

Background: EB1-recruited microtubule +TIP proteins mediate microtubule functions in interphase and mitosis.
Results: CLASP2 binding to EB1 requires electrostatic interactions that are inhibited by CDK- and GSK3-mediated multisite phosphorylation, and CLASP2 plus-end-tracking is switched off during mitosis.
Conclusion: Arginine-glutamate salt bridges contribute considerably to the binding energy between CLASP2 and EB1.
Significance: Multisite phosphorylation may be a general mechanism by which interactions of intrinsically disordered proteins are controlled.
Keywords: CDK (Cyclin-dependent Kinase), Glycogen Synthase Kinase 3, Intrinsically Disordered Proteins, Microtubules, Mitosis, Molecular Dynamics, CLASP2, EB1, Multisite Phosphorylation
Abstract
A group of diverse proteins reversibly binds to growing microtubule plus ends through interactions with end-binding proteins (EBs). These +TIPs control microtubule dynamics and microtubule interactions with other intracellular structures. Here, we use cytoplasmic linker-associated protein 2 (CLASP2) binding to EB1 to determine how multisite phosphorylation regulates interactions with EB1. The central, intrinsically disordered region of vertebrate CLASP proteins contains two SXIP EB1 binding motifs that are required for EB1-mediated plus-end-tracking in vitro. In cells, both EB1 binding motifs can be functional, but most of the binding free energy results from nearby electrostatic interactions. By employing molecular dynamics simulations of the EB1 interaction with a minimal CLASP2 plus-end-tracking module, we find that conserved arginine residues in CLASP2 form extensive hydrogen-bond networks with glutamate residues predominantly in the unstructured, acidic C-terminal tail of EB1. Multisite phosphorylation of glycogen synthase kinase 3 (GSK3) sites near the EB1 binding motifs disrupts this electrostatic “molecular Velcro.” Molecular dynamics simulations and 31P NMR spectroscopy indicate that phosphorylated serines participate in intramolecular interactions with and sequester arginine residues required for EB1 binding. Multisite phosphorylation of these GSK3 motifs requires priming phosphorylation by interphase or mitotic cyclin-dependent kinases (CDKs), and we find that CDK- and GSK3-dependent phosphorylation completely disrupts CLASP2 microtubule plus-end-tracking in mitosis.
Introduction
Dynamic regulation of the microtubule cytoskeleton is essential for many cell functions. A diverse class of plus-end-tracking proteins, +TIPs, that display a characteristic association with growing microtubule ends in cells has been implicated in the control of intracellular microtubule dynamics, signaling, chromosome segregation, and cell migration (1–3). Most +TIPs do not directly bind to growing microtubule ends but rely on interactions with end-binding proteins (EBs)3 that have emerged as central components of +TIP protein interaction networks. EBs are small dimeric proteins consisting of an N-terminal calponin homology domain that directly recognizes a structural feature of growing microtubule ends (4, 5) and a C-terminal EB homology domain that mediates binding to other +TIPs (6). A pocket between the two C-terminal helices in EB1 forms a hydrophobic cavity that binds to a short SXIP sequence motif present in most +TIPs (7). In addition, less well characterized electrostatic interactions between positively charged residues near the SXIP motif and negatively charged amino acids near the C terminus of EB1 likely contribute to +TIP binding. Functional SXIP motifs are embedded in intrinsically disordered regions, and the C-terminal tail of EB1 itself is unstructured and not resolved by x-ray crystallography (7). This has made direct structural investigation of the contribution and regulation of potential electrostatic interactions very difficult.
Except for the presence of one or multiple SXIP motifs, +TIPs are functionally and structurally extremely heterogeneous, and +TIP interactions with EB1 and hence microtubule ends must be spatially and temporally regulated in cells. Phosphorylation has a large impact on net protein charge and is thus a likely regulator of electrostatic interactions (8). Although there is currently no evidence that mammalian EBs are phosphorylated, phosphorylation has been reported for several +TIPs (9–14). Although it is unclear in most cases whether +TIP phosphorylation regulates microtubule plus-end-tracking in cells, we recently demonstrated that multisite phosphorylation of the EB1 binding region of CLASP2 by GSK3 inhibits EB1 and microtubule binding (9). An intriguingly similar regulation of microtubule binding by GSK3 was later described for the spectraplakin ACF7 (12). Both CLASP2 and ACF7 are involved in pathways stabilizing microtubules at the leading edge of migrating cells.
GSK3α and -β play essential roles in intracellular signaling, neuronal development, regulation of cell migration, cell division, and protein degradation pathways and are responsible for multisite phosphorylation of a large number of proteins (15, 16). Despite this central importance of GSK3 and its involvement in numerous disease processes, the molecular mechanisms by which multisite phosphorylation regulates protein functions are not well understood. Highly dynamic electrostatic interactions between disordered protein regions are likely central to many intracellular processes (17, 18), and here we use CLASP2 and EB1 as a model system to investigate how intrinsically disordered proteins can be regulated by multisite phosphorylation.
EXPERIMENTAL PROCEDURES
Plasmids, Protein Expression, and in Vitro Plus-end-tracking
pEGFP-CLASP2-(497–794) was made by cloning a PCR-amplified fragment between EcoRI and SalI restriction sites in pEGFP-C3. Point mutations to inactivate SXIP motifs and phosphorylation sites were generated by QuikChange II site-directed mutagenesis (Agilent Technologies). SXIP mutations in non-phosphorylatable CLASP2 constructs were first generated in published pEGFP-CLASP2-(497–1238) constructs (9) and subcloned into pEGFP-C3 using ScaI and SalI restriction sites. All CLASP2 residue numbering is based on NCBI reference sequence NP_055912.2.
For bacterial expression, His6-CLASP2-(497–794) was subcloned into pHAT2 between EcoRI and BamHI restriction sites. pHAT2-EB1 was constructed by amplifying the EB1 open reading frame from pEGFP-EB1 and cloned in pHAT2 between NcoI and BamHI restriction sites. Proteins were expressed in Escherichia coli BL21 (DE3) and purified using standard protocols. Briefly, after induction with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside, proteins were expressed for 4–6 h, cell lysates were prepared using a Microfluidizer (Microfluidics), and proteins were purified by affinity chromatography on TALON resin (Clontech) in 25 mm HEPES, 400 mm KCl, 5% glycerol, 5 mm MgCl2, 0.1% Triton X-100, 10 μg/ml protease inhibitors (aprotinin, pepstatin, leupeptin), and 1 mm β-mercaptoethanol.
For the in vitro microtubule plus-end-tracking assay, double-sided tape flow chambers using poly-l-lysine-PEG-passivated slides and biotin-PEG-functionalized coverslips were prepared exactly as described (19). Brightly Alexa-568-labeled, biotinylated guanylyl-(α,β)-methylene-diphosphonate (GMPCPP)-stabilized microtubule seeds prepared as described (20) were immobilized in these flow chambers. After blocking and washing steps (20), 15 μm cycled tubulin (21) containing <10% Alexa-568-labeled tubulin, 150 nm unlabeled EB1, and 325 nm EGFP-CLASP2-(497–794) in an oxygen-scavenging system containing assay buffer (20) were perfused into the chamber. The chambers were sealed with VaLaP (1:1:1, vaseline:lanoline:paraffin) and imaged by spinning disk confocal microscopy at 37 °C.
Cell Culture, Live Cell Imaging, and Analysis
HeLa cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C and 5% CO2. For imaging, HeLa cells were grown on 35-mm, #1.5 thickness glass-bottom dishes (Mattek), and EGFP-CLASP2 constructs were introduced by transfection with FuGENE HD transfection reagent (Roche Applied Science). Cells were arrested in metaphase either with 5 μm S-trityl-l-cysteine (STLC) (Tocris Bioscience) or 10 μm MG132 (Calbiochem). The CDK1 inhibitor RO-3306 (EMD Biosciences) was added at 9 μm. For biochemistry, STLC-arrested cells were harvested by mitotic shake-off, and metabolic labeling was performed as described (9).
Live cell spinning disk confocal microscopy was performed as described (9). Fluorescence intensity of EGFP-CLASP2-(497–794) constructs along microtubule plus ends was measured using the “Intensity Profile” function in NIS Elements (Nikon) for three microtubules per cell keeping illumination, exposure, and measurement tool settings constant. Intensity profiles were corrected for local background by subtracting an intensity profile adjacent to the measured microtubule end and normalized for expression level by dividing the background-corrected profile by the average of the background intensity profile minus camera offset. These normalized profiles were fitted with an exponentially modified Gaussian function (22),
 |
Profiles from 10 cells were aligned to the maximum of the fit and averaged. Least square curve-fitting was done using the Solver function in Microsoft Excel.
Molecular Dynamics Simulations
The crystal structure of the C-terminal domain of human EB1 bound to microtubule actin crosslinking factor (MACF) peptides (PBD ID 3gjo) was used as the template to build the homology model of EB1-bound CLASP2 peptides with in-house software (Protein Localization Optimization Program PLOP; distributed as Prime by Schrodinger, LLC) (23) by aligning SXIP motifs in CLASP2 and MACF. The N and C termini of the CLASP2 peptides were capped to neutralize charges. The final model was energy-minimized by a Truncated Newton method (24). Ionizable groups were assumed to have model compound-like pKa values. Each model was placed in an orthorhombic water box with a minimum of 10 Å from any protein heavy atom to the edge of the box. Counterions were added to make the system electrically neutral, and NaCl concentration was set to 0.1 m. Molecular dynamics simulations were performed with the Desmond simulation package (D.E. Shaw Research) (25). The OPLS_2005 forcefield (26) was used for protein, and the TIP3P model was used for waters (27). Partial charges on the phosphate groups were as follows: +1.91 (phosphorus), −0.76 (ester oxygen), and −1.11 (acidic oxygens). A two-stage approach was used to energy-minimize the simulation systems, with a steepest-descent algorithm followed by a conjugate gradient method with the solute restrained. In the first stage, the protein complex was fixed, and only the water and ions were energy-minimized. In the second stage, the entire system was energy-minimized. The minimization was then repeated without restraints on the solute. Long-range electrostatic forces were treated with the Particle Mesh Ewald method (28), subject to a 9 Å cutoff. For all simulations, the SHAKE algorithm (29) was used to constrain hydrogen-heavy atom bonds.
Equilibration was accomplished in four stages. First, the system was simulated in the canonical (NVT) ensemble for 12 ps at a temperature of 10 K with a fast temperature relaxation constant and non-hydrogen solute atoms restrained. Second, the system was simulated in the isothermal-isobaric (NPT) ensemble for 12 ps at 10 K and 1 atm with a fast temperature relaxation constant, a slow pressure relaxation constant, and non-hydrogen atoms restrained. Next, the temperature was increased to 300 K, and the system was simulated for 24 ps. Finally, the system was simulated for 24 ps at 300 K and a pressure of 1 atm with a fast temperature relaxation constant and a normal pressure relaxation constant. For production molecular dynamics, the system was simulated for 20 ns with a 2-fs time step. Independent simulations (at 300 K and 1 atm) were performed with different initial velocities for each system.
DELPHI, which solves the Poission-Boltzmann equation by finite difference methods (30), was employed to calculate the electrostatic potential of the system at different points in the trajectories of the non-phosphorylated and phosphorylated states. Interior dielectric was set at 4, and exterior dielectric was set at 80.0. A box size of 300 Å was chosen in which the molecule occupied 50% of the box volume. Boundary conditions were set to “coulombic,” wherein the boundary is approximated by the sum of Debye-Huckel potentials of all the charges. Scaling was set to 2 grids/Å.
Fluorescence Anisotropy Binding
EB1 binding of N-terminally fluorescein isothiocyanate (FITC)-labeled CLASP2 peptides (Biomatik) was monitored by the increase in fluorescence polarization upon titration of concentrated EB1 protein. The assay was performed in 384-well plates containing 10 nm concentrations of labeled peptide in each well in 20 mm HEPES, pH 7.4, 50 mm (or 250 mm) NaCl, and 2 mm DTT at room temperature. 0.01 mg/ml BSA was added to minimize nonspecific binding. Fluorescence polarization was measured using an Analyst HT Fluorometer (Molecular Devices) with 485-nm excitation and 530-nm emission, and data were fit to the binding equation,
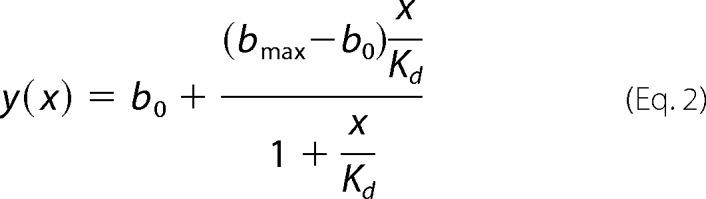 |
31P NMR Spectroscopy
Peptides were dissolved to a final concentration of 600 μm in 20 mm HEPES, 50 mm NaCl, and 10% D2O. 500 μl were placed in Wilmad 535 round-bottom tubes (Wilmad). One-dimensional spectra were acquired at 25 °C with between 500 and 1000 transients on a Varian Inova 600-MHz spectrometer with broadband cryoprobe located in the UCSF NMR facility at Mission Bay. pH was adjusted with 1 n HCl, and NaOH added in 1-μl aliquots at room temperature and measured using a custom NMR glass combination electrode (Mettler Toledo). 31P spectra were referenced indirectly to 4,4-dimethyl-4-silapentane-1-sulfonic acid (31).
In Vitro Phosphorylation Assays
Recombinant human cycB/CDK1 (PV3292), cycA/CDK2 (PV3267), and p25/CDK5 (PV4676) with specific activities of 1.4, 1.8, and 8.4 μmol/min/mg were from Invitrogen. cycD1/CDK4 (7530) with a specific activity 0.154 μmol/min/mg was obtained from Cell Signaling. In vitro kinase reactions were performed with 5 μg of CLASP2-(497–794), 50 ng of CDKs, 100 μm ATP, 5 μCi of [γ-32P]ATP (NEG502A, PerkinElmer Life Sciences) in 100 μl of 50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 0.1 mm EDTA, 2 mm DTT, 0.01% Brij 35 at 30 °C. For autoradiography, 20-μl aliquots were removed after 10 min, and the reaction was stopped by the addition of 20 μl of 2× SDS sample buffer (161-0737, Bio-Rad). Autoradiographs were quantified using a STORM PhosphorImager (GE Healthcare). For progress curve analysis, 10 μl of the kinase reaction were spotted onto P81 phosphocellulose squares (20-134, Millipore), and samples were washed and quantified in a scintillation counter as described (32).
For analysis of priming-dependent GSK3β phosphorylation, 100 μm CLASP2 peptides were phosphorylated in 10 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 2.5 mm dithioerythritol, 4 mm ATP, and EDTA-free protease inhibitor mixture (Roche Applied Science) with a 1:1000 molar ratio of GSK3β (New England Biolabs). The reaction was stopped after 30 min by the addition of 50% methanol, subsequently diluted 1:10 with 50% methanol and 1% formic acid, and applied to an LCQ advantage electrospray ionization mass spectrometer (Thermo Fisher Scientific). Mass spectra were analyzed using Xcalibur software (Thermo Fisher Scientific), and charge states were deconvolved using MagTran software (33).
RESULTS
EB1 Is Necessary and Sufficient for CLASP2 Microtubule Plus-end-tracking
The central, disordered region of vertebrate CLASP proteins contains two partially conserved SXIP sequence motifs that are required to bind to EB1 (Fig. 1, A and B). Although it has been shown that SXIP motifs in other +TIPs are sufficient to mediate plus-end-tracking in the presence of EB1 (7), this has not been directly demonstrated for CLASPs. We, therefore, reconstituted plus-end-tracking of the central CLASP2 domain using an in vitro assay in which dynamic microtubules are polymerized from biotinylated GMPCPP microtubule seeds (19). Consistent with published in vitro microtubule binding data of the central CLASP2 domain (9, 34, 35), purified, recombinant EGFP-tagged CLASP2-(497–794) bound weakly along microtubules in this assay. In the presence of EB1 protein, EGFP-CLASP2-(497–794) accumulated at growing microtubule plus ends and rapidly dissociated from microtubule ends when microtubules paused or transitioned to shortening (Fig. 1, C and D; supplemental Video 1). In contrast, we observed no accumulation at growing microtubule ends without EB1 or with EGFP-CLASP2-(497–794) protein in which both SXIP motifs were mutated to SXNN (data not shown). Thus, EB1 is both necessary and sufficient to mediate CLASP2 plus-end-tracking.
FIGURE 1.

CLASP2 microtubule plus end association is EB1-dependent. A, PONDR-FIT prediction of intrinsically disordered regions in human CLASP2 shows good agreement with known folded domains. B, the domain structure of human CLASP2 and sequence comparison of the EB1 binding region in chordate and insect species highlights the high degree of conservation of SXIP motif-associated arginines (blue) and GSK3 (red) and CDK (yellow) phosphorylation sites. SXIP motifs or GSK3 phosphorylation sites are not easily identifiable in plant or fungal CLASP homologues. Amino acid numbering in this paper is based on the most recent NCBI reference sequence NP_055912.2 and is different from the numbering we previously used (9). C, shown are three representative examples of in vitro microtubule plus-end-tracking of the central SXIP motif-containing CLASP domain, EGFP-CLASP2-(497–794). Panels on the left are overlays of the EGFP channel (green) and X-rhodamine-labeled tubulin (purple; the bright region is the GMPCPP-stabilized microtubule seed). Panels on the right are kymographs of the EGFP channel showing EGFP-CLASP2-(497–794) accumulation at growing microtubule plus ends. Scale bar, 5 μm. D, shown is average fluorescence intensity profile of EGFP-CLASP2-(497–794) along growing microtubules (n = 20). The dashed line indicates the 95% confidence interval, and the solid line is an exponentially modified Gaussian fit. a.u., absorbance units.
Distributed Multisite Phosphorylation Inhibits SXIP-mediated Microtubule Plus-end-tracking
In addition to the SXIP motif, less well characterized electrostatic interactions of +TIPs with EB1 contribute to EB1 binding and EB1-mediated plus-end-tracking (7). CLASP SXIP motifs are embedded in an intrinsically disordered, highly positively charged, serine- and arginine-rich region that is highly conserved across species (Fig. 1, A and B). Each SXIP motif is C-terminally flanked by conserved GSK3 phosphorylation sites that negatively regulate microtubule binding (9), suggesting that phosphorylation may also directly modulate the interaction with EB1.
To determine the relative contribution of phosphorylation of these two EB1 binding modules to the regulation of CLASP2 plus-end-tracking in cells, we quantitatively analyzed the intracellular localization of transiently expressed EGFP-CLASP2-(497–794) constructs in which either one or both SXIP motifs and/or associated phosphorylation sites were inactivated by mutation (7, 9). In cells, these constructs localized to growing microtubule ends and did not bind along microtubules. Occasional nuclear localization was highly variable and of unclear functional relevance (9, 35). An exponentially modified Gaussian function, which approximates the convolution of a single exponential decay with the microscope point spread function, provided a high quality fit for microtubule plus-end-associated EGFP-CLASP2-(497–794) fluorescence intensity profiles both in vitro and in cells (Figs. 1D and 2D). This is consistent with first order decay kinetics of EGFP-CLASP2-(497–794) binding sites at growing microtubule ends (36). The integrated area underneath such fits normalized to EGFP-CLASP2-(497–794) expression levels provided a relative measure for the amount of plus end-associated CLASP2 and thus equilibrium binding to EB1 in cells (Fig. 2D; Table 1).
FIGURE 2.

CLASP2 plus end association is negatively regulated by distributed multisite phosphorylation. A–C, shown are representative images of the localization of the indicated EGFP-CLASP2-(497–794) constructs in HeLa cells. Insets show intracellular regions at higher magnification. Scale bar, 10 μm. A, shown is a comparison of constructs with wild-type GSK3 phosphorylation sites in which one or both SXIP motifs were inactivated by site-directed mutagenesis. B, shown is a comparison of constructs in which all GSK3 phosphorylation sites were replaced with non-phosphorylatable alanine residues (9× S/A), and one or both SXIP motifs were inactivated by site-directed mutagenesis. Either SXIP motif can support plus-end-tracking in the non-phosphorylatable background. C, shown is a comparison of constructs in which different combinations of SXIP and GSK3 motifs were inactivated. All constructs support plus-end-tracking to a similar extent, indicating large flexibility in phospho-regulation of the SXIP motifs. D, shown are average fluorescence intensity profiles of the indicated EGFP-CLASP2-(497–794) constructs along growing microtubules normalized to the intracellular expression level (n = 10 cells; 3 microtubules per cell). The solid lines are exponentially modified Gaussian fits.
TABLE 1.
Quantification of intracellular EGFP-CLASP2-(497–794) microtubule plus end association relative to the level of the wild-type construct
Mutations of SXIP motifs or phosphorylation sites are indicated in bold.
| CLASP construct | Relative plus end association (mean ± S.D.; n = 10 cells) |
|---|---|
| SKIP-S-S-S—SRIP-S-S-S-S-S | 1.00 ± 0.24 |
| SKNN-S-S-S—SRIP-S-S-S-S-S | 0.68 ± 0.11 |
| SKIP-S-S-S—SRNN-S-S-S-S-S | 0.44 ± 0.16 |
| SKNN-S-S-S—SRNN-S-S-S-S-S | No plus-end-tracking |
| SKIP-A-A-A—SRIP-A-A-A-A-A | 2.73 ± 0.54 |
| SKNN-A-A-A—SRIP-A-A-A-A-A | 1.74 ± 0.26 |
| SKIP-A-A-A—SRNN-A-A-A-A-A | 1.68 ± 0.40 |
| SKNN-A-A-A—SRNN-A-A-A-A-A | No plus-end-tracking |
| SKNN-A-A-A—SRIP-S-S-S-S-S | 1.75 ± 0.39 |
| SKNN-S-S-S—SRIP-A-A-A-A-A | 1.93 ± 0.29 |
| SKIP-A-A-A—SRNN-S-S-S-S-S | 1.70 ± 0.33 |
| SKIP-S-S-S—SRNN-A-A-A-A-A | 1.75 ± 0.43 |
A CLASP2 construct in which all GSK3 sites were replaced with non-phosphorylatable alanine residues, EGFP-CLASP2-(497–794) 9× S/A, and in which both SXIP motifs were intact, displayed maximum association with microtubule plus ends (Fig. 2, B and D). Plus-end-tracking was reduced in constructs with only one functional SXIP motif, but both constructs associated equally well with microtubule plus ends, indicating that both SXIP motifs are functionally equivalent in the absence of phosphorylation. As expected (7), mutation of both SXIP motifs completely eliminated plus-end-tracking. Compared with the non-phosphorylatable construct, plus-end-tracking of wild-type EGFP-CLASP2-(497–794) was reduced ∼3-fold (Fig. 2, A and D; Table 1). Because the GSK3 motifs are partially phosphorylated in cells (9, 14), this indicates that phosphorylation directly inhibits EB1 binding. In addition, inactivation of either SXIP motif reduced plus-end-tracking, but the two SXIP motifs were not equal. Mutation of the N-terminal SKIP motif had little effect, whereas mutation of the more C-terminally located SRIP motif alone almost completely abolished plus-end-tracking. We previously reported that mainly GSK3 sites C-terminal of the SRIP motif are phosphorylated (9, 14), and this suggests that inhibition of SXIP-mediated EB1-association is not directly correlated to the distance of phosphorylation sites to the SXIP motif. Alternatively, more complex phosphorylation patterns may exist in vivo.
To further test phosphoregulation of either SXIP motif, we generated constructs with only one functional SXIP motif and either one or the other GSK3 phosphorylation motif replaced by non-phosphorylatable alanine residues. Surprisingly, all four of these constructs supported plus end association equally well (Fig. 2, C and D; Table 1) and to a very similar extent as constructs with one functional SXIP motif and all phosphorylation sites mutated (Fig. 2, B and D). Thus, within the CLASP2 plus-end-tracking domain a subset of possible electrostatic interactions is sufficient to mediate maximal EB1 binding of either SXIP motif. Taken together, these data confirm that SXIP motifs are required for CLASP2 plus-end-tracking in cells (7) and that CLASP2 plus-end-tracking is inhibited by phosphorylation (9). However, these data further demonstrate that the two CLASP SXIP motifs and associated phosphorylation sites do not function as independent EB1 binding modules. Electrostatic interactions that contribute to EB1 binding are complex and exhibit a large degree of flexibility, which is consistent with the interaction of intrinsically disordered protein domains.
Multisite Phosphorylation Tunes CLASP2-EB1 Interaction Strength
To more directly test how phosphorylation affects binding to EB1, we devised an in vitro assay to determine equilibrium binding constants of the CLASP2-EB1 interaction. Our in vivo data showed that EB1 binding could be mediated by either SXIP motif and supported by positive charges distributed throughout the CLASP2 plus-end-tracking domain. For simplicity, we chose the smallest EB1 binding module consisting of a 25-amino acid peptide encompassing the N-terminal SKIP and the associated GSK3 phosphorylation motif. Because GSK3 multisite phosphorylation requires phosphorylation of priming sites at the C-terminal end of GSK3 motifs (15, 16), we compared EB1 binding to CLASP2 peptides that were either non-phosphorylated or synthesized with phosphoserine residues only at the priming site (Ser-741) or all phosphorylated positions (Ser-733, Ser-737, and Ser-741). Binding of FITC-labeled peptides to bacterially produced EB1 protein was monitored by fluorescence anisotropy (37).
At low salt concentrations, the non-phosphorylated peptide bound EB1 with moderately high affinity with a dissociation constant (Kd) of around 1 μm. Binding affinity decreased substantially with increasing salt concentration, which is consistent with a large portion of the binding free energy resulting from electrostatic interactions (Fig. 3). Affinity of the mono-phosphorylated peptide was reduced by ∼5-fold. In contrast, we did not detect any binding of the triple-phosphorylated peptide to EB1 even at the lowest salt concentration tested. These data demonstrate that phosphorylation directly controls binding to EB1, likely by modulating electrostatic interactions, and that multisite phosphorylation is necessary and sufficient to completely disrupt the interaction in a physiological range of salt concentrations.
FIGURE 3.

CLASP2 multisite phosphorylation directly tunes EB1-binding. A, shown are normalized binding curves of the non-phosphorylated and mono-phosphorylated CLASP2 peptide to EB1 at the indicated salt concentrations. Error bars indicate S.D. Dissociation constants summarized in B were obtained by direct curve fitting (solid lines).
Phosphorylation Induces Intramolecular Salt Bridges in the CLASP2 Plus-end-tracking Domain
The modulation of CLASP-EB1 binding by phosphorylation could be mediated by phosphorylation-induced conformational changes in the unbound CLASP peptide, the CLASP-EB1 complex, or both. To further elucidate the mechanism by which phosphorylation regulates CLASP2-EB1 binding, we performed in silico molecular dynamics simulations of the same SKIP peptide as used in the in vitro binding experiments. Starting from an extended conformation, multiple independent 40–50-ns molecular dynamics simulations of either the non-phosphorylated or triple-phosphorylated peptide were carried out in explicit solvent, which yields good agreement with experimental results for phosphopeptide simulations (38). Although the simulation of disordered proteins in explicit solvent suffers from a potential under-sampling problem, the simulations did explore a sufficiently large conformational space to allow interactions between arginine and phosphoserine side chains. In all simulations of the triple-phosphorylated peptide 4-6 long-lived bidentate salt bridges between arginine and phosphoserine residues emerged rapidly (Fig. 4B). In contrast, the arginines remained in bulk water in the non-phosphorylated peptide simulations and only formed few transient intramolecular hydrogen bonds with other residues (Fig. 4A).
FIGURE 4.

CLASP2 phosphorylation induces intramolecular phosphoserine-arginine salt bridges. Representative structures of molecular dynamics simulations of the non-phosphorylated (A) or triple-phosphorylated CLASP2 peptide (B) are shown. Arginine residues, phosphoserines, and hydrogen bonds formed are shown. C, titration of the phosphorylated serine residue in the mono-phosphorylated CLASP2 peptide by 31P NMR spectroscopy is shown. The solid line represents a modified Hill equation fit to determine the acid dissociation constant. The inset shows 31P NMR spectra at high and low pH.
To test whether phosphoserine participates in salt bridges in the CLASP2 plus-end-tracking domain, we determined the acid dissociation constant of the phosphate group in the monophosphorylated CLASP2 peptide by 31P NMR spectroscopy. As expected, one-dimensional NMR spectra revealed a single, narrow peak at a chemical shift of −2.4 ppm at pH 3.1. At higher pH, this peak broadened slightly but was still well resolved and shifted downfield to 1.4 ppm at pH 8.7 (Fig. 4C). Titration between these pH values yielded a pKa of 5.4 with a Hill coefficient of 0.97 corresponding to the −2 to −1 charge transition of the phosphate group. Thus, the pKa is downshifted by ∼0.7 units compared with published pKa values of 6.0–6.1 of solvent-exposed phosphoserine residues in short model peptides or ovalbumin (39–41). This downshifted pKa value is consistent with strong salt bridge interactions that favor the −2 charged state of the phosphate group and would, therefore, make it a stronger acid (42). Taken together with the molecular dynamics data, the pKa downshift strongly supports intramolecular salt bridge formation in the phosphorylated CLASP2 plus-end-tracking domain.
Phosphorylation Disrupts Arginine-Glutamate Hydrogen Bond Networks between CLASP2 and EB1
Although the structure of the interaction between a SXIP motif containing +TIP fragment and EB1 has been solved by x-ray crystallography (7), the highly negatively charged C-terminal tail of EB1 is unstructured and is not present in the crystal structure. To better understand how electrostatic interactions between negatively charged residues in EB1 and positively charged residues surrounding the SXIP motifs contribute to EB1 binding and are regulated by phosphorylation, we performed molecular dynamics simulations of the CLASP2-EB1 interaction.
A homology model was built so that the N-terminal CLASP2 SKIP motif was aligned with the homologous sequence in the MACF-EB1 crystal structure in which the SXIP motif occupies a hydrophobic binding pocket between the two C-terminal α-helices of EB1. The C-terminal eight amino acids of EB1 and residues N- and C-terminal to the SXIP motif that are unstructured and missing from the crystal structure were built in a fully extended conformation to minimize bias in the molecular dynamics simulation. Because EB1 is a dimer, the model contained two CLASP2 peptides bound to either SXIP binding pocket. To test how phosphorylation affects interactions with EB1, both simulations were started from an EB1-bound state. For the triple-phosphorylated CLASP2 peptide that does not bind EB1 in in vitro binding experiments (Fig. 3), this is a somewhat hypothetical situation that, however, does not affect the thermodynamics of the system. Molecular dynamics simulations were carried out in explicit solvent, and over the course of 20 ns simulations the portion of the non-phosphorylated CLASP2 peptide outside of the actual SXIP binding pocket folded into more compact structures that optimized ion-pairing and hydrogen bonding interactions with EB1 (Fig. 5A). Calculation of the electrostatic potential by solving the Poisson-Boltzmann equation utilizing finite difference methods showed a rapid decrease of the total coulombic energy of the system toward an electrostatically more favorable state within the first 5 ns.
FIGURE 5.

CLASP2 phosphorylation disrupts electrostatic interactions with glutamate residues in the EB1 C terminus. Representative structures of molecular dynamics simulations of EB1-bound non-phosphorylated (A) or triple-phosphorylated CLASP2 peptide (B) are shown. The EB1 dimer is shown in orange/red; CLASP2 peptides are cyan/blue. The structures on the right highlight observed arginine-glutamate and arginine-phosphoserine salt bridges from different simulation runs. Only relevant residues are shown. C and E, shown is the total number of hydrogen bonds formed between the EB1 dimer and the non-phosphorylated or triple-phosphorylated CLASP2 peptide (C) or CLASP2 peptides in which the middle three arginines were replaced with lysine residues (E). Data points are the average of three independent simulation runs each; solid lines represent the S.D. Data were smoothed with a 20-frame running average. D and F, shown is the average number of hydrogen bonds formed over the last 5 ns of the simulation by each individual arginine residue (D) or substituted lysine residues (F) in the CLASP2 peptide with residues in EB1 (left graph) or residues in the CLASP2 peptide itself (right graph). Error bars indicate 95% confidence intervals.
The number of hydrogen bonds between the CLASP2 peptide and EB1 also rapidly converged toward a steady state plateau (Fig. 5C), and inspection of the molecular dynamics trajectories revealed that conserved arginine residues in the CLASP2 peptide were responsible for forming the majority of intermolecular hydrogen bonds with EB1 (Fig. 5A). These arginines preferentially formed bidentate salt bridges with conserved glutamate residues in EB1, and single arginine residues often interacted with pairs of glutamate residues in the EB1 C terminus. In contrast, in simulations with the triple-phosphorylated SKIP peptide, the same arginine residues preferentially formed intramolecular bidentate salt bridges with phosphoserines (Fig. 5B). All five arginines contributed to 5.1 ± 1.7 intramolecular hydrogen bonds with phosphoserines (compared with only 0.8 ± 0.9 intramolecular hydrogen bonds between arginines and other residues in the non-phosphorylated simulations; n = 6 peptides from 3 independent simulations; Fig. 5D). In parallel, the number of intermolecular hydrogen bonds between the SKIP peptide and EB1 was significantly reduced, mostly due to fewer arginine salt bridges with EB1 (4.0 ± 1.3 compared with 6.5 ± 1.7 hydrogen bonds in the non-phosphorylated simulations; p = 0.014). Non-arginine intermolecular hydrogen bonds were less affected by phosphorylation (3.2 ± 0.7 compared with 4.5 ± 0.3 in the non-phosphorylated simulations), and these were mostly contributed by a core group of backbone interactions in the SXIP binding pocket (7). We also performed simulations in which arginines at positions 732, 738, and 744 were substituted with lysines. Although this conserves the overall charge of the peptide at neutral pH, the total number of intermolecular hydrogen bonds formed with EB1 was substantially reduced, and the lysine residues contributed only very little to interactions with EB1 (Fig. 5F). In addition, the number of intermolecular hydrogen bonds was nearly identical for both the non-phosphorylated and triple-phosphorylated peptide (Fig. 5E).
Together these molecular dynamics simulations demonstrate that distributed arginine residues in the CLASP2 plus-end-tracking domain are required to form strong bidentate salt bridge interactions with glutamate residues in the EB1 C terminus. These interactions can be disrupted by phosphate groups at GSK3 phosphorylation sites, which efficiently compete for arginine salt bridge interactions.
Cyclin-dependent Kinases Phosphorylate CLASP2 GSK3 Priming Sites
Although multisite phosphorylation by GSK3 disrupts CLASP2-EB1 interactions, the intracellular signals controlling this phosphorylation are incompletely understood. In the CLASP2 sequence, priming sites for GSK3 phosphorylation are highly conserved (S/T)PX(R/K) cyclin-dependent kinase (CDK) phosphorylation motifs (Fig. 1B). To test whether the GSK3 phosphorylation pattern observed in cells can be explained by differential phosphorylation of these priming sites, we examined in vitro phosphorylation of CLASP2-(497–794) by different CDKs.
We first analyzed phosphorylation by cycB/CDK1. In an end-point assay in which we determined phosphate incorporation after 10 min, mutation of either Ser-741 or Ser-775 to alanine reduced phosphorylation slightly. Mutation of both predicted priming sites decreased phosphorylation by ∼60%, indicating that these sites are phosphorylated, but that additional CDK1 phosphorylation occurred elsewhere. Further examination of the CLASP2-(497–794) peptide sequence revealed an additional consensus site at Ser-697, and triple mutation of Ser-697, Ser-741, and Ser-775 completely abolished CDK1 phosphorylation in vitro (Fig. 6A). Although Ser-697 is part of a third potential GSK3 motif, this motif does not appear to be phosphorylated, as mutation of the two other GSK3 motifs completely abolishes GSK3-dependent phosphorylation (see Fig. 8B) (9). Ser-697 is also not associated with a SXIP motif, and its functional significance remains unclear.
FIGURE 6.

Mitotic and interphase CDKs phosphorylate priming sites in the CLASP2 GSK3 motifs. A, shown is in vitro phosphorylation of CLASP2-(497–794) and the indicated phosphorylation site mutations by cycB/CDK1. Top panel, autoradiograph; bottom panel, corresponding Coomassie-stained gel. Sizes of molecular mass markers (kDa) are indicated on the left. B, shown is quantification of phosphate incorporation from three independent experiments (indicated by different shaped symbols) normalized to the average wild-type value. Three serine residues are phosphorylated by cycB/CDK1 in the CLASP2 plus-end-tracking domain, and Ser-741 appears to be the preferred site. C and D, shown are progress curves of phosphorylation of the indicated double priming site mutant constructs by cycB/CDK1 (n = 4) and p25/CDK5 (n = 3). Data are normalized to the wild-type protein in each experiment. Error bars indicate 95% confidence intervals, and the progress curves were approximated by an exponential fit (solid lines). As judged by the initial rates, p25/CDK5 appears to prefer Ser-741 even more than cycB/CDK1.
FIGURE 8.

CLASP2 plus-end-tracking is inhibited during mitosis. A, shown are interphase cells expressing either wild-type EGFP-CLASP2-(497–1238) or the double-priming site mutant (S741A/S775A) together with constitutively active mRFP-GSK3β(S9A). Insets indicate GSK3β expression. B, shown is metabolic labeling of HeLa cells with 32P-labeled phosphate. EGFP-CLASP2-(497–1238) was immunoprecipitated and analyzed by SDS-PAGE and autoradiography. Note that radioactivity incorporation is not directly comparable between non-synchronized and STLC-arrested cells because of differences in loading due to limited recovery of mitotic cells. However, the increased gel shift in STLC-arrested cells indicates increased mitotic phosphorylation. In addition, decreased radioactivity incorporation and downshift in the presence of a GSK3 inhibitor (20 μm SB216763) or of the non-phosphorylatable mutant (9× S/A) demonstrates GSK3-mediated phosphorylation in both interphase and mitosis. C, shown are HeLa cells in metaphase expressing wild-type (WT) EGFP-CLASP2-(497–1515) or constructs in which all GSK3 sites (9× S/A) or only SXIP motif-associated CDK priming sites (S741A/S775A) were mutated. Insets show the indicated regions at higher magnification. D, shown is metaphase cell-expressing EB1-EGFP. E, HeLa cells expressing the indicated EGFP-CLASP2-(497–1238) constructs that lack the kinetochore binding domain were treated with STLC, which results in monopolar spindles arrested in a metaphase-like state. F, quantification of data in E demonstrates that mutation of the priming sites alone restores CLASP2 plus-end-tracking during mitosis. Each symbol represents the average EGFP-CLASP2-(497–1238) fluorescence intensity in the cytoplasm of one cell (n = 80). a.u., arbitrary units. G, EGFP-CLASP2-(497–1515)-expressing HeLa cells arrested in metaphase with MG132 and subsequently treated with the CDK1 inhibitor RO-3306 results in the reappearance of EGFP-CLASP2-(497–1515) on microtubules. Scale bars, 10 μm.
In several experiments, S741A was noticeably less phosphorylated than S697A and S775A (Fig. 6, A and B), suggesting that these three sites are not completely equal. To test whether Ser-741 is preferentially utilized by CDK1, we analyzed phosphorylation of double mutant constructs in which only one of the three CDK sites was functional and the other two were replaced by non-phosphorylatable alanines. CLASP2-(497–794) S697A/S775A protein, in which Ser-741 is the only functional CDK site, was phosphorylated significantly more efficiently than both other constructs (Fig. 6B). Phosphorylation of the S697A/S775A protein also exhibited a slightly increased initial rate in phosphorylation time-course experiments compared with the other double phosphorylation site mutants (Fig. 6C).
In interphase cells, the N-terminal GSK3 motif associated with the CDK priming site at Ser-741 does not appear to be phosphorylated (9). This suggests that Ser-741 may not be phosphorylated by interphase CDKs. To test whether this could be due to different priming site utilization by different CDKs, we analyzed phosphorylation of the double-mutant constructs by p25/CDK5 that has proposed functions in migrating interphase cells (43). Surprisingly, CDK5 showed an even more pronounced preference for Ser-741. The initial rate of phosphorylation was ∼2-fold increased over the other two CDK sites and indistinguishable from the wild-type construct containing all three functional CDK sites (Fig. 6D). Thus, the site specificity of CDK5 cannot explain the pattern of GSK3 phosphorylation observed in interphase cells (9).
Priming Site Phosphorylation Is Required for CLASP2 Phosphorylation by GSK3β
To test whether phosphorylation of the CDK sites is needed for efficient phosphorylation by GSK3β, we compared GSK3β phosphorylation of CLASP2 peptides synthesized with phosphoserines in different positions. Because phosphorylation causes a characteristic increase in molecular mass of ∼80 Da, we analyzed the products of these in vitro kinase reactions by mass spectrometry.
No phosphorylated peptide species were detected in GSK3β kinase reactions in which the non-phosphorylated CLASP2 peptide was used as substrate (Fig. 7A). In contrast, GSK3β phosphorylation of the CLASP2 peptide that had a phosphoserine at the priming position only (Ser-741) or at all three previously confirmed phosphorylated positions (Ser-733, Ser-737, and Ser-741) resulted in a mass increase of ∼240 Da (Fig. 7B) or ∼80 Da (Fig. 7C), indicating incorporation of three or one additional phosphates. This demonstrates that priming site phosphorylation is required for phosphorylation by GSK3β. However, it also indicates that at least in vitro, GSK3β phosphorylates an additional residue. Remarkably, we also did not detect any double- or triple-phosphorylated intermediate products (Fig. 7B), suggesting processivity of the GSK3β phosphorylation reaction.
FIGURE 7.

GSK3β phosphorylation requires priming site phosphorylation. Charge-state-deconvolved mass spectra of GSK3β in vitro kinase reaction products using either non-phosphorylated (A), mono-phosphorylated (B), or triple-phosphorylated CLASP2 peptide as substrate (C). Representative spectra of three independent experiments are shown. The indicated molecular masses include the peptide FITC tag. a.u., arbitrary units.
Priming phosphorylation is also required in cells. Expression of constitutively active GSK3β(S9A) results in hyperphosphorylation and CLASP2 dissociation from microtubules (9). Mutation of only the CDK priming positions at Ser-741 and Ser-775 restored CLASP2 microtubule plus-end-tracking in all analyzed cells (Fig. 8A), indicating that the S741A/S775A double priming site mutant is not a GSK3β substrate.
CLASP2 Plus-end-tracking Is Inhibited during Mitosis
Because cycB/CDK1 is specifically active during mitosis, we examined CLASP2-(497–1238) phosphorylation by metabolic labeling in cells arrested in mitosis with the Eg5 kinesin inhibitor STLC (44). STLC-arrested cells displayed increased CLASP2 phosphorylation compared with non-synchronized cells as indicated by an increased upshift by gel electrophoresis (Fig. 8B). Similar to non-synchronized cells, phosphorylation was substantially decreased in STLC-arrested cells either treated with a GSK3 inhibitor, SB216763, or expressing a CLASP2 construct with mutated GSK3 phosphorylation sites as evidenced by both a downshift of the CLASP2 band and decreased incorporation of radioactive phosphate. Thus, a large fraction of mitotic CLASP2 phosphorylation occurs on the same GSK3 sites as in interphase. However, in both cases the CLASP band remained slightly upshifted compared with non-synchronized cells, which is expected due to increased phosphorylation of mitotic CDK sites.
Next, we tested how increased phosphorylation during mitosis regulated CLASP2 plus end association. Although EB1-EGFP was clearly present on microtubule ends in metaphase spindles (Fig. 8D), we could not detect wild-type EGFP-CLASP2 constructs on microtubule ends in metaphase (Fig. 8C). In contrast, mutation of all GSK3 sites (9× S/A) or of only the two SXIP motif-associated CDK priming sites (S741A/S775A) fully restored CLASP2 plus-end-tracking in metaphase cells. In addition, this mitotic regulation of CLASP2 plus-end-tracking did not depend on the presence of the C-terminal kinetochore binding domain (Fig. 8, C, E, and F) (45).
Finally, to test whether the lack of CLASP2 plus-end-tracking in mitotic cells was due to priming phosphorylation by CDK1, we treated cells that had been arrested in metaphase with the proteasome inhibitor MG132 (46) with a highly specific CDK1 inhibitor, RO-3306 (47). Although these cells entered cytokinesis without chromosome segregation after ∼30 min, rescue of CLASP2 microtubule association was evident within a few minutes of the addition of the CDK1 inhibitor (Fig. 8G). Together these data indicate that down-regulation of CLASP2 plus-end-tracking during mitosis depends on phosphorylation of the Ser-741 and Ser-775 priming sites by CDK1 and subsequent phosphorylation by GSK3 and thus likely results from inhibition of the interaction between EB1 and CLASP2.
DISCUSSION
Multisite phosphorylation is a common posttranslational modification in eukaryotic cells and adds a layer of combinatorial complexity to intracellular signaling networks. However, the underlying mechanisms by which multisite phosphorylation regulates protein interactions and activity are incompletely understood. GSK3α and -β are ubiquitous protein kinases that are responsible for the majority of sequential multisite phosphorylation in eukaryotic cells (15, 16). We previously reported that CLASP2, which is involved in microtubule stabilization at the cell cortex in migrating interphase cells, is a substrate of GSK3 phosphorylation and identified two phosphorylation motifs that regulate CLASP2-microtubule interactions (9, 35). These GSK3 phosphorylation motifs are in close proximity to SXIP sequence motifs that have recently been identified to mediate +TIP interactions with EB1 (7). This region is nearly identical in CLASP1 and CLASP2 and highly conserved in CLASP proteins from other species (Fig. 1B).
Because the C terminus of EB1 is highly negatively charged and sequences surrounding SXIP motifs in CLASP2 are interspersed with positively charged arginine residues, electrostatic interactions likely contribute to binding to EB1. However, both the C-terminal tail of EB1 and the region surrounding the CLASP2 SXIP motifs are intrinsically disordered, preventing direct structural analysis of this interaction. Based on molecular dynamics simulations of an EB1-bound CLASP2 model peptide containing one SXIP motif followed by the arginine-rich GSK3 phosphorylation motif, we propose a model of how electrostatic interactions mediate CLASP2-EB1 binding and are perturbed by phosphorylation. Because of the large possible conformational space, molecular dynamics simulations of intrinsically disordered proteins are inherently incomplete (48). Thus, the simulation outcomes likely represent possible local energy minima and may not completely represent the ensemble at thermodynamic equilibrium. Nevertheless, comparison of multiple simulation runs identified common types of interactions. In all simulations of the non-phosphorylated CLASP2 peptide bound to EB1, conserved arginine residues in CLASP2 formed bidentate salt bridges preferentially with glutamate residues near the unstructured C terminus of EB1. Such coplanar arginine-glutamate (R-E) salt bridges are geometrically and energetically highly favorable (49). We find that lysine residues cannot substitute for arginines likely because of the less favorable lysine-glutamate hydrogen bond geometry (49), which may explain the strong conservation of these arginines in CLASPs (Fig. 1B). Thus, although hydrophobic interactions of the SXIP motif itself determine the specificity of binding to EB1, molecular Velcro formed by multiple coplanar R-E salt bridges likely contributes significantly to binding strength. Although our molecular dynamics analysis focused on the N-terminal SKIP motif, the sequence around the more C-terminal SRIP motif is very similar, and we obtained qualitatively similar results in molecular dynamics simulations.4
In simulations of EB1-bound phosphorylated CLASP2 peptides, these intermolecular R-E interactions were mostly replaced by intramolecular salt bridges between the arginine residues and phosphorylated serines (R-pS). Such coplanar R-pS salt bridges are predicted to be significantly more favorable than even the bidentate R-E side chain interaction (49), and we propose that phosphoserines outcompete R-E interactions. Because our simulations are short on a molecular time-scale and start from an artificial situation in which the phosphorylated CLASP2 peptide is bound to EB1, we did not observe complete dissociation of the interaction. In reality, CLASP2 phosphorylation likely occurs when it is not bound to EB1. We expect that pre-existing intramolecular R-pS salt bridges prevent binding to EB1, which is supported both by our cell and in vitro experiments (Figs. 2 and 3). Intramolecular R-pS bridges also rapidly formed in simulations of the phosphorylated CLASP2 peptide alone and are supported by 31P NMR spectroscopy data (Fig. 4). In addition, the formation of similar stable R-pS salt bridges has been proposed in hyperphosphorylated RS dipeptide repeat motifs (50).
Both GSK3 kinases recognize a characteristic (S/T)XXX(pS/pT) repeat sequence in which the C-terminal serine or threonine residue has already been phosphorylated by a different priming kinase or by GSK3 itself. Interestingly, our data show that GSK3β phosphorylation motifs do not always conform to this consensus sequence. At least in vitro, additional phosphorylation occurs most likely at Ser-728 because it is N-terminal to the GSK3β phosphorylation motif even though the distance between Ser-733 and Ser-728 does not conform to the consensus. In addition, the absence of phosphorylation intermediates in in vitro GSK3β kinase reactions strongly suggests that phosphorylation by GSK3β proceeds processively, which would indicate a switch-like response of GSK3β-mediated CLASP2 regulation (51).
The priming sites of the GSK3 phosphorylation motifs in CLASP2 are highly conserved cyclin-dependent kinase consensus motifs. Although phosphorylation of Ser-775 by CDK5 has been reported earlier (14), we now demonstrate that both priming sites are efficiently phosphorylated by mitotic (cycB/CDK1) and interphase CDKs (p25/CDK5). CLASP2 was also phosphorylated by cycA/CDK2 that is active during the S/G2 transition but not by cycD1/CDK4, which is active in late G1 (52), indicating potentially complex cell cycle regulation of CLASP2.5 Phosphorylation of the same GSK3 motifs regulates CLASP2-microtubule interactions in both interphase and mitosis. In interphase, CLASPs are only partially phosphorylated (9), yet we find that CDK5 phosphorylates both priming sites equally well. CDK5 is thought to be mostly functional in neuronal cells (53). Thus, a different kinase may phosphorylate Ser-775 in other interphase cells, or competing phosphatase activity could result in partial dephosphorylation of the GSK3 motifs.
In contrast, in mitosis CDK1- and GSK3-dependent phosphorylation globally switches off CLASP2 plus-end-tracking. In mammalian cells, CLASPs are localized to kinetochores independent of microtubules. CLASPs may regulate the dynamics of kinetochore microtubules, and CLASP depletion results in mitotic defects (45, 54–57). GSK3 activity is also required for accurate chromosome segregation (58), and it is thus likely that mitotic CLASP function depends on highly localized regulation of kinetochore-associated CLASP phosphorylation as CLASPs would need to be dephosphorylated to mediate kinetochore-microtubule interactions (59). Mitotic inhibition of microtubule plus-end-tracking has also been observed for other +TIPs such as SLAIN2 and Kebab, although phosphorylation sites involved have not been identified (11, 60). The functional relevance of mitotic phosphorylation of these proteins remains to be determined.
In conclusion, phosphoregulation of +TIPs is likely common and may explain the abundance of arginine residues around SXIP motifs in CLASP proteins and other +TIPs. Spectraplakins contain GSK3 phosphorylation motifs in an arginine-rich region near a functional SXIP motif at the C terminus (7, 12), and phosphorylation by Aurora B kinase in an arginine-rich region inhibits plus-end-tracking of the mitotic centromere-associated kinesin (MCAK) (61). Interestingly, GSK3 phosphorylation also directly inhibits EB1-independent microtubule binding of CLASP2 and ACF7 (9, 12), and the acidic C terminus of EB1 is highly similar to the C-terminal tail of most α/β tubulin isoforms (62). In addition, most microtubule-associated proteins contain large intrinsically disordered regions. Microtubule binding often depends on the acidic tubulin tail and is inhibited by phosphorylation (63). Thus, molecular mechanisms similar to that proposed here for regulation by multisite phosphorylation may also apply to other microtubule-binding proteins. Furthermore, it has become increasingly evident that a large percentage of the proteome does not form stably folded domain structures but exists in an unfolded, intrinsically disordered state, and multisite phosphorylation often occurs in such disordered regions (64). The strong electrostatic perturbation introduced by multiple phosphate residues provides one mechanism for regulating such interactions by perturbing favorable but potentially nonspecific electrostatic interactions involving unstructured peptides.
Supplementary Material
Acknowledgments
We thank Peter Bieling for advice on the plus-end-tracking assay, Colin Smith and Cristina Melero for helpful discussions and assistance with fluorescence polarization assays, and Mark Kelly for assistance with NMR spectroscopy. This research was conducted in part in a facility constructed with support from Research Facilities Improvement Program Grant C06 RR16490 from the National Center for Research Resources of the National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM079139 (to T. W.). This work was also supported by American Heart Association Predoctoral Fellowship 11PRE7590115 (to H. P.).

This article contains supplemental Video 1.
M. Chimenti and T. Wittmann, unpublished results.
P. Kumar and T. Wittmann, unpublished results.
- EB
- ending binding
- CLASP2
- cytoplasmic linker-associated protein 2
- GSK3
- glycogen synthase kinase 3
- CDK
- cyclin-dependent kinase
- STLC
- S-trityl-l-cysteine
- R-E
- arginine-glutamate
- R-pS
- arginine-phosphorylated serine
- CDK
- cyclin-dependent kinase
- EGFP
- enhanced GFP
- GMPCPP
- guanylyl(α,β)methylene diphosphonate.
REFERENCES
- 1. Akhmanova A., Steinmetz M. O. (2008) Tracking the ends. A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 9, 309–322 [DOI] [PubMed] [Google Scholar]
- 2. Galjart N. (2010) Plus-end-tracking proteins and their interactions at microtubule ends. Curr. Biol. 20, R528–R537 [DOI] [PubMed] [Google Scholar]
- 3. Slep K. C. (2010) Structural and mechanistic insights into microtubule end-binding proteins. Curr. Opin. Cell Biol. 22, 88–95 [DOI] [PubMed] [Google Scholar]
- 4. Hayashi I., Ikura M. (2003) Crystal structure of the amino-terminal microtubule binding domain of end-binding protein 1 (EB1). J. Biol. Chem. 278, 36430–36434 [DOI] [PubMed] [Google Scholar]
- 5. Bieling P., Kandels-Lewis S., Telley I. A., van Dijk J., Janke C., Surrey T. (2008) CLIP-170 tracks growing microtubule ends by dynamically recognizing composite EB1/tubulin-binding sites. J. Cell Biol. 183, 1223–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Honnappa S., John C. M., Kostrewa D., Winkler F. K., Steinmetz M. O. (2005) Structural insights into the EB1-APC interaction. EMBO J. 24, 261–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Honnappa S., Gouveia S. M., Weisbrich A., Damberger F. F., Bhavesh N. S., Jawhari H., Grigoriev I., van Rijssel F. J., Buey R. M., Lawera A., Jelesarov I., Winkler F. K., Wüthrich K., Akhmanova A., Steinmetz M. O. (2009) An EB1 binding motif acts as a microtubule tip localization signal. Cell 138, 366–376 [DOI] [PubMed] [Google Scholar]
- 8. Narayanan A., Jacobson M. P. (2009) Computational studies of protein regulation by post-translational phosphorylation. Curr. Opin. Struct. Biol. 19, 156–163 [DOI] [PubMed] [Google Scholar]
- 9. Kumar P., Lyle K. S., Gierke S., Matov A., Danuser G., Wittmann T. (2009) GSK3β phosphorylation modulates CLASP-microtubule association and lamella microtubule attachment. J. Cell Biol. 184, 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Näthke I. S. (2004) The adenomatous polyposis coli protein. The Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 20, 337–366 [DOI] [PubMed] [Google Scholar]
- 11. van der Vaart B., Manatschal C., Grigoriev I., Olieric V., Gouveia S. M., Bjelic S., Demmers J., Vorobjev I., Hoogenraad C. C., Steinmetz M. O., Akhmanova A. (2011) SLAIN2 links microtubule plus end-tracking proteins and controls microtubule growth in interphase. J. Cell Biol. 193, 1083–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu X., Shen Q. T., Oristian D. S., Lu C. P., Zheng Q., Wang H. W., Fuchs E. (2011) Skin stem cells orchestrate directional migration by regulating microtubule-ACF7 connections through GSK3β. Cell 144, 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang X., Ems-McClung S. C., Walczak C. E. (2008) Aurora A phosphorylates MCAK to control ran-dependent spindle bipolarity. Mol. Biol. Cell 19, 2752–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watanabe T., Noritake J., Kakeno M., Matsui T., Harada T., Wang S., Itoh N., Sato K., Matsuzawa K., Iwamatsu A., Galjart N., Kaibuchi K. (2009) Phosphorylation of CLASP2 by GSK-3β regulates its interaction with IQGAP1, EB1, and microtubules. J. Cell Sci. 122, 2969–2979 [DOI] [PubMed] [Google Scholar]
- 15. Hur E. M., Zhou F. Q. (2010) GSK3 signaling in neural development. Nat. Rev. Neurosci. 11, 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jope R. S., Yuskaitis C. J., Beurel E. (2007) Glycogen synthase kinase-3 (GSK3). Inflammation, diseases, and therapeutics. Neurochem. Res. 32, 577–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Uversky V. N. (2011) Multitude of binding modes attainable by intrinsically disordered proteins. A portrait gallery of disorder-based complexes. Chem. Soc. Rev. 40, 1623–1634 [DOI] [PubMed] [Google Scholar]
- 18. Mittag T., Kay L. E., Forman-Kay J. D. (2010) Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 23, 105–116 [DOI] [PubMed] [Google Scholar]
- 19. Bieling P., Telley I. A., Hentrich C., Piehler J., Surrey T. (2010) Fluorescence microscopy assays on chemically functionalized surfaces for quantitative imaging of microtubule, motor, and +TIP dynamics. Methods Cell Biol. 95, 555–580 [DOI] [PubMed] [Google Scholar]
- 20. Telley I. A., Bieling P., Surrey T. (2011) Reconstitution and quantification of dynamic microtubule end tracking in vitro using TIRF microscopy. Methods Mol. Biol. 777, 127–145 [DOI] [PubMed] [Google Scholar]
- 21. Castoldi M., Popov A. V. (2003) Purification of brain tubulin through two cycles of polymerization-depolymerization in a high molarity buffer. Protein Expr. Purif. 32, 83–88 [DOI] [PubMed] [Google Scholar]
- 22. Di Marco V. B., Bombi G. G. (2001) Mathematical functions for the representation of chromatographic peaks. J. Chromatogr. A 931, 1–30 [DOI] [PubMed] [Google Scholar]
- 23. Jacobson M. P., Pincus D. L., Rapp C. S., Day T. J., Honig B., Shaw D. E., Friesner R. A. (2004) A hierarchical approach to all-atom protein loop prediction. Proteins 55, 351–367 [DOI] [PubMed] [Google Scholar]
- 24. Zhu K., Shirts M. R., Friesner R. A., Jacobson M. P. (2007) Multiscale optimization of a truncated Newton minimization algorithm and application to proteins and protein-ligand complexes. J. Chem. Theory Comput. 3, 640–648 [DOI] [PubMed] [Google Scholar]
- 25. Shivakumar D., Williams J., Wu Y. J., Damm W., Shelley J., Sherman W. (2010) Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 6, 1509–1519 [DOI] [PubMed] [Google Scholar]
- 26. Jorgensen W. L., Maxwell D. S., Tirado-Rives J. (1996) Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 [Google Scholar]
- 27. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926 [Google Scholar]
- 28. Darden T. A., York D. M., Pedersen L. G. (1993) Particle mesh Ewald: An log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089 [Google Scholar]
- 29. Ryckaert J. P., Ciccotti G., Berendsen H. J. C. (1976) Numerical integration of the cartesian equations of motion of a system with constraints. Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 30. Nicholls A., Honig B. (1991) A rapid finite difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J. Comput. Chem. 12, 435–445 [Google Scholar]
- 31. Maurer T., Kalbitzer H. R. (1996) Indirect Referencing of 31P and 19F NMR Spectra J. Magn. Reson. B 113, 177–178 [DOI] [PubMed] [Google Scholar]
- 32. Hastie C. J., McLauchlan H. J., Cohen P. (2006) Assay of protein kinases using radiolabeled ATP. A protocol. Nat. Protoc. 1, 968–971 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z., Marshall A. G. (1998) A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass Spectrom. 9, 225–233 [DOI] [PubMed] [Google Scholar]
- 34. Wittmann T., Waterman-Storer C. M. (2005) Spatial regulation of CLASP affinity for microtubules by Rac1 and GSK3β in migrating epithelial cells. J. Cell Biol. 169, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mimori-Kiyosue Y., Grigoriev I., Lansbergen G., Sasaki H., Matsui C., Severin F., Galjart N., Grosveld F., Vorobjev I., Tsukita S., Akhmanova A. (2005) CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J. Cell Biol. 168, 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wittmann T. (2008) EBs clip CLIPs to growing microtubule ends. J. Cell Biol. 183, 1183–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jameson D. M., Sawyer W. H. (1995) Fluorescence anisotropy applied to biomolecular interactions. Methods Enzymol. 246, 283–300 [DOI] [PubMed] [Google Scholar]
- 38. Wong S. E., Bernacki K., Jacobson M. (2005) Competition between intramolecular hydrogen bonds and solvation in phosphorylated peptides. Simulations with explicit and implicit solvent. J. Phys Chem. B 109, 5249–5258 [DOI] [PubMed] [Google Scholar]
- 39. Vogel H. J., Bridger W. A. (1982) Phosphorus-31 nuclear magnetic resonance studies of the two phosphoserine residues of hen egg white ovalbumin. Biochemistry 21, 5825–5831 [DOI] [PubMed] [Google Scholar]
- 40. Hoffmann R., Reichert I., Wachs W. O., Zeppezauer M., Kalbitzer H. R. (1994) 1H and 31P NMR spectroscopy of phosphorylated model peptides. Int. J. Pept. Protein Res. 44, 193–198 [DOI] [PubMed] [Google Scholar]
- 41. Bienkiewicz E. A., Lumb K. J. (1999) Random-coil chemical shifts of phosphorylated amino acids. J. Biomol. NMR 15, 203–206 [DOI] [PubMed] [Google Scholar]
- 42. Li H., Robertson A. D., Jensen J. H. (2004) The determinants of carboxyl pKa values in turkey ovomucoid third domain. Proteins 55, 689–704 [DOI] [PubMed] [Google Scholar]
- 43. Lalioti V., Pulido D., Sandoval I. V. (2010) Cdk5, the multifunctional surveyor. Cell Cycle 9, 284–311 [DOI] [PubMed] [Google Scholar]
- 44. Skoufias D. A., DeBonis S., Saoudi Y., Lebeau L., Crevel I., Cross R., Wade R. H., Hackney D., Kozielski F. (2006) S-Trityl-l-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 281, 17559–17569 [DOI] [PubMed] [Google Scholar]
- 45. Maiato H., Fairley E. A., Rieder C. L., Swedlow J. R., Sunkel C. E., Earnshaw W. C. (2003) Human CLASP1 is an outer kinetochore component that regulates spindle microtubule dynamics. Cell 113, 891–904 [DOI] [PubMed] [Google Scholar]
- 46. Genschik P., Criqui M. C., Parmentier Y., Derevier A., Fleck J. (1998) Cell cycle-dependent proteolysis in plants. Identification of the destruction box pathway and metaphase arrest produced by the proteasome inhibitor mg132 Plant Cell 10, 2063–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vassilev L. T., Tovar C., Chen S., Knezevic D., Zhao X., Sun H., Heimbrook D. C., Chen L. (2006) Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. U.S.A. 103, 10660–10665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rauscher S., Pomès R. (2010) Molecular simulations of protein disorder. Biochem. Cell Biol. 88, 269–290 [DOI] [PubMed] [Google Scholar]
- 49. Mandell D. J., Chorny I., Groban E. S., Wong S. E., Levine E., Rapp C. S., Jacobson M. P. (2007) Strengths of hydrogen bonds involving phosphorylated amino acid side chains. J. Am. Chem. Soc. 129, 820–827 [DOI] [PubMed] [Google Scholar]
- 50. Hamelberg D., Shen T., McCammon J. A. (2007) A proposed signaling motif for nuclear import in mRNA processing via the formation of arginine claw. Proc. Natl. Acad. Sci. U.S.A. 104, 14947–14951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Salazar C., Höfer T. (2009) Multisite protein phosphorylation. From molecular mechanisms to kinetic models. FEBS J. 276, 3177–3198 [DOI] [PubMed] [Google Scholar]
- 52. Morgan D. O. (1997) Cyclin-dependent kinases. Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 13, 261–291 [DOI] [PubMed] [Google Scholar]
- 53. Jessberger S., Gage F. H., Eisch A. J., Lagace D. C. (2009) Making a neuron. Cdk5 in embryonic and adult neurogenesis. Trends Neurosci. 32, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pereira A. L., Pereira A. J., Maia A. R., Drabek K., Sayas C. L., Hergert P. J., Lince-Faria M., Matos I., Duque C., Stepanova T., Rieder C. L., Earnshaw W. C., Galjart N., Maiato H. (2006) Mammalian CLASP1 and CLASP2 cooperate to ensure mitotic fidelity by regulating spindle and kinetochore function. Mol. Biol. Cell 17, 4526–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mimori-Kiyosue Y., Grigoriev I., Sasaki H., Matsui C., Akhmanova A., Tsukita S., Vorobjev I. (2006) Mammalian CLASPs are required for mitotic spindle organization and kinetochore alignment. Genes Cells 11, 845–857 [DOI] [PubMed] [Google Scholar]
- 56. Hannak E., Heald R. (2006) Xorbit/CLASP links dynamic microtubules to chromosomes in the Xenopus meiotic spindle. J. Cell Biol. 172, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Maffini S., Maia A. R., Manning A. L., Maliga Z., Pereira A. L., Junqueira M., Shevchenko A., Hyman A., Yates J. R., 3rd, Galjart N., Compton D. A., Maiato H. (2009) Motor-independent targeting of CLASPs to kinetochores by CENP-E promotes microtubule turnover and poleward flux. Curr. Biol. 19, 1566–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tighe A., Ray-Sinha A., Staples O. D., Taylor S. S. (2007) GSK-3 inhibitors induce chromosome instability. BMC Cell Biol. 8, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Welburn J. P., Vleugel M., Liu D., Yates J. R., 3rd, Lampson M. A., Fukagawa T., Cheeseman I. M. (2010) Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol. Cell 38, 383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meireles A. M., Dzhindzhev N. S., Ohkura H. (2011) Kebab. Kinetochore and EB1 associated basic protein that dynamically changes its localization during Drosophila mitosis. PLoS ONE 6, e24174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Moore A. T., Rankin K. E., von Dassow G., Peris L., Wagenbach M., Ovechkina Y., Andrieux A., Job D., Wordeman L. (2005) MCAK associates with the tips of polymerizing microtubules. J. Cell Biol. 169, 391–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Komarova Y., Lansbergen G., Galjart N., Grosveld F., Borisy G. G., Akhmanova A. (2005) EB1 and EB3 control CLIP dissociation from the ends of growing microtubules. Mol. Biol. Cell 16, 5334–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cassimeris L., Spittle C. (2001) Regulation of microtubule-associated proteins. Int. Rev. Cytol. 210, 163–226 [DOI] [PubMed] [Google Scholar]
- 64. Pearlman S. M., Serber Z., Ferrell J. E., Jr. (2011) A mechanism for the evolution of phosphorylation sites. Cell 147, 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.