
Figure 1.
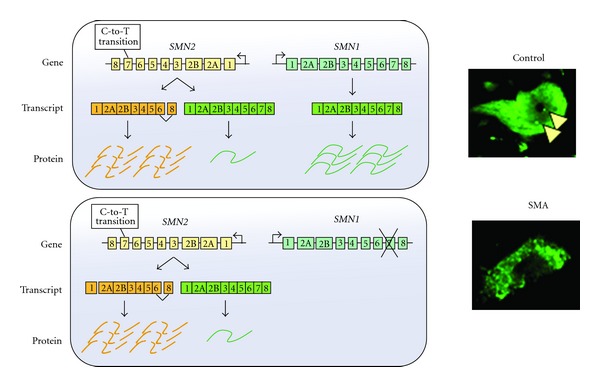
Schematic diagram of the SMN1 and SMN2 genes. Humans are the only species that carry both SMN1 and SMN2 genes, located in the human 5q11.2–13.3 region [5, 168]. The SMN1 and SMN2 genes differ by five nucleotide exchanges [6]. Among them, a translationally silent cytosine to thymidine exchange at position 6 of exon 7 is responsible for the skipping of exon 7 during splicing of the SMN2 gene [6]. The C-to-T transition abolishes an exonic splice enhancer site and generates a new exonic splicing silencer domain for the last coding exon [169, 170]. Subsequently, through alternative splicing, most of the translating SMN protein from the SMN2 gene lacks the C-terminal residue and becomes less stable and relatively inactive [171]. In normal situation, abundant SMN protein is produced mainly from SMN1 gene with a little amount from SMN2 gene. The spinal motor neuron from a wild-type mouse thus expresses a high level of SMN in both cytoplasm and nucleus with several gems (arrow head) as compared to that in an SMA mouse. With homozygous mutation of the SMN1 genes, all SMA patients still have at least one SMN2 gene copy [6]. While complete loss of SMN expression is embryonically lethal [172], the small amount of full-length SMN protein produced by the SMN2 gene (about 20%) prevents lethality in SMA patients, but has insufficient SMN levels to assist in recovery from spinal motor neuron death [28].