Cytochrome c Release
Cytosolic extracts were prepared as described (30). Briefly, cells were lysed in 40 μl of hypotonic buffer (20 mm HEPES (pH 7.4), 10 mm KCl, 2 mm MgCl2, 1 mm EDTA) supplemented with 1 mm PMSF and 0.75 mg/ml digitonin (all from Sigma-Aldrich) for 3 min. Thereafter, debris was pelleted by centrifugation, supernatants were subjected to SDS-PAGE, and Western blot analysis was performed as described above.
Detection of Caspase Activation on the Single-cell Level
Activation of caspase-9-like (LEHDases) activities was determined by using a cell-permeable, carboxyfluorescein (FAM)-labeled derivative of LEHDfmk from Immunochemistry Technologies (Bloomington, MN) according to the manufacturer's protocol as described (21). Caspase-9-like (LEHDases) activities were quantified by flow cytometry.
Detection of Bax and Bak N-terminal Conformational Change
N-terminal conformational change of Bax (BaxNT*) or Bak (BakNT*) was analyzed as described previously (22). Briefly, cells were permeabilized by incubation in PBS-supplemented 0.5% paraformaldehyde and were incubated with antibodies specifically detecting BaxNT* or BakNT*. Thereafter, cells were incubated with a secondary FITC-labeled goat anti-rabbit antibody or a FITC-labeled anti-mouse antibody. Detection of BaxNT* or BakNT* was performed by flow cytometry using unlabeled cells or cells incubated with either primary or secondary antibodies as negative controls.
Analysis of Bak and Bax Clustering
Bax and Bak oligomerization was determined by fluorescence microscopy in DU145 cells stably expressing GFP-Bax or GFP-Bak as described (30). Treatment with cisplatin (10 μg/ml) served as a positive control. Bax and Bak localization and clustering were studied using a Zeiss Axiovert 200 microscope (Carl Zeiss, Jena, Germany). Images were acquired by using the Openlab Deconvolution Software (Improvision, Coventry, UK).
RNA Interference
On-target plus siRNA against Nbk, Puma, Noxa, and control siRNA were purchased from Dharmacon (Lafayette, CO). Transfection was performed using DharmaFECT according to the instructions of the manufacturer as described (33). After 24 h, cells were infected with adenoviral vectors as described above.
Stable Transfection and Establishment of Single Cell Clones
A Mcl-1 cDNA was introduced into the pRTS1 vector in the right origin under the control of the Tet-On system to achieve conditional expression in the presence of doxycycline (tetracycline). DU145WT cells were transfected with the pRTS1-Mcl-1 construct (10 μg) by using DharmaFECT transfection reagent. Clones were selected with 500 μg/ml hygromycin B (Calbiochem). Selected cells were induced for 24 h with doxycycline, and GFP-positive cells were sorted by FACS. DU145 cells stably transfected with a vector expressing a full-length human Bcl-xL cDNA were described earlier (34).
RESULTS
Expression of p14ARF Triggers p53/Bax-independent Mitochondrial Apoptosis
We showed previously that forced expression of p14ARF by adenovirus triggers p53-independent apoptosis in the Bax-mutated human prostate cancer cell line DU145 (25). As shown in Fig. 1, re-expression of functional Bax does not alter the propensity of p14ARF to induce a breakdown of the mitochondrial potential (ΔΨm) (Fig. 1A), the release of cytochrome c into the cytosol (B), as well as a dose-dependent activation of caspase-9-like (LEHDase) activities (data not shown). Consequently, adenovirus-mediated expression of p14ARF induces apoptotic DNA fragmentation in a dose-dependent manner, regardless of the presence or absence of functional Bax (Fig. 1C). As shown in Fig. 1D, apoptotic DNA fragmentation was detectable as early as 48 h after infection with Ad-ARF, further increased to 72 h, and remained stable thereafter. Although not fully redundant, a number of previous reports indicate that the pro-apoptotic Bax Homolog Bak may compensate for the loss of Bax to activate the mitochondrial apoptosis signaling cascade in response to diverse stimuli (20, 35–37). Therefore, we reasoned that Bak, which is readily expressed in Bax-mutated DU145 cells (Fig. 1B), mediates mitochondrial permeability shift and apoptosis upon expression of p14ARF in these cells. Like Bax, activation of Bak is initiated by a N-terminal conformational change (NT*), which precedes the formation of oligomeric clusters and allows for the permeabilization of the outer mitochondrial membrane (38, 39). As shown in Fig. 1E, expression of p14ARF by adenovirus (50 m.o.i.) resulted in an NT* of Bak (BakNT*) in both Bax-deficient and Bax-reconstituted DU145 cells at 48 h. To our surprise, expression of p14ARF did not trigger an NT* of Bax (BaxNT*) (Fig. 1F). In contrast, treatment of Bax-proficient DU145 cells with epirubicine induced an NT* of both Bak (Fig. 1E) and Bax (F). Taken together, these results underline that the activation of the mitochondrial apoptosis signaling cascade following forced expression of p14ARF is fully independent from Bax and coincides with the activation of Bak in p53-deficient DU145 cells (20, 25).
FIGURE 1.

P14ARF-induced cell death is Bax-independent and coincides with activation of Bak. A, p53-mutated, Bax-deficient (mock) or Bax-reconstituted (bax) DU145 cells were infected with the indicated adenoviral vectors at an m.o.i. of 50 or mock-treated for 48 h. Loss of the mitochondrial membrane potential (ΔΨm) was determined by flow-cytometry using the cationic dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanin iodide. The relative number of cells displaying ΔΨm is given. B, DU145 cells were infected with the indicated adenoviral vectors (50 m.o.i.) or mock-treated. Total cellular proteins (25 μg/lane) were separated by SDS-PAGE and subjected to Western blot analysis using the indicated antibodies. C and D, cells were infected with Ad-p14ARF or mock-treated for 72 h. Apoptosis was determined by flow cytometric detection of fragmented nuclear DNA. Cells displaying a sub-G1 DNA content were less than 10% upon infection with an Ad-lacZ control adenovirus (50 m.o.i., data not shown). E and F, DU145 cells were infected with Ad-ARF or Ad-lacZ (50 m.o.i.) or mock-treated (Co) for 48 h. In parallel, cells were exposed to epirubicine (Epi, 1 μg/μl) as a positive control. An N-terminal conformational change of Bax (BaxNT*) (E) or Bak (BakNT*) (F) was studied by flow cytometry using conformation-specific antibodies. Error bars represent mean ± S.D. from triplicates.
Apoptosis Induction by p14ARF Is Mediated by the Bax Homolog Bak
To further investigate the role of Bax and Bak in mediating the activation of the mitochondrial apoptosis signaling cascade following expression of p14ARF, Bax or Bak coupled to GFP were stably introduced into DU145 cells (30). As shown in Fig. 2A, Western blot analysis revealed comparable levels of either GFP-Bax or GFP-Bak in the respective cell line as compared with untransfected parental DU145 cells. Overall, levels of GFP protein are higher in the GFP-Bax cells as compared with the GFP-Bak cells, which may explain their more intense background fluorescence pattern. As shown in Fig. 2B, expression of p14ARF by adenovirus (50 m.o.i.) in DU145 cells expressing GFP-tagged Bak (a–d) induced the formation of Bak clusters, indicated by the occurrence of a punctuated fluorescence pattern, as compared with mock-treated or Ad-lacZ control vector-infected cells. In contrast, expression of p14ARF did not trigger Bax-clustering in DU145 cells expressing GFP-tagged Bax (Fig. 2B, e–h). As expected, treatment with cisplatin resulted in clustering of both Bak and Bax (Fig. 2B, d and h). Similar data were obtained at later time points (72 h, data not shown), which underscores that mitochondrial apoptosis in p53-deficient DU145 cells following expression of p14ARF primarily depends on Bak but not Bax. To test whether the enforced expression of Bak sensitizes toward p14ARF-induced apoptosis in DU145 cells, DU145 cells stably transfected with GFP-Bak were infected with either Ad-ARF or Ad-lacZ (both 50 m.o.i.) or treated mock for 72 h. Fig. 2C shows that the presence of GFP-Bak sensitizes toward p14ARF-induced apoptosis, as evidenced by an increase in the relative number of cells displaying apoptotic DNA-fragmentation, when compared with empty vector transduced or mock-treated controls. We next tested whether the knockdown of functional Bak in the Bax-deficient DU145 parental cells by RNA interference attenuates p14ARF-induced apoptosis. As shown in figure 2D, RNA interference resulted in a substantial reduction of Bak protein as examined by Western blot analysis. This lead to a marked decrease in the number of cells displaying apoptotic DNA fragmentation following expression of p14ARF as compared with mock treatment or infection with an Ad-lacZ control adenovirus. However, small amounts of residual Bak appear sufficient to allow for some apoptotic response. These data corroborate that p14ARF-induced mitochondrial apoptosis in p53-/Bax-deficient DU145 cancer cells is primarily mediated by Bak whereas Bax remains inactive.
FIGURE 2.

P14ARF-induced mitochondrial apoptosis is mediated by Bak but not Bax. A, DU145 cells stably expressing GFP-tagged Bax or GFP-tagged Bak were generated as described (30). Total cellular proteins (25 μg/lane) were separated by SDS-PAGE and subjected to Western blot analysis using the indicated antibodies. B, DU145 Bax-GFP (left column, a–d) or Bak-GFP (right column, e–h) were either mock-treated (control) or infected with Ad-ARF or Ad-lacZ (both 50 m.o.i.). Treatment with cisplatin (10 μg/ml) was used as a positive control. The distribution of GFP-Bak and GFP-Bax was determined by fluorescence microscopy. In contrast to Bax, Bak shows a more reticular localization because of its association with mitochondria. In apoptotic cells, Bak (c and d) or Bax (h) coalesce into large clusters. Representative high-power fields at a ×400 magnification are shown. Scale bar = 50 μm). C, control vector (mock) or Bak overexpressing DU145 cells were infected with Ad-p14ARF or Ad-lacZ (both 50 m.o.i.) or mock-treated (Co), and apoptotic DNA-fragmentation was assessed by flow cytometry at 72 h after infection. D, expression of Bak was down-regulated by RNA interference (insert, confirmation by Western blot analysis). 24 h later, cells were infected with the indicated adenoviral vectors (50 m.o.i.) or treated mock for 48 h. Apoptotic DNA fragmentation was determined by flow cytometry. Error bars represent mean ± S.D. from triplicates.
P14ARF-induced Mitochondrial Apoptosis Is Not Preceded by Up-regulation of Pro-apoptotic BH3-only Proteins
BH3-only proteins are the major upstream regulators of pro-apoptotic Bax and Bak (19, 40, 41). We reported previously that the transcriptional activation of the BH3-only protein Puma in p53-proficient cancer cells is a prerequisite for triggering a Bax-mediated mitochondrial apoptosis signaling pathway upon expression of p14ARF (22). Therefore, p53-independent activation of one or several distinct BH3-only proteins may trigger Bak-mediated apoptosis in p53-deficient cells following expression of p14ARF. To investigate this hypothesis, we systematically analyzed the BH3-only protein expression at the mRNA level across the family by the use of quantitative real-time PCR (qPCR) in DU145 cells following expression of p14ARF. As shown in Fig. 3A, expression of p14ARF by adenovirus (50 m.o.i.) induced the transcriptional up-regulation of Puma, Nbk, and Noxa mRNA over time. In contrast, the mRNA levels of Bid, Bim, or Bad (and, not shown, Hrk, Bmf, Bnip3, and Spike) remained unaltered. However, Western blot analysis (Fig. 3B) revealed that p14ARF did not induce the expression of any of the BH3-only proteins investigated before by qPCR. In particular, and in contrast to the mRNA data, we did not detect a marked up-regulation of Puma, Nbk, or Noxa protein following expression of p14ARF as compared with mock treatment or control vector infection at 48 h. Consistent results were obtained at 72 h (data not shown), suggesting that the regulation of specific (sets of) BH3-only proteins is not primarily involved in activating the mitochondrial apoptosis signaling pathway in p53-deficient cancer cells following expression of p14ARF. To functionally address that neither Puma nor Nbk or Noxa are relevant for mediating p14ARF-induced apoptosis in this setting, we used RNA-interference by siRNA. As shown in Fig. 3C, the siRNA-mediated knockdown of Puma, Nbk, and/or Noxa is effective as confirmed by qPCR (lower panel). As shown in the upper panel, knockdown of Puma, Nbk, or Noxa neither alone nor in combination alters the apoptosis sensitivity of the cells, as measured by apoptotic DNA fragmentation. Taken together, these results indicate that, in contrast to p53-proficient cells, the differential regulation of BH3-only proteins appears dispensable for mediating p14ARF-induced mitochondrial apoptosis in this setting of p53-deficient human cancer cells.
FIGURE 3.

Expression of p14ARF induces Noxa, Puma, and Nbk mRNA expression. A, DU145 cells were infected with Ad-p14ARF at an m.o.i. of 50. In parallel, cells were mock-treated (control) or infected with a control vector (Ad-lacZ, 50 m.o.i.). Cells were harvested at 24, 48, or 72 h after infection, and mRNA levels of the indicated BH3-only proteins were analyzed by quantitative PCR. B, DU145 prostate carcinoma cells were infected with the indicated adenoviral vectors (50 m.o.i.) or mock-treated for 48 h. Total cellular proteins (25 μg/lane) were separated by SDS-PAGE and subjected to Western blot analysis using the indicated antibodies. C, upper panel, DU145 cells were transfected with the indicated siRNA and cultured for 24 h. Thereafter, cells were infected with either Ad-ARF or Ad-lacZ (both 50 m.o.i.) or mock-treated (control) for 48 h. Apoptosis was assessed by flow cytometric detection of fragmented nuclear DNA. Lower panel, cells treated as described above and mRNA levels of the indicated BH3-only proteins were analyzed by quantitative PCR using cells transfected with control siRNA as reference. Error bars represent mean ± S.D. from triplicates.
P14ARF-induced Mitochondrial Apoptosis via Bak Is Facilitated by Down-regulation of Anti-apoptotic Mcl-1 and Bcl-xL
The pro-apoptotic activity of Bak is primarily counteracted by the anti-apoptotic Bcl-2 proteins Mcl-1 and, at least in part, Bcl-xL (42–45). As DU145 cells do not express relevant levels of Bcl-2 (31, 33), we reasoned that a down-modulation of Mcl-1 and/or Bcl-xL may be sufficient to allow for the activation of Bak following expression of p14ARF, which has been reported previously for apoptosis induction by the BH3-only protein Nbk or following TRAIL exposure (31, 43). Therefore, we examined whether the expression of p14ARF alters the levels of anti-apoptotic Mcl-1 or Bcl-xL in DU145 prostate cancer cells. As shown in Fig. 4A, Western blot analysis revealed that both Mcl-1 and, at least to some extent, Bcl-xL protein in DU145 cells decreases following expression of p14ARF (50 m.o.i.) as compared with mock-treated or control vector-infected cells at 48 h. Cleavage of Mcl-1 or Bcl-xL by activated caspases was ruled out by addition of the pan-caspase inhibitor Q-VD-OPh, which did not prevent the deprivation of Mcl-1 or Bcl-xL protein following expression of p14ARF. As shown in Fig. 4B, the decrease in Mcl-1 protein upon expression of p14ARF occurred in a dose-dependent manner. Similarly, but to a lesser degree, expression of p14ARF induced a dose-dependent decrease of Bcl-xL.
FIGURE 4.

Expression of p14ARF triggers down-regulation of anti-apoptotic Mcl-1 and Bcl-xL. A, DU145 cells were either mock-treated or infected with the indicated adenoviral vectors (50 m.o.i.) in the presence or absence of the broad-spectrum caspase inhibitor Q-VD-OPh (final concentration 10 μm) for 48 h. Total cellular proteins (25 μg/lane) were separated by SDS-PAGE and subjected to Western blot analysis using the indicated antibodies. B, dose response. Cells were infected with Ad-ARF (10, 25, 50, 100 m.o.i.). In parallel, cells were either mock-treated or infected with an Ad-lacZ (100 m.o.i.). Total cellular proteins (25 μg/lane) were subjected to SDS-PAGE and Western blot analysis with the indicated antibodies.
To confirm these findings in an independent system of p53-deficient human cancer cells, p14ARF was expressed in p53-deficient, Bax/Bak-proficient SAOS-2 osteosarcoma cells. As displayed in Fig. 5A, adenovirus-mediated expression of p14ARF (50 m.o.i.) induced a decrease of both Mcl-1 and, to a lesser extent, Bcl-xL protein levels as detected by Western blot analysis as early as 48 h (Mcl-1) or 72 h (Bcl-xL). In analogy to DU145 cells, expression of p14ARF triggered BakNT* (Fig. 5B) as well as apoptotic DNA fragmentation (Fig. 5C). In turn, and as depicted in Fig. 5D, knockdown of Bax by RNA interference does not alter the apoptosis sensitivities of SAOS-2 cells at 72 h after expression of p14ARF (50 m.o.i.). Similar data were obtained at 48 h (data not shown).
FIGURE 5.

Expression of p14ARF in p53-deficient SAOS-2 cells triggers Mcl-1 and Bcl-xL deprivation and apoptosis. A, p53-deficient SAOS-2 osteosarcoma cells were infected with indicated adenoviral vectors at 50 m.o.i. or mock-treated (control). Total cellular proteins were separated by SDS-PAGE and subjected to Western blot analysis after 24, 48, and 72 h. B, SAOS-2 cells were treated as described in A. An N-terminal conformational change of Bak (BakNT*) was detected by flow cytometry using a conformation-specific antibody at 48 h after infection. C, cells were treated as described in A. Apoptotic cells were analyzed by flow cytometry to detect fragmented nuclear DNA 72 h after infection. D, expression of Bax was down-regulated by RNA interference (right panel, confirmation by Western blot analysis). 24 h later, cells were infected with the indicated adenoviral vectors (50 m.o.i.) or mock-treated for 48 h. Apoptotic DNA fragmentation was determined by flow cytometry. Error bars represent the means ± S.D. from triplicates.
To address the functional relevance of Mcl-1 and Bcl-xL in p14ARF-induced apoptosis, we investigated whether overexpression of Mcl-1 or Bcl-xL attenuates apoptosis induction in this setting. To this end, Mcl-1 was conditionally expressed in DU145 cells by the use of the doxycycline (tetracycline)-inducible (Tet-On) expression vector pRTS1 (46), designated pRTS1-Mcl-1. As shown in Fig. 6A, addition of doxycycline (+Dox: On) triggered the up-regulation of Mcl-1 protein expression, which fully abrogated p14ARF-induced apoptotic DNA fragmentation as compared with vehicle-treated cells (-Dox: Off). Likewise, overexpression of Bcl-xL blocked apoptosis in DU145 cells following infection with Ad-p14ARF (50 m.o.i.) (Fig. 6B) to background levels. In contrast, expression of Bcl-2 provides only reduced protection, i.e. a ∼50% reduction in apoptotic DNA fragmentation as compared with empty vector transfected cells (data not shown). These results indicate that the down-modulation of anti-apoptotic Mcl-1 and Bcl-xL plays a key role in facilitating p14ARF-induced Bak-mediated apoptosis in p53/Bax-deficient DU145 cancer cells.
FIGURE 6.

Overexpression of Mcl-1 or Bcl-xL attenuates p14ARF-induced apoptosis. A, an pRTS1-Mcl-1 vector was stably introduced into DU145 cells to allow for the conditional expression of Mcl-1 in the presence of tetracycline (doxycycline, Dox). Cells were infected with indicated adenoviral vectors at 50 m.o.i. or mock-treated (Co). B, DU145 cells stably overexpressing Bcl-xL were infected with Ad-ARF or Ad-lacZ (both 50 m.o.i.) or mock-treated in parallel with empty vector-transduced controls (mock). Apoptosis was measured by flow cytometric detection of fragmented nuclear DNA at 72 h. The relative number of apoptotic cells is given. Error bars represent mean ± S.D. from triplicates.
Deprivation of Mcl-1 and Bcl-xL Expression by p14ARF Involves a Posttranscriptional Mechanism
Finally, we addressed whether the down-regulation of Mcl-1 and Bcl-xL resides at the transcriptional or the posttranscriptional level. As shown in Fig. 7A, quantification of mRNA expression by qPCR does not reveal a decrease in Mcl-1 or Bcl-xL in mock-treated versus p14ARF-expressing cells, suggesting that the regulation occurs at the posttranscriptional level, e.g. by enhanced proteasomal degradation. Therefore, we next tested whether inhibition of the proteasome attenuates the p14ARF-induced deprivation of Mcl-1 or Bcl-xL. To this end, DU145 cells were infected with Ad-ARF or Ad-lacZ (both 50 m.o.i.), and expression of Mcl-1 and Bcl-xL was determined by Western blot analysis in the presence or absence of MG132 (Fig. 7B). As expected, expression of p14ARF induces a decrease in Mcl-1 and Bcl-xL protein levels at 48 h. In the presence of MG132, proteasomal degradation of Mcl-1 is blocked, as indicated by an increase in total and Ser-159/Thr-163-phosphorylated Mcl-1 protein. Likewise, inhibition the ubiquitin ligases ARF-BP1/mule or β-TrCP, both of which have been implicated in the posttranslational regulation/degradation of Mcl-1 (47–49), by RNA interference does not alter Mcl-1 protein levels (data not shown), indicating that p14ARF does not accelerate the proteasomal degradation of Mcl-1. In contrast, Bcl-xL was somewhat stabilized in MG132-treated cells expressing p14ARF.
FIGURE 7.

Regulation of Mcl-1 and Bcl-xL. A, DU145 cells were infected with Ad-p14ARF or Ad-lacZ (both 50 m.o.i.) or mock-treated (control). Cells were harvested at 24 h or 48 h after infection, and mRNA expression of Mcl-1 (left panel) or Bcl-xL (right panel) was analyzed by quantitative real-time PCR. B, DU145 cells were infected with the indicated adenoviral vectors (50 m.o.i.) or mock-treated for 24 h. Thereafter, cells were cultured in the presence or absence of the proteasome inhibitor MG132 (final concentration 10 μm). Total cellular proteins were separated by SDS-PAGE and subjected to Western blot analysis using the indicated antibodies. Error bars represent the mean ± S.D. from triplicates.
DISCUSSION
Human cancer cells deficient for p53 and Bax are highly resistant toward a broad range of cell death signals, e.g. DNA damaging agents, ionizing radiation, the death-receptor ligand TRAIL, or the forced expression of BH3-only proteins such as Nbk (25, 30, 31, 50–52). Nonetheless, a number of reports indicate that the forced expression of the p14ARF tumor suppressor triggers the mitochondrial cell death pathway in a p53/Bax-independent manner as well (25–27, 29, 53). This is remarkable and implies that high-level expression of ARF is capable of circumventing the Hdm-2/p53-mediated induction of Puma and the subsequent activation of Bax in case any of these signaling mediators are mutated or functionally inactivated (22). We show here that the pro-apoptotic Bcl-2 protein Bak not only complements for the loss of Bax in p53-deficient human cancer cells, as it does in p53-proficient cells forced to express p14ARF (20). Rather, our data indicate that Bak is the principal mediator of mitochondrial apoptosis upon expression of p14ARF in this setting. This is underscored by the finding that overexpression of Bak sensitizes toward p14ARF-induced apoptots, whereas re-expression of Bax has no impact on the apoptosis sensitivity of the cell upon expression of p14ARF. Vice versa, knockdown of Bax in p53-deficient, Bax/Bak-proficient cells by RNA interference does not attenuate p14ARF-induced apoptosis. In contrast, knockdown of Bak does, which further underscores that apoptosis induced by p14ARF solely depends on Bak activation irrespective of the presence or absence of functional Bax. In fact, p14ARF fails to trigger exposure of the N-terminal Bax epitope as well as Bax re-distribution to the mitochondria and oligomerization in a p53-deficient cells, which delineates that Bak, and not Bax, is required for the activation of the mitochondrial apoptosis pathway upon expression of p14ARF in p53-deficient human cancer cells. In this regard, our data are in line with previous reports pointing out the differential regulation of Bak versus Bax in mediating apoptosis (31, 54–56). In general, our data underscore that the redundancy of Bax and Bak is dictated by the type of death stimulus and the cellular context, i.e. cell type and its developmental stage as well as environmental factors (57). This may also serve to explain why exogenous p14ARF does not trigger Bax activation despite low levels of anti-apoptotic Bcl-2, the main antagonist of Bax, present in DU145 cells (31, 33). In this regard, we found that re-expression of Bcl-2 provided only limited protection toward p14ARF-induced apoptosis as opposed to what is achieved by overexpression of Mcl-1 or Bcl-xL. Although we did not specifically address this point, this suggests that Bax is primarily kept in check by Bcl-2 proteins other than Bcl-2, which may prevent its activation following expression of p14ARF (16, 23). Likewise, overexpression of Bcl-2 in the absence of Bax may result in Bak inhibition to some degree.
However, this brings up the question: what determines the exclusive activation of Bak and not Bax in this situation? Although Bax and Bak appear functionally equivalent in many settings (35, 36, 58), our findings support previous reports delineating that Bax and Bak are not fully redundant. Irrespective of the model, i.e. direct activator versus “inhibitor/derepressor,” this may be reflected at the molecular level by the fact that BH3-only proteins preferentially activate (either directly or indirectly) Bax- or Bak-dependent events. Furthermore, the activity of Bax and Bak is blocked by distinct (sets of) anti-apoptotic Bcl-2 proteins (33, 42, 44, 59). In particular, Bak interacts with Mcl-1, which is degraded rapidly at the onset of apoptosis (60, 61). In the same vein, DNA damage was shown to trigger the dissociation of Mcl-1 (and Bcl-xL) from Bak to allow for its self-association, i.e. the formation of homo-oligomers that then trigger the permeabilization of the outer mitochondrial membrane (42, 59). Likewise, recent work from our group (33, 43) illustrates that Mcl-1 and Bcl-xL are major determinants of Bak-dependent apoptosis following expression of Nbk or exposure to TRAIL. High levels of Mcl-1 or Bcl-xL specifically guard against the activation of Bak until counteracted by another BH3-only protein such as Noxa or down-regulated by small molecules such as sorafenib or roscovitine. In keeping with these data, we show here that p14ARF-induced mitochondrial apoptosis in p53-deficient cells proceeds via Bak and is facilitated by a simultaneous decrease of Mcl-1 and, to some degree, Bcl-xL. A model for p53-dependent and independent induction of apoptosis by p14ARF is depicted in Fig. 8. In fact, loss of Mcl-1 and Bcl-xL strongly correlates with the apoptosis sensitivity of the target cells. As such, DU145 cells re-expressing Bak, which exhibit a strong reduction in Mcl-1 and Bcl-xL protein levels following expression of p14ARF, are most sensitive. In contrast, cells exhibiting only a moderate reduction of Mcl-1 and Bcl-xL are considerably less sensitive. Similar results were obtained in SAOS-2 cells as an alternative model. Furthermore, expression of Mcl-1 or Bcl-xL rescues from ARF-induced apoptosis. Taken together, this suggests that the de-repression of Bak through down-regulation of Mcl-1 and Bcl-xL upon expression of p14ARF is sufficient to activate the mitochondrial apopotis pathway (59, 62, 63). This is supported by the notion that conditional knockout of both Mcl-1 and Bcl-xL renders hepatocytes highly susceptible to spontaneous apoptosis in a gene dose-dependent manner (45).
FIGURE 8.
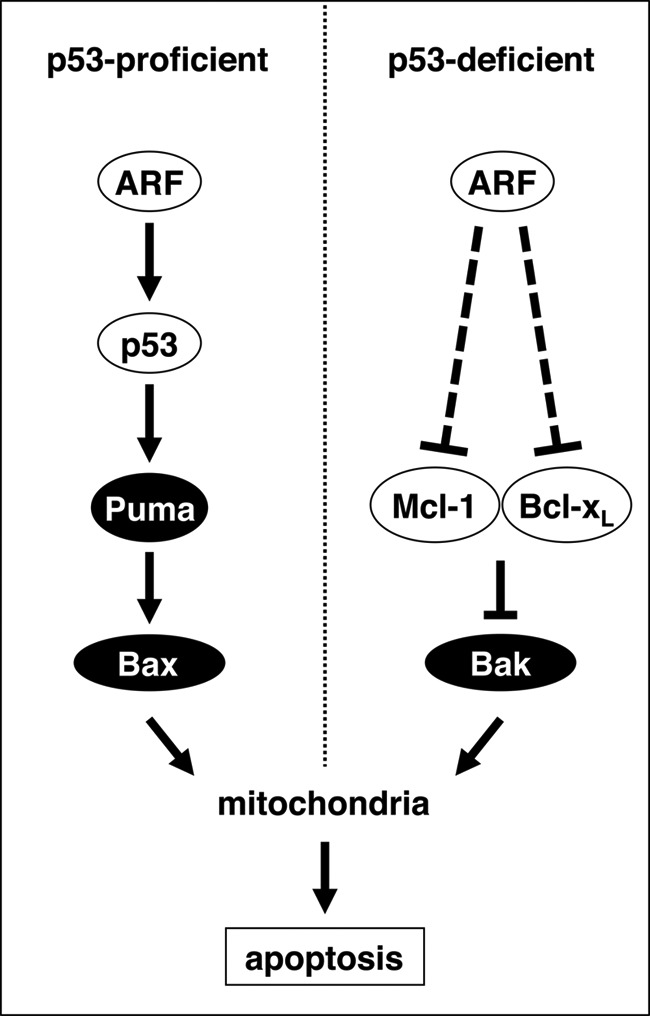
Schematic model. Expression of the p14ARF tumor suppressor triggers both p53-dependent (left panel) and p53-independent (right panel) apoptosis signaling cascades. In p53-proficient cells, p14ARF induces the physical stabilization of p53 and the consecutive up-regulation of the BH3-only protein Puma (22). Bax and Bak then trigger the permeabilization of the outer mitochondrial membrane, the release of cytochrome c, and the activation of caspases such as caspase-9 and caspase-3/7. In p53-deficient cells, p14ARF triggers mitochondrial apoptosis via down-regulation of the anti-apoptotic Bcl-2 homologs Mcl-1 and Bcl-xL. This attenuates their inhibitory effect on pro-apoptotic Bak, which then undergoes an N-terminal conformational change to allow for the activation of mitochondria in the absence of BH3-only protein induction.
The simultaneous activation of two BH3-only proteins, e.g. Noxa and Bad, would allow for the targeting of both Mcl-1 and Bcl-xL (42, 59). Therefore, and in analogy to our previous approach by which we identified Puma as a critical player in activating mitochondrial apoptosis upon expression of p14ARF (22), we investigated whether p14ARF triggers the up-regulation of BH3-only proteins in a p53-indepedent manner. Moreover, knockdown of Noxa, Puma, or Nbk by RNA interference, either alone or in combination by RNA interference, did not alter the apoptotic response of the target cells upon expression of p14ARF. Our data suggest that BH3-only proteins are not primarily involved in the activation of Bak in this setting. In turn, the down-regulation of Mcl-1 and Bcl-xL seems to be the major mechanism to facilitate the activation of Bak. This is well in line with the inhibitor/derepressor model proposed by Willis et al. (42) where the role of BH3-only proteins would be to rerepress anti-apoptotic Bcl-2 family members to allow for the activation of Bax and/or Bak as opposed to the direct activator model, where BH3-only proteins would be required to physically interact with Bax/Bak to promote their activation. This also indirectly argues against the idea that loss of Bax protects cells from apoptosis by decreasing the abundance of apoptosis-promoting BH3-only proteins under a critical threshold (64). Instead, the decrease in Mcl-1 expression most likely occurs through a mechanism that interferes with protein synthesis. One key function of ARF in this regard might be its propensity to modulate ribosomal biogenesis (65, 66). This may be particularly relevant for Mc-1, which was reported to have a short half-life (67). In contrast, transcriptional repression of Mcl-1 or Bcl-xL, which was described to occur via an NK-κB-dependent pathway, is not relevant in our model (9). Likewise, we were unable to detect enhanced proteasomal degradation of Mcl-1 and/or Bcl-xL in p14ARF-expressing cells. Nonetheless, we cannot formally rule out that BH3-only proteins are fully dispensable for p14ARF-induced apoptosis in p53/Bax-deficient human cancer cells. As such, degradation of Mcl-1 and/or Bcl-xL might cause the release a range of BH3-only proteins that could then directly or indirectly trigger the activation of Bak, which will have to be addressed more thoroughly. Further work will be needed to clarify this issue.
The preferential activation of Bak in p53/Bax-deficient cancer cells might have important implications in cancer therapy. Tumor cells frequently lose p53 and Bax during malignant progression, which renders them highly resistant toward anticancer therapies such as chemodrugs or ionizing irradiation. In this regard, the p53-independent effects triggered by ARF may represent a putative “fail-safe” mechanism that allows cells to escape malignant transformation even in the absence of a functional p53/Bax pathway. In this regard, it will be important to address the mechanism by which p14ARF represses expression of Mcl-1 and Bcl-xL, which will have to be addressed in more detail.
Acknowledgments
We thank Anja Richter for expert technical assistance and Monika Dejewska for providing the pRTS1-Mcl-1 vector.
Footnotes
- BH
- Bcl-2 homolog
- Ad
- adenovirus
- m.o.i.
- multiplicity of infection
- qPCR
- quantitative PCR
- TRAIL
- TNF-related apoptosis inducing ligand
- ARF
- alternative reading frame product.
REFERENCES
- 1. Mao L., Merlo A., Bedi G., Shapiro G. I., Edwards C. D., Rollins B. J., Sidransky D. (1995) A novel p16INK4A transcript. Cancer Res. 55, 2995–2997 [PubMed] [Google Scholar]
- 2. Duro D., Bernard O., Della Valle V., Berger R., Larsen C. J. (1995) A new type of p16INK4/MTS1 gene transcript expressed in B-cell malignancies. Oncogene 11, 21–29 [PubMed] [Google Scholar]
- 3. Quelle D. E., Zindy F., Ashmun R. A., Sherr C. J. (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83, 993–1000 [DOI] [PubMed] [Google Scholar]
- 4. Stone S., Jiang P., Dayananth P., Tavtigian S. V., Katcher H., Parry D., Peters G., Kamb A. (1995) Complex structure and regulation of the P16 (MTS1) locus. Cancer Res. 55, 2988–2994 [PubMed] [Google Scholar]
- 5. Zindy F., Eischen C. M., Randle D. H., Kamijo T., Cleveland J. L., Sherr C. J., Roussel M. F. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 12, 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Palmero I., Pantoja C., Serrano M. (1998) p19ARF links the tumour suppressor p53 to Ras. Nature 395, 125–126 [DOI] [PubMed] [Google Scholar]
- 7. Radfar A., Unnikrishnan I., Lee H. W., DePinho R. A., Rosenberg N. (1998) p19(Arf) induces p53-dependent apoptosis during Abelson virus-mediated pre-B cell transformation. Proc. Natl. Acad. Sci. U.S.A. 95, 13194–13199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Stanchina E., McCurrach M. E., Zindy F., Shieh S. Y., Ferbeyre G., Samuelson A. V., Prives C., Roussel M. F., Sherr C. J., Lowe S. W. (1998) E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev. 12, 2434–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rocha S., Garrett M. D., Campbell K. J., Schumm K., Perkins N. D. (2005) Regulation of NF-κB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. EMBO J. 24, 1157–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Inoue R., Asker C., Klangby U., Pisa P., Wiman K. G. (1999) Induction of the human ARF protein by serum starvation. Anticancer Res. 19, 2939–2943 [PubMed] [Google Scholar]
- 11. Williams R. T., Roussel M. F., Sherr C. J. (2006) Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. U.S.A. 103, 6688–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sherr C. J. (2006) Divorcing ARF and p53. An unsettled case. Nat. Rev. Cancer 6, 663–673 [DOI] [PubMed] [Google Scholar]
- 13. Ozenne P., Eymin B., Brambilla E., Gazzeri S. (2010) The ARF tumor suppressor: structure, functions and status in cancer. Int. J. Cancer 127, 2239–2247 [DOI] [PubMed] [Google Scholar]
- 14. Reef S., Zalckvar E., Shifman O., Bialik S., Sabanay H., Oren M., Kimchi A. (2006) A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol. Cell 22, 463–475 [DOI] [PubMed] [Google Scholar]
- 15. Pimkina J., Humbey O., Zilfou J. T., Jarnik M., Murphy M. E. (2009) ARF induces autophagy by virtue of interaction with Bcl-xl. J. Biol. Chem. 284, 2803–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adams J. M., Cory S. (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Youle R. J., Strasser A. (2008) The BCL-2 protein family. Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 18. van Delft M. F., Huang D. C. (2006) How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 16, 203–213 [DOI] [PubMed] [Google Scholar]
- 19. Lomonosova E., Chinnadurai G. (2008) Oncogene 27, S2–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hemmati P. G., Güner D., Gillissen B., Wendt J., von Haefen C., Chinnadurai G., Dörken B., Daniel P. T. (2006) Bak functionally complements for loss of Bax during p14ARF-induced mitochondrial apoptosis in human cancer cells. Oncogene 25, 6582–6594 [DOI] [PubMed] [Google Scholar]
- 21. Hemmati P. G., Normand G., Verdoodt B., von Haefen C., Hasenjäger A., Güner D., Wendt J., Dörken B., Daniel P. T. (2005) Loss of p21 disrupts p14 ARF-induced G1 cell cycle arrest but augments p14 ARF-induced apoptosis in human carcinoma cells. Oncogene 24, 4114–4128 [DOI] [PubMed] [Google Scholar]
- 22. Hemmati P. G., Müer A., Gillissen B., Overkamp T., Milojkovic A., Wendt J., Dörken B., Daniel P. T. (2010) Systematic genetic dissection of p14ARF-mediated mitochondrial cell death signaling reveals a key role for p21CDKN1 and the BH3-only protein Puma/bbc3. J. Mol. Med. 88, 609–622 [DOI] [PubMed] [Google Scholar]
- 23. Brunelle J. K., Letai A. (2009) Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci. 122, 437–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Czabotar P. E., Colman P. M., Huang D. C. (2009) Bax activation by Bim? Cell Death Differ. 16, 1187–1191 [DOI] [PubMed] [Google Scholar]
- 25. Hemmati P. G., Gillissen B., von Haefen C., Wendt J., Stärck L., Güner D., Dörken B., Daniel P. T. (2002) Adenovirus-mediated overexpression of p14(ARF) induces p53 and Bax-independent apoptosis. Oncogene 21, 3149–3161 [DOI] [PubMed] [Google Scholar]
- 26. Normand G., Hemmati P. G., Verdoodt B., von Haefen C., Wendt J., Güner D., May E., Dörken B., Daniel P. T. (2005) p14ARF induces G2 cell cycle arrest in p53- and p21-deficient cells by down-regulating p34cdc2 kinase activity. J. Biol. Chem. 280, 7118–7130 [DOI] [PubMed] [Google Scholar]
- 27. Eymin B., Leduc C., Coll J. L., Brambilla E., Gazzeri S. (2003) p14ARF induces G2 arrest and apoptosis independently of p53 leading to regression of tumours established in nude mice. Oncogene 22, 1822–1835 [DOI] [PubMed] [Google Scholar]
- 28. Eymin B., Karayan L., Séité P., Brambilla C., Brambilla E., Larsen C. J., Gazzéri S. (2001) Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene 20, 1033–1041 [DOI] [PubMed] [Google Scholar]
- 29. Tsuji K., Mizumoto K., Sudo H., Kouyama K., Ogata E., Matsuoka M. (2002) p53-independent apoptosis is induced by the p19ARF tumor suppressor. Biochem. Biophys. Res. Commun. 295, 621–629 [DOI] [PubMed] [Google Scholar]
- 30. von Haefen C., Gillissen B., Hemmati P. G., Wendt J., Güner D., Mrozek A., Belka C., Dörken B., Daniel P. T. (2004) Multidomain Bcl-2 homolog Bax but not Bak mediates synergistic induction of apoptosis by TRAIL and 5-FU through the mitochondrial apoptosis pathway. Oncogene 23, 8320–8332 [DOI] [PubMed] [Google Scholar]
- 31. Gillissen B., Essmann F., Graupner V., Stärck L., Radetzki S., Dörken B., Schulze-Osthoff K., Daniel P. T. (2003) Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 22, 3580–3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sturm I., Stephan C., Gillissen B., Siebert R., Janz M., Radetzki S., Jung K., Loening S., Dörken B., Daniel P. T. (2006) Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 13, 619–627 [DOI] [PubMed] [Google Scholar]
- 33. Gillissen B., Essmann F., Hemmati P. G., Richter A., Oztop I., Chinnadurai G., Dörken B., Daniel P. T. (2007) Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J. Cell Biol. 179, 701–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hasenjäger A., Gillissen B., Müller A., Normand G., Hemmati P. G., Schuler M., Dörken B., Daniel P. T. (2004) Smac induces cytochrome c release and apoptosis independently from Bax/Bcl-x(L) in a strictly caspase-3-dependent manner in human carcinoma cells. Oncogene 23, 4523–4535 [DOI] [PubMed] [Google Scholar]
- 35. Wei M. C., Zong W. X., Cheng E. H., Lindsten T., Panoutsakopoulou V., Ross A. J., Roth K. A., MacGregor G. R., Thompson C. B., Korsmeyer S. J. (2001) Proapoptotic BAX and BAK. A requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Degenhardt K., Sundararajan R., Lindsten T., Thompson C., White E. (2002) Bax and Bak independently promote cytochrome C release from mitochondria. J. Biol. Chem. 277, 14127–14134 [DOI] [PubMed] [Google Scholar]
- 37. Korsmeyer S. J., Wei M. C., Saito M., Weiler S., Oh K. J., Schlesinger P. H. (2000) Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 7, 1166–1173 [DOI] [PubMed] [Google Scholar]
- 38. Nechushtan A., Smith C. L., Lamensdorf I., Yoon S. H., Youle R. J. (2001) Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 153, 1265–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Westphal D., Dewson G., Czabotar P. E., Kluck R. M. (2011) Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 1813, 521–531 [DOI] [PubMed] [Google Scholar]
- 40. Brenner D., Mak T. W. (2009) Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 21, 871–877 [DOI] [PubMed] [Google Scholar]
- 41. Giam M., Huang D. C., Bouillet P. (2008) BH3-only proteins and their roles in programmed cell death. Oncogene 27, S128–136 [DOI] [PubMed] [Google Scholar]
- 42. Willis S. N., Chen L., Dewson G., Wei A., Naik E., Fletcher J. I., Adams J. M., Huang D. C. (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gillissen B., Wendt J., Richter A., Müer A., Overkamp T., Gebhardt N., Preissner R., Belka C., Dörken B., Daniel P. T. (2010) Endogenous Bak inhibitors Mcl-1 and Bcl-xL. Differential impact on TRAIL resistance in Bax-deficient carcinoma. J. Cell Biol. 188, 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhai D., Jin C., Huang Z., Satterthwait A. C., Reed J. C. (2008) Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J. Biol. Chem. 283, 9580–9586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hikita H., Takehara T., Shimizu S., Kodama T., Li W., Miyagi T., Hosui A., Ishida H., Ohkawa K., Kanto T., Hiramatsu N., Yin X. M., Hennighausen L., Tatsumi T., Hayashi N. (2009) Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology 50, 1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bornkamm G. W., Berens C., Kuklik-Roos C., Bechet J. M., Laux G., Bachl J., Korndoerfer M., Schlee M., Hölzel M., Malamoussi A., Chapman R. D., Nimmerjahn F., Mautner J., Hillen W., Bujard H., Feuillard J. (2005) Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic Acids Res. 33, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen D., Kon N., Li M., Zhang W., Qin J., Gu W. (2005) ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 121, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 48. Zhong Q., Gao W., Du F., Wang X. (2005) Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 49. Ding Q., He X., Hsu J. M., Xia W., Chen C. T., Li L. Y., Lee D. F., Liu J. C., Zhong Q., Wang X., Hung M. C. (2007) Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell Biol. 27, 4006–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wendt J., von Haefen C., Hemmati P., Belka C., Dörken B., Daniel P. T. (2005) TRAIL sensitizes for ionizing irradiation-induced apoptosis through an entirely Bax-dependent mitochondrial cell death pathway. Oncogene 24, 4052–4064 [DOI] [PubMed] [Google Scholar]
- 51. Theodorakis P., Lomonosova E., Chinnadurai G. (2002) Critical requirement of BAX for manifestation of apoptosis induced by multiple stimuli in human epithelial cancer cells. Cancer Res. 62, 3373–3376 [PubMed] [Google Scholar]
- 52. LeBlanc H., Lawrence D., Varfolomeev E., Totpal K., Morlan J., Schow P., Fong S., Schwall R., Sinicropi D., Ashkenazi A. (2002) Tumor cell resistance to death receptor–induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat. Med. 8, 274–281 [DOI] [PubMed] [Google Scholar]
- 53. Suzuki H., Kurita M., Mizumoto K., Nishimoto I., Ogata E., Matsuoka M. (2003) p19ARF-induced p53-independent apoptosis largely occurs through BAX. Biochem. Biophys. Res. Commun. 312, 1273–1277 [DOI] [PubMed] [Google Scholar]
- 54. Cartron P. F., Juin P., Oliver L., Martin S., Meflah K., Vallette F. M. (2003) Nonredundant role of Bax and Bak in Bid-mediated apoptosis. Mol. Cell Biol. 23, 4701–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lindenboim L., Kringel S., Braun T., Borner C., Stein R. (2005) Bak but not Bax is essential for Bcl-xS-induced apoptosis. Cell Death Differ. 12, 713–723 [DOI] [PubMed] [Google Scholar]
- 56. Pardo J., Urban C., Galvez E. M., Ekert P. G., Müller U., Kwon-Chung J., Lobigs M., Müllbacher A., Wallich R., Borner C., Simon M. M. (2006) The mitochondrial protein Bak is pivotal for gliotoxin-induced apoptosis and a critical host factor of Aspergillus fumigatus virulence in mice. J. Cell Biol. 174, 509–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tait S. W., Green D. R. (2010) Mitochondria and cell death. Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632 [DOI] [PubMed] [Google Scholar]
- 58. Lindsten T., Ross A. J., King A., Zong W. X., Rathmell J. C., Shiels H. A., Ulrich E., Waymire K. G., Mahar P., Frauwirth K., Chen Y., Wei M., Eng V. M., Adelman D. M., Simon M. C., Ma A., Golden J. A., Evan G., Korsmeyer S. J., MacGregor G. R., Thompson C. B. (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Willis S. N., Fletcher J. I., Kaufmann T., van Delft M. F., Chen L., Czabotar P. E., Ierino H., Lee E. F., Fairlie W. D., Bouillet P., Strasser A., Kluck R. M., Adams J. M., Huang D. C. (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859 [DOI] [PubMed] [Google Scholar]
- 60. Cuconati A., Mukherjee C., Perez D., White E. (2003) DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 17, 2922–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. (2003) Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leu J. I., Dumont P., Hafey M., Murphy M. E., George D. L. (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 6, 443–450 [DOI] [PubMed] [Google Scholar]
- 63. Gélinas C., White E. (2005) BH3-only proteins in control. Specificity regulates MCL-1 and BAK-mediated apoptosis. Genes Dev. 19, 1263–1268 [DOI] [PubMed] [Google Scholar]
- 64. Gallenne T., Gautier F., Oliver L., Hervouet E., Noël B., Hickman J. A., Geneste O., Cartron P. F., Vallette F. M., Manon S., Juin P. (2009) Bax activation by the BH3-only protein Puma promotes cell dependence on antiapoptotic Bcl-2 family members. J. Cell Biol. 185, 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Itahana K., Bhat K. P., Jin A., Itahana Y., Hawke D., Kobayashi R., Zhang Y. (2003) Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol. Cell 12, 1151–1164 [DOI] [PubMed] [Google Scholar]
- 66. Bertwistle D., Sugimoto M., Sherr C. J. (2004) Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol. Cell Biol. 24, 985–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Warr M. R., Shore G. C. (2008) Unique biology of Mcl-1. Therapeutic opportunities in cancer. Curr. Mol. Med. 8, 138–147 [DOI] [PubMed] [Google Scholar]