

Abstract
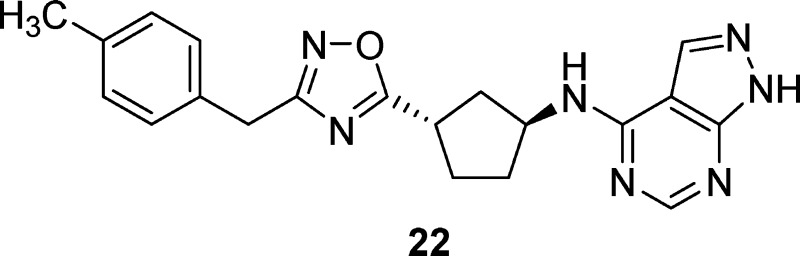
A series of 3-substituted aminocyclopentanes has been identified as highly potent and selective NR2B receptor antagonists. Incorporation of a 1,2,4-oxadiazole linker and substitution of the pendant phenyl ring led to the discovery of orally bioavailable analogues that showed efficient NR2B receptor occupancy in rats. Unlike nonselective NMDA antagonists, the NR2B-selective antagonist 22 showed no adverse affects on motor coordination in the rotarod assay at high dose. Compound 22 was efficacious following oral administration in a spinal nerve ligation model of neuropathic pain and in an acute model of Parkinson’s disease in a dose dependent manner.
Keywords: NR2B antagonist, NMDA, neuropathic pain, Parkinson’s disease
The N-methyl-d-aspartate (NMDA) receptor is highly expressed throughout the mammalian central nervous system (CNS). This type of ionotropic glutamate receptor mediates excitatory synaptic transmission following activation by the amino acids glutamate and glycine.1,2 Excessive stimulation of NMDA receptors has been implicated in a number of neurological disorders including stroke,1,3,4 Alzheimer’s disease,3−5 Parkinson’s disease,6−8 and neuropathic pain.9,10 The nonselective NMDA channel blocker memantine was recently approved for the treatment of medium-to-severe Alzheimer’s disease.11,12 Preclinical and clinical studies with ketamine, another nonselective antagonist, have demonstrated efficacy for pain; however, a narrow therapeutic window has severely limited the use of this agent due to undesirable motor and cognitive side effects associated with inhibition of all subtypes of NMDA receptors.13 The NMDA receptor complex comprises three subunits, designated NR1(a-h), NR2(A-D), and NR3(A-B).14 While a pharmacological role for the NR3 subunit has not been fully elucidated,15 the functional NMDA receptor is composed of a heteromultimeric complex containing both NR1 subunits and at least one of the four NR2 subunits.16 The NR2 subunits influence the physiological and functional properties of the NMDA receptors, including sensitivity to magnesium blockade, channel kinetics, and ligand affinities,2,17,18 and are regionally distributed throughout the CNS. The NR2B subunit is concentrated in structures of the forebrain and dorsal horn of the spinal cord,16,19,20 suggesting NR2B subtype-selective NMDA receptor antagonists may be effective without the side effects associated with nonselective NMDA channel blockers.20−23 NR2B subtype-selective antagonists, such as 1 (ifenprodil)24 and 2 (traxoprodil, CP-101,606),25 have shown efficacy with diminished CNS side effects26,27 in animal models of cerebral ischemia,28 neuropathic pain,20,29 and Parkinson’s disease.30−32 CP-101,606 (2) is reported to be well tolerated in patients with traumatic head injury33 and to be efficacious for pain in patients with spinal cord injury and monoradiculopathy.34
Research efforts have focused on identifying novel structural classes of NR2B subtype-selective antagonists with improved potency and selectivity profiles over the early leads.35 Several NR2B inhibitors that contain the classical phenol terminal ring or phenol isostere linked to a basic amine (typically a piperidine ring) have been reported, including ifenprodil (1), traxoprodil (2), and Ro-25-6981 (3)36 (Figure 1). More recently, new classes of NR2B selective antagonists have been described which show significant departure from the classical NR2B pharmacophore, such as aminoquinoline 4,37 benzamidine 5,38 and carbamate 6.39 Carbamate 6 was reported by Merck to be a potent NR2B-selective antagonist (Ki = 3.4 nM) that was efficacious in preclinical models of both neuropathic pain and Parkinson’s disease following oral dosing. Our goal at Merck was to identify structurally distinct classes of NR2B-selective inhibitors from 6 that were subnanomolar against NR2B, highly selective over other NR2 subtypes and hERG activity, and active in preclinical models of pain and Parkinson’s following oral dosing. Historically, achieving high levels of selectivity over hERG activity has been particularly challenging with NR2B antagonists. As a result, hERG activity was determined on all active compounds using an MK499 binding assay, a known ligand for the channel, and only compounds with greater than 10 μM binding in the hERG binding assay were considered for advancement. In this paper, we report the design and optimization of a unique series of highly potent and selective NR2B antagonists which demonstrate robust oral efficacy in preclinical models of neuropathic pain and Parkinson’s disease.
Figure 1.

NR2B-selective NMDA receptor antagonists.
Results and Discussion
An analysis of several classes of NR2B subtype-selective antagonists in the Merck sample collection identified prototypical phenol-containing structures related to carbamate 6, such as phenol 7, from which a novel series of NR2B antagonists could be developed.40 We hypothesized that the 4-aminomethyl-piperidine could be replaced with an alternate ring system in which the number of atoms between the phenol and the phenyl ring would remain constant. For example, as shown in Figure 2, cyclization between the aminomethyl carbon and the C3 carbon of the piperidine in 7, and removal of the C5 and the C6 carbons of the piperidine, led to a 3-substituted aminocyclopentane as a novel central constraint. In addition to replacing the 4-aminomethylpiperidine core, we planned to substitute the phenol carboxamide in 7 with the 4-amino-1H-pyrazolo[3,4-d]pyrimidine.41 Based on this hypothesis, we prepared a series of structurally novel 3-substituted N-1H-pyrazolo[3,4-d]pyrimidin-4-amine cyclopentanes for evaluation as NR2B subtype-selective NMDA receptor antagonists.
Figure 2.
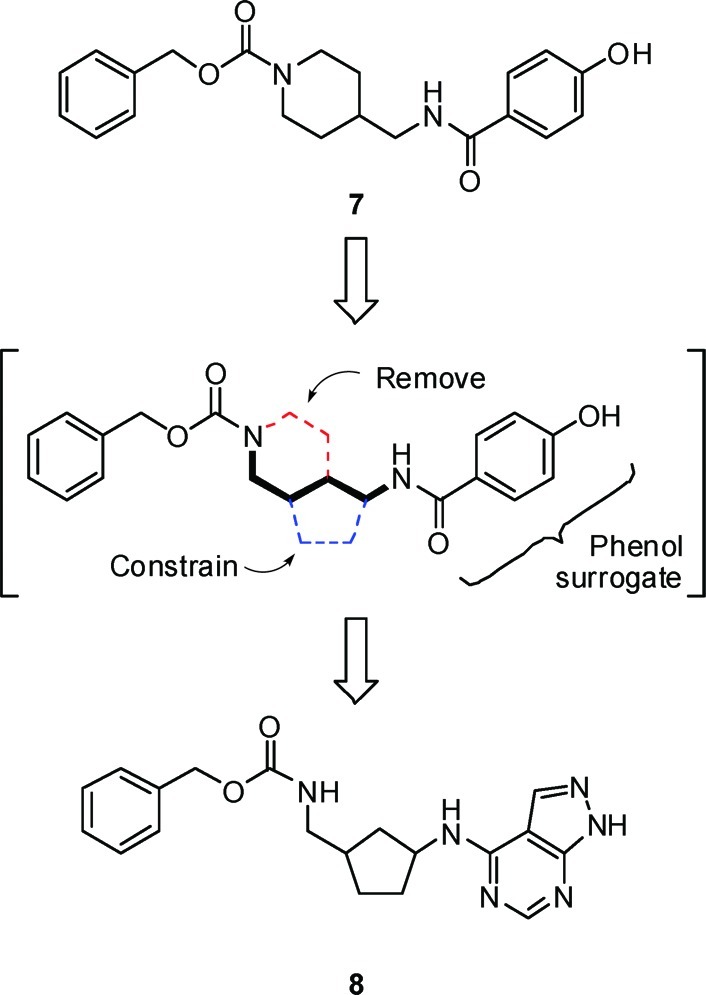
Origin of aminocyclopentane as a novel central constraint.
Gratifyingly, in support of the original hypothesis, two isomers of 8 were comparably potent against NR2B as the benchmark Merck compound (6). As shown in Table 1, evaluation of the absolute stereochemical requirements for compound 8 showed a strong preference for S configuration at C1 ((R,R)-8 versus (S,R)-8). On the other hand, NR2B potency was relatively insensitive to the configuration at C3 ((S,R)-8 versus (S,S)-8). Although (S,R)-8 and (S,S)-8 were similarly potent for NR2B, compound (S,R)-8 was less selective for hERG. Furthermore, (S,R)-8 and (S,S)-8 were susceptible to human Pgp efflux in vitro (MDR1 B–A/A–B ratios = 3.8 and 5.4, respectively)42 which could limit CNS exposure to these compounds. As a result, identification of a potent analogue that was not susceptible to Pgp efflux was essential.
Table 1. In Vitro Binding Data for Aminocyclopentanesa.

Compounds (S,R)-8 and (S,S)-8 each contain three hydrogen bond donors. We hypothesized that replacement of the carbamate with a suitable heterocycle lacking a hydrogen bond donor would lower the propensity for Pgp efflux.45 We first targeted the readily accessible 1,2,4-oxadiazole as a suitable isostere for the carbamate lacking a hydrogen bond donor. Incorporation of 3-phenyl-1,2,4-oxadiazole as a carbamate replacement gave compound 9, which was approximately 10-fold less potent against NR2B than the carbamates and modestly selective over hERG binding (Table 2). As anticipated, reducing the number of hydrogen bond donors was sufficient to mitigate the Pgp liability and 9 was not subject to human Pgp efflux in vitro (MDR1 B–A/A–B ratio = 0.9).42 Given that 9 represented a promising new series of NR2B inhibitors with the potential to be CNS penetrant, a more detailed evaluation of the compound was undertaken. Compound 9 showed modest NR2B potency in a functional assay with cells expressing NR2B (IC50 = 7.5 nM) and did not show significant reversible inhibition of CYP3A4, 2D6, or 2C9 (IC50 > 26 μM). Evaluation of pharmacokinetics of 9 in rats showed modest oral bioavailability and moderate to low clearance with a 1.2 h half-life (Table 7). NR2B receptor occupancy was determined in an ex vivo binding assay using a radiolabeled-ligand displacement assay on temporal cortex harvested after dosing of the test agent. Compound 9 effectively engaged the target in vivo and showed 50% receptor occupancy in the cortex at 7.2 and 21 mpk following IV and PO dosing, respectively. Overall, compound 9 represented a promising new lead as a brain-penetrant, oral NR2B antagonist. On the basis of this promising early lead, evaluation of the structure–activity relationship (SAR) around 9 was undertaken to further improve in vitro and in vivo potency.
Table 2. In Vitro Binding Data for Phenyl-Oxadiazolesa.
Table 7. In Vivo Evaluation for Selected Compoundsa,47.
| pharmacokinetic Profileb |
receptor occupancy ED50c |
|||||
|---|---|---|---|---|---|---|
| compd | species | %F | T1/2 (hr) | Cl (mL/min/kg) | IV dosing (mg/kg) | PO dosing (mg/kg) |
| 6 | rat | 45 | 2.7 | 26 | 2.0 | 4.9 |
| 9 | rat | 14 | 1.2 | 8.7 | 7.2 | 21 |
| 14 | rat | 67 | 11.2 | 0.8 | <10 | |
| 16 | rat | 23 | 1.4 | 10 | 0.6 | 8.1 |
| 22 | rat | 34 | 0.7 | 24 | 0.9 | 4.8 |
| 22 | dog | 83 | 7.5 | 3.6 | ||
| 22 | rhesus | 17 | 1.5 | 12 | ||
| 23 | rat | 13 | 2.1 | 13 | 1.8 | 3.5 |
| 30 | rat | 3 | 0.3 | 18 | >3 | |
| 37 | rat | 3.1 | 10.8 | |||
Abbreviations: %F, oral bioavailability; T1/2, half-life; Cl, clearance.
Sprague–Dawley rats (n = 3); oral dose =10 mg/kg, intravenous dose = 2 mg/kg; Beagle dogs (n = 2), intravenous dose = 0.3 mg/kg; Rhesus monkeys (n = 2), oral dose = 1 mg/kg, intravenous dose = 1 mg/kg.
Sprague–Dawley rats, n = 4 at each dose of 1, 3, and 10 mg/kg; <10 = 70% inhibition at 10 mpk; >3 = 34% inhibition at 3 mpk.
Unlike the carbamates (8), the C3 stereochemistry of the cyclopentane was critical with the 1,2,4-oxadiazoles and a significant loss of potency was observed with the (3R)-diastereomer (10) over the (3S)-diastereomer (9) (Table 2). While maintaining the S-configuration at C3, substitution on the pendant phenyl ring of 9 was explored. A loss of potency was observed by installation of a methyl substituent in either the 2- or 3-positions on the phenyl ring (11 and 12, respectively). Methylation of the 4-position on the phenyl ring (13) resulted in a 5-fold enhancement of NR2B potency over 9 without improving the hERG binding. Substitution with a chlorine in the 4-position gave 14, which showed comparable NR2B potency (Ki = 8.5 nM) and >2000-fold selectivity over hERG. In rat, 14 showed substantial improvements over 9 in pharmacokinetic parameters including clearance (0.8 mL/min/kg), half-life (11.2 h) and oral bioavailability (F = 67%), and modest ex vivo receptor occupancy (70% inhibition at 10 mpk IV) (Table 7).
In parallel with the 3-phenyl-1,2,4-oxadiazole SAR, we began investigating optimal linkers between the cyclopentane, 1,2,4-oxadiazole, and phenyl moieties in 9. Insertion of a methylene unit between the cyclopentane and the oxadiazole of 9 had a minimal effect on potency and hERG binding (15, Table 3). On the other hand, insertion of a methylene unit between the oxadiazole and the phenyl ring to give 16 resulted in both enhanced potency at NR2B and selectivity over hERG binding relative to 9. Further extension of the alkyl chain with two methylene units (17) resulted in a significant loss of potency, indicating a benzyl-1,2,4-oxadiazole was the optimal linker length to maximize potency and selectivity. Compound 16 showed excellent selectivity over NR2A (IC50 > 300 μM), showed no significant reversible inhibition of CYP3A4, 2D6, 2C9 (IC50 > 50 μM), and was not a substrate for human Pgp efflux (MDR1 B–A/A–B ratio = 2.4). Compound 16 showed a similar profile to 9 in rat with respect to plasma protein binding (4.9% free fraction) and pharmacokinetic parameters (Table 7). As a result, the enhanced in vitro potency of 16 translated to improved ED50 values over 9 in the ex vivo receptor assay following IV and PO dosing (0.6 and 8.1 mpk, respectively). Given the in vitro profile and markedly improved in vivo activity observed with 16, we focused on optimizing the benzyl-1,2,4-oxadiazoles.
Table 3. In Vitro Binding Data for Cyclopentyl-Oxadiazolesa.

As shown in Table 4, incorporation of fluorine in the 3-position resulted in a loss of potency (19), while both the 2- and 4-fluoro derivatives were well tolerated in terms of potency and hERG selectivity (18, 20). When the size of the ortho substituent was increased to a methyl group (21), a significant loss in potency and selectivity was observed. Substitution with a methyl group on the 4-position of the phenyl ring (22) led to excellent potency at NR2B (Ki = 0.88 nM) and selectivity over hERG binding. In rats, compound 22 showed improved oral bioavailability over 16 and improved receptor occupancy following oral dosing with an ED50 = 4.8 mpk (Table 7). The 4-chloro derivative (23) remained subnanomolar at NR2B; however, the hERG binding worsened (IP = 7200 nM). Compound 23 showed improved clearance and half-life in rats, and achieved an ED50 = 3.5 mpk in the ex vivo receptor occupancy assay following oral dosing (Table 7). Larger alkyl groups such as ethyl (24) and isopropyl (25) were tolerated in the 4-position in terms of NR2B potency; however, the hERG selectivity further deteriorated as the substituent became larger. Fluorinated methyl derivatives of 22 were investigated in an attempt to block potential metabolism on the methyl group in 22. While the trifluoromethyl analogue (26) resulted in a modest loss of potency and the difluoromethyl derivative (27) maintained potency, both compounds suffered from significant loss in selectivity over hERG binding. Interestingly, incorporation of the difluoromethyl group significantly worsened the Pgp liability in rat for 27 versus the methyl derivative 22 (mdr1a B–A/A–B ratios = 8.4 versus 1.3). It was anticipated more polar substituents would improve the hERG selectivity, and the methanol derivative 28 showed minimal hERG binding. Unfortunately, polar substituents were not well tolerated in terms of NR2B potency and 28 suffered a significant loss in potency at NR2B. Even the slightly more polar methoxy group (29) was not well tolerated in terms of NR2B potency. Finally, the 2-fluoro substitution had been well tolerated in 18, so it was combined with the 4-methyl group to give 30, a highly potent and selective compound. Unfortunately, 30 showed poor oral bioavailability in rat pharmacokinetic studies and worse receptor occupancy than 22 following IV dosing in rat (Table 7), precluding further characterization of 30.
Table 4. In Vitro Binding Data for Benzyl-Oxadiazolesa.

| NR2B | hERG | ||
|---|---|---|---|
| compd | R1 | Ki (nM)b | IP (nM)c |
| 16 | H | 6.2 | 28000 |
| 18 | 2-F | 7.6 | 20000 |
| 19 | 3-F | 29 | 15000 |
| 20 | 4-F | 6.5 | 21000 |
| 21 | 2-CH3 | 26 | 3400 |
| 22 | 4-CH3 | 0.88 | 20000 |
| 23 | 4-Cl | 0.99 | 7200 |
| 24 | 4-CH2CH3 | 0.93 | 6200 |
| 25 | 4-CH(CH3)2 | 0.98 | 1500 |
| 26 | 4-CF3 | 3.7 | 5100 |
| 27 | 4-CHF2 | 0.84 | 4000 |
| 28 | 4-CH2OH | 91 | >30000 |
| 29 | 4-OCH3 | 36 | 8600 |
| 30 | 2-F, 4-CH3 | 0.83 | 14000 |
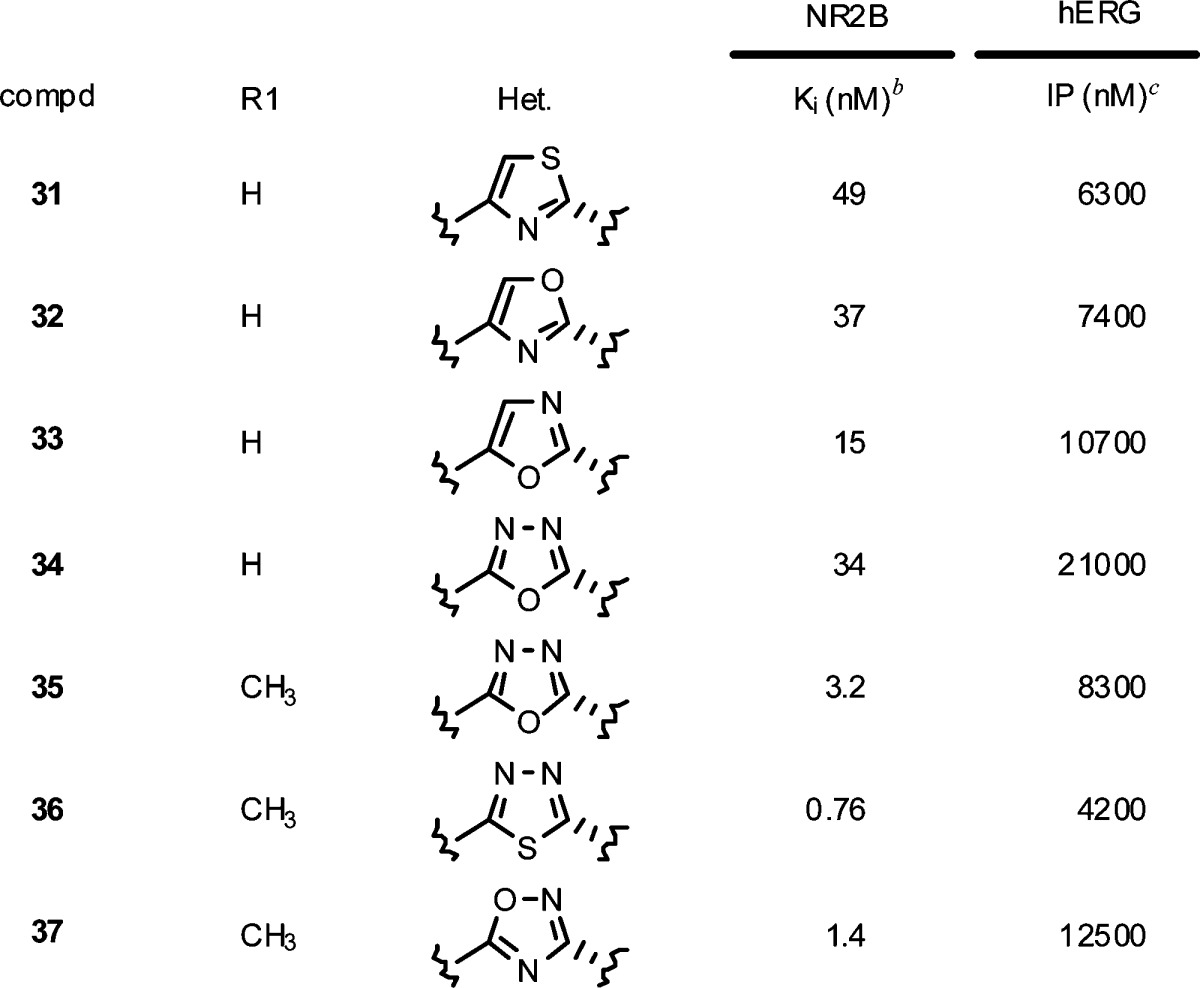
Based on the promising leads identified with the 3-benzyl-1,2,4-oxadiazole ring system, optimization of the heterocycle linker was investigated. Replacing the 1,2,4-oxadiazole with a 4-benzyl-thiazole (31), 4-benzyl-oxazole (32) or 5-benzyl-oxazole (33) resulted in inferior NR2B potency and hERG selectivity to 16. The isomeric 1,3,4-oxadiazole linker (34) also lost 5-fold in potency but maintained an acceptable hERG binding value. Consistent with the earlier SAR, incorporation of a 4-methyl substituent (35) regained much of the potency, though the hERG binding now dropped below the 10 μM target. The subnanomolar potency target was achieved by replacing the oxygen in 35 with a sulfur to give 1,3,4-thiadiazole 36 (Ki = 0.76 nM). Unfortunately, the hERG binding also became more potent and precluded further in vivo characterization of 36. Interestingly, the more polarized 1,3,4-thiadiazole caused 36 to become a substrate for human and rat Pgp efflux (MRD1 and mdr1a B–A/A–B ratio = 5.6 and 8.6, respectively). A better balance between potency, selectivity, and Pgp liability was obtained by preparing the isomeric 5-substituted-1,2,4-oxadiazole (37). Though slightly less potent (Ki = 1.4 nM) and selective (hERG IP = 12.5 μM) than 22, compound 37 was not a substrate for human and rat Pgp efflux (MRD1 and mdr1a B–A/A–B ratio = 0.9 and 2.1, respectively) and was advanced into the ex vivo receptor occupancy study. Compound 37 was less potent in vivo relative to 22 following either IV dosing (ED50 = 3.1 mpk) or oral dosing (ED50 = 10.8 mpk) and therefore was not advanced further.
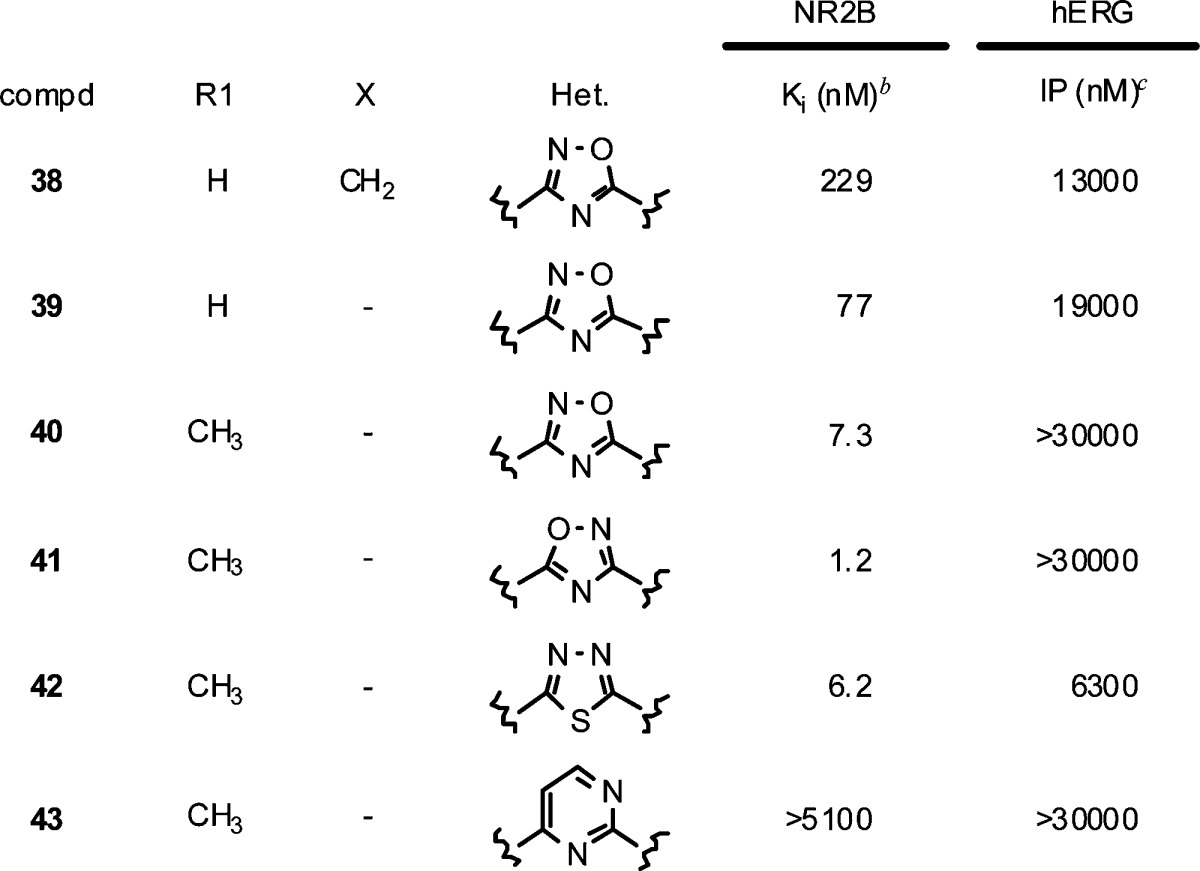
In parallel to investigating the heterocycle linker, we wanted to optimize the cyclopentane core. Though we found the corresponding cyclohexane core maintained most of the NR2B potency, we were most interested in simplifying the cyclopentane core by removing one of the stereocenters. This could be accomplished by incorporating an N-linked (3S)-aminopyrrolidine in place of the (1S,3S)-disubstituted cyclopentane. Initially, we were surprised to see a substantial loss of potency simply by replacing the carbon linked benzyl-1,2,4-oxadiazole in 16 with an N-linkage (38). Reasoning that the trajectory of the terminal phenyl ring changed with the planar N-linkage, we decided to reinvestigate the phenyl-substituted oxadiazole. Gratifyingly, 39 retained much of the potency observed in the earlier C-linked phenyl-oxadiazole series and 4-methyl substitution (40) was only 2-fold less potent than the cyclopentane 9. Unlike the C-linked oxadiazoles, phenyl substituents appended to the 1,2,4-oxadiazole were superior to the benzyl groups when linked through a nitrogen. Interestingly the selectivity over hERG binding also was improved with the N-linkage, likely due to the increased basicity and more polar nature of the central N-linked heterocycle. The isomeric 1,2,4-oxadiazole 41 showed an additional 6-fold improvement in potency (Ki = 1.2 nM) and was highly selective over hERG binding (IP > 30 μM). Unfortunately, the physical properties of 41 were very poor and precluded dosing in the receptor occupancy assay. Incorporation of a more basic linker to improve solubility, such as a 1,3,4-thiadiazole (42), led to a loss in potency, as did 6-membered heterocycles (e.g., 43).
Table 6. In Vitro Binding Data for Amino-Heterocyclesa.
 |
Based on the favorable NR2B potency both in vitro and in vivo following oral dosing, a more detailed evaluation of 22 was undertaken. Compound 22 was highly potent in a functional assay using cells expressing NR2B (IC50 = 1.0 nM) and remained equipotent in a binding assay using a sample of homogenized human temporal cortex (Ki = 0.81 nM). In an electrophysiology assay using NR2B receptors, 22 showed full blockade of ion flux with KD = 0.35 nM. Compound 22 exhibited high levels of selectivity over NR2A (IC50 = 200 μM), hERG binding (IP = 20 μM), α-adrenergic receptors based on Prazosin binding (IC50 > 100 μM),51 and CYP P450s including CYP3A4, 2C9, and 2D6. Compound 22 was not a human or rat Pgp substrate (MDR1 and mdr1 B–A/A–B ratios = 1.4 and 1.3, respectively) and showed a high passive permeability coefficient (Papp = 36 × 10–6 cm/s), indicating free penetration of the blood-brain barrier was likely with this analogue. In pharmacokinetic studies with higher species, 22 showed excellent oral bioavailability, half-life and clearance in dog, and moderate clearance and oral bioavailability in rhesus (Table 7).
Compound 22 was further evaluated in animal models of neuropathic pain, Parkinson’s disease, and motor function. In the spinal nerve ligation model of neuropathic pain in rats (Chung model), surgical ligation of two lumbar nerves in the spinal column induces a state of mechanical allodynia.48 The 50% paw withdrawal threshold was then determined as a function of dose following oral administration of compound 22 and reported as a percent maximum possible effect (% MPE)49 relative to presurgical withdrawal threshold. As shown in Figure 3A, compound 22 significantly inhibited tactile allodynia in a dose dependent manner after oral administration at 10 and 30 mg/kg. Specifically, 22 produced an average improvement in the maximal possible effect of 15% (3 mg/kg), 41% (10 mg/kg), and 69% (30 mg/kg) compared to vehicle treated animals.
Figure 3.

(A) Oral efficacy of 22 in spinal nerve ligation model of neuropathic pain in rats (n = 10). Abbreviation: %MPE = maximum possible effect relative to presurgical animals. (B) Effects on haloperidol-induced catalepsy with increasing oral doses of 22 in rats (n = 8).
In addition to reversing mechanical allodynia in rats, compound 22 was efficacious in an acute rodent model of Parkinson’s disease, the haloperidol-induced catalepsy model. In this model, the dopamine antagonist haloperidol is administered at a dose previously shown to elicit an acute cataleptic response in rats,50 and test agents are evaluated for the ability to reverse catalepsy. As shown in Figure 3B, compound 22 reduced catalepsy scores in a dose dependent manner, producing average improvements of 34% (3 mg/kg), 86% (10 mg/kg), and 92% (30 mg/kg) compared to vehicle treated animals. In the rotarod assay, there was no measurable affect on motor coordination when 22 was dosed orally at 100 mg/kg.20 Therefore, in contrast to nonselective NMDA antagonists, compound 22 showed a significant therapeutic margin between efficacy in the pain and Parkinson’s models and locomotor impairment.
In summary, a novel class of potent NR2B-selective NMDA receptor antagonists was designed and optimized to give a compound with a superior profile to that of the benchmark compound 6. Initial assessment of the stereochemical requirements of 3-substituted aminocyclopentanes demonstrated the required (S)-stereochemistry of the amine. Replacement of the carbamate led to the identification of a 1,2,4-oxadiazole as a suitable isostere that was not susceptible to Pgp efflux. Further optimization with respect to selectivity over hERG binding and pharmacokinetics led to the identification of compound 22 as a highly potent and selective oral agent that demonstrated efficient receptor occupancy in rats. Furthermore, 22 did not adversely affect motor function at high dose and demonstrated excellent efficacy following oral administration in the spinal nerve ligation model of neuropathic pain and in an acute model of Parkinson’s disease in a dose dependent manner.
Methods
Chemistry
As shown in Scheme 1, compound 8 was readily prepared from the appropriate N-Boc-1-aminocyclopentane-3-carboxylic acid (44). All the stereoisomers of 44 were commercially available as single enantiomers and were independently converted to 8. Reduction of the carboxylic acid 44 followed by reaction with methanesulfonyl chloride provided mesylate 45. Conversion of mesylate 45 to the Cbz-protected amine 46 was accomplished via azide displacement, reduction, Cbz-protection, and subsequent treatment with acid. Coupling amine 46 to 4-chloro-1-(tetrahydro-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidine (47)41 followed by removal of the tetrahydropyran group gave compound 8.
Scheme 1.

Reagents and conditions: (a) BH3·THF, THF, 0 °C, 1 h; (b) MsCl, TEA, DCM, 0 °C, 0.3 h; (c) NaN3, DMF, 50 °C, 15 h; (d) H2, 10%Pd/C, ethanol, RT, 3.5 h; (e) N-(benzyloxycarbonyloxy) succinimide, DCM, RT, 12 h; (f) TFA, RT, 0.5 h; (g) 4-chloro-1-(tetrahydro-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidine (47), 2-propanol, DIPEA, 85 °C, 7 h; (h) 6N HCl, methanol, 60 °C, 1 h.
3-Substituted 1,2,4-oxadiazoles were prepared from the corresponding hydroxyamidines in a straightforward manner (Scheme 2). Synthesis of the 1,2,4-oxadiazole ring was accomplished via a two-step procedure involving acylation of 48 with (S,S)-44 under standard EDC coupling conditions followed by cyclodehydration with sodium acetate.51 Boc-deprotection under acidic conditions yielded amines 49, which were then coupled to 47. Final deprotection of the tetrahydropyran group gave compounds 9–14 and 16–30.
Scheme 2.

Reagents and conditions: (a) (S,S)-44, EDC, HOBt, DCM, RT, 3–12 h; (b) NaOAc, ethanol, water, 85 °C, 5–14 h; (c) HCl, EtOAc RT, 0.5 h; (d) 47, 1-butanol, DIPEA, 90 °C, 3–7 h; (e) HCl, EtOAc, RT, 1–2 h.
Preparation of the homologated phenyl-1,2,4-oxadiazole 15 is shown in Scheme 3. Cyanide displacement of mesylate (S,S)-45 and hydrolysis provided homologated acid 50. As described previously, condensation with hydroxyamidine 48 and cyclization provided the 1,2,4-oxadiazole linker. Amine deprotection, coupling with 47, and acid deprotection then gave compound 15.
Scheme 3.

Reagents and conditions: (a) NaCN, DMSO, 70 °C, 24 h; (b) NaOH, methanol, 70 °C, 15 h; (c) 48, EDC, HOBt, DCM, RT, 3 h; (d) NaOAc, ethanol, water, 85 °C, 5 h; (e) TFA, RT, 0.5 h; (f) 47, 2-propanol, DIPEA, 85 °C, 7 h; (g) 6N HCl, methanol, 60 °C, 0.5 h.
The various heterocycle linkers in Table 5 were available from (S,S)-44 (Scheme 4). Formation of thiazole 31 was achieved by first reacting primary amide 52 with Lawesson’s reagent, followed by treatment of the resultant thioamide with 1-chloro-3-phenylpropan-2-one, to form the thiazole ring. Amine deprotection, coupling with 47, and final acid deprotection gave compound 31. In a similar fashion, oxazole 32 was prepared by treatment of amide 52 with 1-chloro-3-phenylpropan-2-one, followed by amine deprotection, coupling with 47, and final acid deprotection. The isomeric 1,2,4-oxadiazole ring in 37 was accomplished via dehydration of amide 52 to give a nitrile that was converted to hydroxyamidine 53 using hydroxylamine hydrochloride. Coupling of 53 with [4-(methyl)phenyl]acetic acid and cyclization gave the Boc-precursor to 37. Deprotection of the amine, coupling to 47, and final deprotection of the tetrahydropyran group gave compound 37. The isomeric oxazole 33 was prepared from (S,S)-44 by first coupling with 1-amino-3-phenylpropan-2-ol and then oxidizing to the ketone 54. In this case, deprotection of the Boc-amine and coupling with 47 was performed prior to cyclodehydration using Burgess reagent. Acid deprotection of the THP protecting group provided oxazole 33. The 1,3,4-oxadiazoles (34, 35) and 1,3,4-thiadiazole (36) were prepared first by condensing (S,S)-44 with the corresponding acyl-hydrazides to give 55. Dehydration in the presence of Burgess reagent gave the 1,3,4-oxadiazole linker, while dehydration in the presence of Lawesson’s reagent gave the 1,3,4-thiadiazole linker. Boc-deprotection, coupling with 47, and THP-deprotection then gave compounds 34–36.
Table 5. In Vitro Binding Data for Benzyl Heterocyclesa.
 |
Scheme 4.

Reagents and conditions: (a) NH4OH, EDC, HOBt, DMF, RT, 72 h; (b) Lawesson's reagent, THF, RT, 17 h; (c) 1-chloro-3-phenyl acetone, EtOH, 85 °C, 3 h; (d) 1-chloro-3-phenyl acetone, 130 °C, 2 h; (e) BOC-ON, triethylamine, THF, RT, 3–16 h; (f) TFA, RT, 0.5 h or HCl in EtOAc (∼4M), RT, 20 min; (g) 47, 1-butanol, DIPEA, 150 °C, 10 min in microwave or 47, 2-propanol, DIPEA, 70 °C, 15 h; (h) 6 N HCl, methanol, 60 °C, 1 h or HCl in EtOAc (∼4 M), 1:1 EtOAc/MeOH, RT, 15 min; (i) trifluoroacetic anhydride, pyridine, THF, 0 °C to RT, 2 h; (j) HONH2 HCl, ethanol, Na2CO3, 85 °C, 72 h; (k) [4-(methyl)phenyl]acetic acid, HBTU, DIPEA, HOBt, DMF, RT, 1.5 h; (l) NaOAc, ethanol, water, 85 °C, 5 h; (m) 1-amino-3-phenylpropan-2-ol, EDC, HOBt, DCM, RT, 1.5 h; (n) Dess–Martin periodinane, DCM, RT, 15 min; (o) Burgess reagent, THF, 120 °C, 10 min in microwave; (p) phenylacetohydrazide or 2-(4-methylphenyl)acetohydrazide, HBTU, DCM, RT, 14 h; (q) Lawesson's reagent, toluene, 150 °C, 10 min in microwave.
Preparation of the amino-linked heterocycles is shown in Scheme 5. For the targeted structures (38–42), once the N-linked heterocycles were formed, the Boc-protected precursors were advanced to the final compounds as described previously, involving Boc deprotection, coupling to 47, and final deprotection with acid. The 5-amino-1,2,4-oxadiazole precursors to 38–40 were prepared by reaction of the appropriate hydroxyamidines (48) with trichloroacetic anhydride, followed by displacement using tert-butyl (3S)-pyrrolidin-3-ylcarbamate (57). Treatment of pyrrolidine 57 with cyanogen bromide followed by the addition of hydroxylamine, acylation, and cyclodehydration gave the isomeric 3-amino-1,2,4-oxadiazole precursor to 41. The aminothiadiazole precursor to 42 was prepared by reaction between pyrrolidine 57 and chloride 59. Chloride 59 was available in two steps through condensation between 4-methylbenzonitrile and thiosemicarbazide, followed by standard Sandmeyer halogenation.
Scheme 5.

Reagents and conditions: (a) Trichloroacetic anhydride, HOBt, toluene, 120 °C, 2.5 h; (b) 57, MeOH, RT, 10 days; (c) TFA, RT, 0.5 h; (d) 47, 1-butanol, DIPEA, 150 °C, 15 min in microwave; (e) 6 N HCl, methanol, 60 °C, 1 h; (f) NaOAc, cyanogen bromide, methanol, 0 °C to RT, 5 h; (g) HONH2 HCl, ethanol, Na2CO3, 85 °C, 3 h; (h) 4-methylbenzoic acid, EDC, HOBt, DCM, RT, 14 h; (i) NaOAc, ethanol, water, 85 °C, 3 h; (j) thiosemicarbazide; (k) CuCl, NaNO2, HCl.
Synthesis of N-{(1S,3S)-3-[3-(4-Methylbenzyl)-1,2,4-oxadiazol-5-yl]cyclopentyl}-1H-pyrazolo[3,4-d]pyrimidin-4-amine (22) (Scheme 2)
(1Z)-N′-Hydroxy-2-(4-methylphenyl)ethanimidamide (48)
To a solution of (4-methylphenyl)acetonitrile (31.5 g, 240 mmol) in 95% ethanol (525 mL) was added hydroxylamine hydrochloride (67.8 g, 976 mmol) and sodium carbonate (103 g, 976 mmol), and the resulting solution was heated to 85 °C. After 14 h, the reaction was cooled to room temperature, filtered, and concentrated. The residue was suspended in ether and extracted with 1 M hydrochloric acid. The combined aqueous layers were basified to pH = 9 with ammonium hydroxide, saturated with sodium chloride, and extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered, and concentrated to give 48 (30.9 g) as a waxy solid. MS 165.3 (M + 1).
{(1S,3S)-3-[3-(4-Methylbenzyl)-1,2,4-oxadiazol-5-yl]cyclopentyl}amine (49)
To a solution of (1S,3S)-3-[(tert-butoxycarbonyl)amino]cyclopentanecarboxylic acid (12.9 g, 56.1 mmol) in methylene chloride (150 mL) at room temperature was added HOBt (8.73 g, 57.0 mmol) and EDC (11.0 g, 57.2 mmol). After 20 min, (1Z)-N′-hydroxy-2-(4-methylphenyl)ethanimidamide (11.2 g, 68.4 mmol) was added to the reaction and the reaction was permitted to stir an addition 12 h. The reaction was poured into saturated sodium bicarbonate solution and extracted with ethyl acetate. The combined organic layers were washed with 1 M sodium hydroxide, dried over sodium sulfate, filtered, and concentrated to give tert-butyl {(1S,3S)-3-[({[(1Z)-1-amino-2-(4-methylphenyl)ethylidene]amino}oxy)carbonyl]cyclopentyl}carbamate (19.1 g) as a waxy solid. MS 376.3 (M + 1).
To a solution of tert-butyl {(1S,3S)-3-[({[(1Z)-1-amino-2-(4-methylphenyl) ethylidene]amino}oxy)carbonyl]cyclopentyl}carbamate (19.1 g, 50.9 mmol) in 80% aqueous ethanol (250 mL) was added sodium acetate trihydrate (15.8 g, 116 mmol), and the resulting solution was heated to 85 °C under N2. After 14 h, the reaction was cooled to room temperature, partially concentrated, poured into saturated citric acid, and extracted with ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate, water, brine, dried over sodium sulfate, filtered, and concentrated to give tert-butyl {(1S,3S)-3-[3-(4-methylbenzyl)-1,2,4-oxadiazol-5-yl]cyclopentyl} carbamate (11.4 g) as a yellow solid. HRMS (M + H+): calculated = 358.2125, observed = 358.2135; 1H NMR (400 MHz, DMSO-d6) δ 7.21–7.09 (m, 4H), 7.00 (d, J = 6.4 Hz, 1H), 3.99 (s, 2H), 3.96–3.88 (m, 1H), 3.59–3.48 (m, 1H), 2.27 (s, 3H), 2.20–2.09 (m, 1H), 2.07–1.98 (m, 1H), 1.98–1.85 (m, 2H), 1.79–1.68 (m, 1H), 1.57–1.47 (m, 1H), 1.38 (s, 9H).
To tert-butyl {(1S,3S)-3-[3-(4-methylbenzyl)-1,2,4-oxadiazol-5-yl]cyclopentyl} carbamate (11.37 g, 6.87 mmol) was added anhydrous hydrochloric acid in ethyl acetate (30 mL, ∼4 M), and the resulting solution was stirred at room temperature. After 30 min, the reaction was concentrated. The residue was dissolved in 1 M hydrochloric acid and washed with ethyl acetate three times. The aqueous layer was basified to pH = 8 with ammonium hydroxide, saturated with sodium chloride, and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate, filtered, and concentrated. A solution of the residue in methanol was treated with anhydrous hydrochloric acid in ethyl acetate and then concentrated to yield the hydrochloride salt of 49 (6.87 g) as a white solid. HRMS (M + H+): calculated = 258.1601, observed = 258.1590; 1H NMR (400 MHz, DMSO-d6) δ 8.06 (s, 3H), 7.20–7.09 (m, 4H), 4.00 (s, 2H), 3.74–3.63 (m, 2H), 2.27 (s, 3H), 2.26–2.18 (m, 2H), 2.16–2.06 (m, 2H), 1.87–1.77 (m, 1H), 1.77–1.64 (m, 1H).
N-{(1S,3S)-3-[3-(4-Methylbenzyl)-1,2,4-oxadiazol-5-yl]cyclopentyl}-1H-pyrazolo[3,4-d]pyrimidin-4-amine (22)
To a solution of 49 (6.87 g, 23.4 mmol) in 1-butanol (50 mL) was added DIPEA (50 mL) and 4-chloro-1-(tetrahydro-pyran-2-yl)-1H-pyrazolo[3,4-d]pyrimidine (47) (5.91 g, 24.8 mmol), and the solution was heated to 90 °C for 3 h. The mixture was cooled and concentrated under reduced pressure. The resulting residue was dissolved in methanol (15 mL) and a solution of anhydrous hydrochloric acid in ethyl acetate (30 mL, ∼4 M) was added. The mixture was stirred for 2 h, at which time the reaction was quenched with saturated sodium bicarbonate. The solution was extracted with ethyl acetate, and the organic layer was dried over sodium sulfate, filtered, and concentrated to dryness under reduced pressure. The compound was recrystallized from ethanol (40 mL) to yield 22 (4.51 g) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.14 (s, 1H), 8.24 (s, 1H), 7.14–7.20 (m, 4H), 4.77 (m, 1H), 4.00°(s, 2H), 3.73–3.62 (m, 1H), 2.50–2.39 (m, 1H), 2.39–2.32 (m, 2H), 2.32–2.28 (s, 3H), 2.25–2.16 (m, 1H), 2.07–1.95 (m, 1H), 1.90–1.79 (m, 1H); 1H NMR (400 MHz, DMSO-d6) δ 8.48 (s, 1H), 7.22–7.10 (m, 4H), 4.69 (br s, 1H), 4.02 (s, 2H), 3.81–3.68 (m, 1H), 2.39–2.20 (m, 7H), 1.98–1.80 (m, 2H) ppm. Elemental analysis: Calculated for (C20H21N7O·HCl/H2O): C, 63.98%; H, 5.64%; N, 26.12%. Observed: C, 63.64%; H, 5.42%; N, 25.82%. HRMS (ESI) m/z 376.1881 [(M + H)+ calcd 376.1880].
NR2B in Vitro Activity
All final compounds were evaluated as NR2B antagonists using an NR2B-selective binding assay,43 and select compounds were evaluated in a functional assay measuring Ca2+ flux in cells expressing recombinant NR1/NR2A receptors52 or NR1/NR2B receptors.53 Selectivity over the hERG-channel was evaluated in an MK-499-binding assay.44 Inhibition of NMDA receptor-activated currents at recombinant human NMDA receptor subtypes was determined as described previously.28
Pharmacokinetics
Pharmacokinetic characterization of test agents was conducted in conscious male Sprague–Dawley rats (300–500 g; n = 3–4/study), male and female beagle dogs (13–15 kg; n = 2/study), or male rhesus (4–6 kg, n = 2/study). In all species, single doses of test agents were administered either intravenously in a vehicle of 100% DMSO or orally by gavage in a vehicle of 1% methylcellulose aqueous suspension. Typical test doses were 2 mg/kg IV and 10 mg/kg PO to rats; 0.5–1 mg/kg IV and 1–3 mg/kg PO to dogs; and 1 mg/kg IV and PO to rhesus. Blood samples for the determination of test agent plasma concentration were obtained at multiple time points up to 24 h after single dose test agent administration.
NR2B in Vivo Receptor Occupancy in Rats
Rats were maintained and tested in an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility in strict compliance with all applicable regulations. Male Sprague–Dawley rats weighing 90–115 g (Taconic Farms, Taconic, NY) were housed in 12 h light/dark cycle and fasted overnight (water was provided ad libitum). Compounds were administered orally in a 50/50 PEG200/5% dextrose solution; three doses were tested (3, 1, and 0.3 mg/kg) with four animals per dose. For oral testing, test compounds were dosed by gavage and radiotracer (200 uCi/kg of 3-H-Compound Y) was administered by IV injection 52.5 min after test compound dosing. Animals were euthanized 7.5 min after administration of the radiotracer, and a 100–150 mg sample of cortex was collected. Brain samples were homogenized in ice-cold HEPES buffer for 10 s with a Polytron (Brinkman Instruments, Westbury, NY). Homogenized brain samples were immediately filtered through 25 mm Pall A/E filters (Pall Corporation, East Hills, NY) presoaked in 0.2% polyethyleneimine (PEI) using a Hoefer filter unit (GE Healthcare, Piscataway, NJ). Filters were washed with 25 mL ice-cold HEPES buffer (10 mM HEPES, 150 mM NaCl, and 5 mM KCL pH 7.4). For tail vein injections, rats were placed in a Perspex restrainer. Total binding was determined by oral administration of vehicle (50/50 PEG200/5% Dextrose) 52.5 min prior to IV radiotracer administration. Nonspecific binding was determined by IV administration of 15 mg/kg of test compound 7.5 min prior to administering the radiotracer. A nonfiltered aliquot (0.5 mL) of homogenized tissue (free) and the filtered (bound) were counted using a LS 6000 instrument (Beckman Coulter, Fullerton, California).
Spinal Nerve Ligation in Rats
Male Sprague–Dawley rats (Taconic, Germantown, NY) weighing 200–250 g at the time of testing housed at 22 °C under a 12 h light/12 h dark cycle and with free access to water and food ad libitum were used. All treatment and testing procedures were approved by the Institutional Animal Care and Use Committee of MRL at Merck. Spinal nerve ligation (SNL) injury was induced using the procedure of Kim and Chung.48 Anesthesia was induced with 2% gaseous isofluorane (for induction, 3–5% and O2 500–700 L; for maintenance, 2–3% and O2 400–500 L). Following dorsal skin incision and muscle separation, the posterior interarticular transverse process of L/S1 was exposed and carefully removed with a micro Rongeur. The L5 and L6 spinal nerves were tightly ligated by a square knot with 6-0 silk thread. The muscles were closed with 4-0 absorbable sutures, and the skin was closed with wound clips. Rats that exhibited motor deficiency were excluded from further testing. Preoperation cutoff value is 15 g. Compound 22 was dissolved in 1% methylcellulose and orally administered via gavage. Rats were placed individually in a mesh metal floor and habituated for 20 min before testing. The tactile allodynia was measured as the force of withdrawal threshold to the ipsilateral hind paw in response to probing with a series of von Frey filaments. The von Frey hair was presented perpendicular to the plater surface. The animals with presurgery baseline withdrawal threshold greater than 15 g were used for surgery. Pretreatment baseline testing was conducted at 7 days post surgery just prior to the compound injection. The animals developed allodynia (paw withdrawal threshold smaller than 3.5 g) were used for compound testing. The results were expressed by percent of maximal possible effect (MPE). MPE was calculated as follows:
Behavioral testing was analyzed with t test for difference between 22d treated groups with vehicle group.
Haloperidol-Induced Catalepsy in Rats
Thirty minutes after injecting rats with the dopamine receptor antagonist haloperidol (1.5 mg/kg, i.p.), rats were dosed with either vehicle (1% methylcellulose) or compound 22d (3, 10, 30 mg/kg, PO) and measured for catalepsy 1 h after dosing. Haloperidol was administered at a dose previously shown to elicit an acute cataleptic response in rats.50 Thirty minutes after injecting rats with haloperidol (1.5 mg/kg, i.p., dissolved in 0.2% lactic acid), rats were dosed with either vehicle or 22d in vehicle (1% methylcellulose) via oral gavage at 3, 10, and 30 mg/kg and measured for catalepsy 1 h after dosing using a rectangular wire grid. Latency time (seconds) after treatment was recorded, and differences in mean time values among vehicle, 3, 10, and 30 mg/kg 22d treated groups were compared by one-way ANOVA followed by Dunnett’s post test to assess significance in comparison with vehicle-treated rats. Statistical significance was set at P < 0.05. Data are expressed as mean ± SEM.
Supporting Information Available
Additional experimental details and characterization of 8–21 and 23–40. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Dingledine R.; Borges K.; Bowie D.; Traynelis S. F. (1999) The Glutamate Receptor Ion Channels. Pharmacol. Rev. 51(1), 7–61. [PubMed] [Google Scholar]
- Banke T. G.; Traynelis S. F. (2003) Activation of NR1/NR2B NMDA Receptors. Nat. Neurosci. 6(2), 144–152. [DOI] [PubMed] [Google Scholar]
- Le D. A.; Lipton S. A. (2001) Potential and Current Use of N-Methyl-d-Aspartate (NMDA) Receptor Antagonists in Diseases of Aging. Drugs Aging 18(10), 717–724. [DOI] [PubMed] [Google Scholar]
- Parsons C. G.; Danysz W.; Quack G. (1998) Glutamate in CNS Disorders as a Target for Drug Development: An Update. Drug News Perspect. 11(9), 523–579. [DOI] [PubMed] [Google Scholar]
- Kemp J. A.; McKernan R. M. (2002) NMDA receptor pathways as drug targets. Nat. Neurosci. 5(suppl.), 1039–1042. [DOI] [PubMed] [Google Scholar]
- Hallett P. J.; Standaert D. G. (2004) Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 102, 155–174. [DOI] [PubMed] [Google Scholar]
- Marino M. J.; Valenti O.; Conn P. J. (2003) Glutamate Receptors and Parkinson’s Disease: Opportunities for Intervention. Drugs Aging 20(5), 377–397. [DOI] [PubMed] [Google Scholar]
- Hallett P. J.; Dunah A. W.; Ravenscroft P.; Zhou S.; Bezard E.; Crossman A. R.; Brotchie J. M.; Sandaert D. G. (2005) Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology 48, 503–516. [DOI] [PubMed] [Google Scholar]
- Parsons C. G. (2001) NMDA receptors as targets for drug action in neuropathic pain. Eur. J. Pharmacol. 429, 71–78. [DOI] [PubMed] [Google Scholar]
- Petrenko A. B.; Yamakura T.; Baba H.; Shimoji K. (2003) The Role of N-methyl-d-aspartate (NMDA) Receptors in Pain: A Review. Anesth. Analg. 97(4), 1108–1116. [DOI] [PubMed] [Google Scholar]
- Reisberg B.; Doody R.; Stoffler A.; Schmitt F.; Ferris S.; Modius H. J. (2003) Memantine in Moderate-to-Severe Alzheimer’s Disease. N. Engl. J. Med. 348, 1333–1341. [DOI] [PubMed] [Google Scholar]
- Witt A.; Macdonald N.; Kirkpatrick P. (2004) Memantine hydrochloride. Nat. Rev. Drug Discovery 3(2), 109–110. [DOI] [PubMed] [Google Scholar]
- Hocking G.; Cousins M. J. (2003) Ketamine in chronic pain management: an evidence-based review. Anesth. Analg. 97(6), 1730–1739. [DOI] [PubMed] [Google Scholar]
- a McBain C. J.; Mayer M. L. (1994) N-Methyl-d-Aspartic Acid Receptor Structure and Function. Physiol. Rev. 74(3), 723–760. [DOI] [PubMed] [Google Scholar]; b Bendel O.; Meijer B.; Hurd Y.; Von Euler G. (2005) Cloning and expression of the human NMDA receptor subunit NR3B in the adult human hippocampus. Neurosci. Lett. 377, 31–36. [DOI] [PubMed] [Google Scholar]
- Chatterton J. E.; Awobuluyl M.; Premkumar L. S.; Takahashi H.; Talantova M.; Shin Y.; Cui J.; Tu S.; Sevarino K. A.; Nakanishi N.; Tong G.; Lipton S. A.; Zhang D. (2002) Excitatory glycine receptor containing the NR3 family of NMDA receptor subunits. Nature (London) 415(6873), 793–798. [DOI] [PubMed] [Google Scholar]
- Monyer H.; Burnashev N.; Laurie D. J.; Sakmann B.; Seeburg P. H. (1994) Developmental and Regional Expression in the Rat Brain and Functional Properties of Four NMDA Receptors. Neuron 12, 529–540. [DOI] [PubMed] [Google Scholar]
- Lynch D. R.; Guttman R. P. (2001) NMDA Receptor Pharmacology: Perspectives from Molecular Biology. Curr. Drug Targets 2(3), 215–231. [DOI] [PubMed] [Google Scholar]
- Erreger K.; Dravid S. M.; Banke T. G.; Wyllie D. J. A.; Traynelis S. F. (2005) Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signaling profiles. J. Physiol. 563(2), 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftis J. M.; Janowsky A. (2003) The N-methyl-d-aspartate receptor subunit NR2B: localization, functional properties, regulation, and clinical implications. Pharmacol. Ther. 97(1), 55–85. [DOI] [PubMed] [Google Scholar]
- Boyce S.; Wyatt A.; Webb J. K.; O’Donnell R.; Mason G.; Rigby M.; Sirinathsinghji D.; Hill R. G.; Rupniak N. M. J. (1999) Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology 38, 611–623. [DOI] [PubMed] [Google Scholar]
- Chazot P. L. (2004) The NMDA Receptor NR2B Subunit: A Valid Therapeutic Target for Multiple CNS Pathologies. Curr. Med. Chem. 11(3), 389–396. [DOI] [PubMed] [Google Scholar]
- Higgins G. A.; Ballard T. M.; Huwyler J.; Kemp J. A.; Gill R. (2003) Evaluation of the NR2B-selective NMDA receptor antagonist Ro-63-1908 on rodent behavior: evidence for an involvement of NR2B NMDA receptors in response inhibition. Neuropharmacology 44(3), 324–341. [DOI] [PubMed] [Google Scholar]
- Chizh B. A.; Headley P. M.; Tzschentke T. M. (2001) NMDA Receptor Antagonists as Analgesics: Focus on the NR2B Subtype. Trends Pharmacol. Sci. 22(12), 636–642. [DOI] [PubMed] [Google Scholar]
- Williams K. (2001) Ifenprodil, a Novel NMDA Receptor Antagonist: Site and Mechanism of Action. Curr. Drug Targets 2, 285–298. [DOI] [PubMed] [Google Scholar]
- Chenard B. L.; Bordner J.; Butler T. W.; Chambers L. K.; Collins M. A.; De Costa D. L.; Ducat M. F.; Dumont M. L.; Fox C. B.; Mena E. E.; Menniti F. S.; Nielsen J.; Pagnozzi M. J.; Richter K. E. G.; Ronau R. T.; Shalaby I. A.; Stemple J. Z.; White W. F. (1995) (1S,2S)-1-(4-Hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: A Potent New Neuroprotectant Which Blocks N-Methyl-d-Aspartate Responses. J. Med. Chem. 38(16), 3138–3145. [DOI] [PubMed] [Google Scholar]
- Doyle K. M.; Feerick S.; Kirby D. L.; Eddleston A.; Higgins G. A. (1998) Comparison of various N-methyl-d-aspartate receptor antagonists in a model of short-term memory and on overt behavior. Behav. Pharmacol. 9(8), 671–681. [DOI] [PubMed] [Google Scholar]
- Guscott M. R.; Clarke H. F.; Murray F.; Grimwood S.; Bristow L. J.; Hutson P. H. (2003) The effect of CP-101,606, an NMDA receptor NR2B subunit selective antagonist, in the Morris watermaze. Eur. J. Pharmacol. 476(3), 193–199. [DOI] [PubMed] [Google Scholar]
- Kundrotiene J.; Cebers G.; Wagner A.; Liljequist S. (2004) The NMDA NR2B Subunit-Selective Receptor Antagonist, CP-101,606, Enhances the Functional Recovery and Reduces Brain Damage after Cortical Compression-Induced Brain Ischemia. J. Neurotrauma 21(1), 83–93. [DOI] [PubMed] [Google Scholar]
- Tanaguchi K.; Shinjo K.; Mizutani M.; Shimada K.; Ishikawa T.; Menniti F. S.; Nagahishi A. (1997) Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br. J. Pharmacol. 122(5), 809–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash J. E.; Brotchie J. M. (2002) Characterisation of Striatal NMDA Receptors Involved in the Generation of Parkinsonian Symptoms: Intrastrial Microinjection Studies in the 6-OHDA-Lesioned Rat. Mov. Disord. 17(3), 455–466. [DOI] [PubMed] [Google Scholar]
- Wessell R. H.; Ahmed S. M.; Menniti F. S.; Dunbar G. L.; Chase T. N.; Oh J. D. (2004) NR2B selective NMDA receptor antagonist CP-101,606 prevents levodopa-induced motor response alterations in hemi-parkinsonian rats. Neuropharmacology 47, 184–194. [DOI] [PubMed] [Google Scholar]
- Steece-Collier K.; Chambers L. K.; Jaw-Tsai S. S.; Menniti F. S.; Greenamyre J. T. (2000) Antiparkinsonian Actions of CP-101,606, an Antagonist of NR2B Subunit-Containing N-Methyl-d-Aspartate Receptors. Exp. Neurol. 163(1), 239–243. [DOI] [PubMed] [Google Scholar]
- a Merchant R. E.; Bullock M. R.; Carmack C. A.; Shah A. K.; Wilner K. D.; Ko G.; Williams S. A. (1999) A double-blind, placebo-controlled study of the safety, tolerability and pharmacokinetics of CP-101,606 in patients with a mild or moderate traumatic brain injury. Ann. N.Y. Acad. Sci. 890, 42–50. [DOI] [PubMed] [Google Scholar]; b Bullock R. M.; Merchant R. E.; Carmack C. A.; Doppenberg E.; Shah A. K.; Wilner K. D.; Ko G.; Williams S. A. (1999) An open-label study of CP-101,606 in subjects with a severe traumatic head injury or spontaneous intracerebral hemorrhage. Ann. N.Y. Acad. Sci. 890, 51–58. [DOI] [PubMed] [Google Scholar]
- Sang C. N.; Weaver J. J.; Jinga L.; Wouden J.; Saltarelli M. D. (2003) The NR2B subunit-selective NMDA receptor antagonist, CP-101,606, reduces spontaneous pain intensity in patients with central and peripheral neuropathic pain. Soc. Neurosci. Abstr. 814, 9. [Google Scholar]
- a Layton M. E.; Kelly M. J.; Rodzinak K. J. (2006) Recent Advances in the Development of NR2B Subtype-selective NMDA Receptor Antagonists. Curr. Top. Med. Chem. 6, 697–709. [DOI] [PubMed] [Google Scholar]; b McCauley J. A. (2005) NR2B subtype-selective NMDA receptor antagonists: 2001–2004. Expert Opin. Ther. Pat. 15(4), 389–407. [Google Scholar]
- Fischer G.; Mutel V.; Trube G.; Malherbe P.; Key J. N. C.; Mohacsi E.; Heitz M. P.; Kemp J. A. (1997) Ro 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterization in vitro. J. Pharm. Exp. Ther. 283, 1285–1292. [PubMed] [Google Scholar]
- Pinard E.; Alanine A.; Bourson A.; Buttelmann B.; Heitz M.-P.; Mutel V.; Gill R.; Trube G.; Wyler R. (2002) 4-Aminoquinolines as a Novel Class of NR1/2B Subtype Selective NMDA Receptor Antagonists. Bioorg. Med. Chem. Lett. 12, 2615–2619. [DOI] [PubMed] [Google Scholar]
- a Claiborne C. F.; McCauley J. A.; Libby B. E.; Curtis N. R.; Diggle H. J.; Kulagowski J. J; Michelson S. R.; Anderson K. D.; Claremon D. A.; Freidinger R. M.; Bednar R. A.; Mosser S. D.; Gaul S. L.; Connolly T. M.; Condra C. L.; Bednar B.; Stump G. L.; Lynch J. J.; Macaulay A.; Wafford K. A.; Koblan K. S.; Liverton N. J. (2003) Orally Efficacious NR2B-Selective NMDA Receptor Antagonists. Bioorg. Med. Chem. Lett. 13, 697–700. [DOI] [PubMed] [Google Scholar]; b Nguyen K. T.; Claiborne C. F.; McCauley J. A.; Libby B. E.; Claremon D. A.; Bednar R. A.; Mosser S. D.; Gaul S. L.; Connolly T. M.; Condra C. L.; Bednar B.; Stump G. L.; Lynch J. J.; Koblan K. S.; Liverton N. J. (2007) Cyclic benzamidines as orally efficacious NR2B-selective NMDA receptor antagonists. Bioorg. Med. Chem. Lett. 17, 3997–4000. [DOI] [PubMed] [Google Scholar]
- Liverton N. J.; Bednar R. A.; Bednar B.; Butcher J. W.; Claiborne C. F.; Claremon D. A.; Cunningham M.; DiLella A. G.; Gaul S. L.; Libby B. E.; Lyle E. A.; Lynch J. J.; McCauley J. A.; Mosser S. D.; Nguyen K. T.; Stump G. L.; Sun H.; Wang H.; Yergey J.; Koblan K. S. (2007) Identification and Characterization of 4-Methylbenzyl 4-[(Pyrimidin-2-ylamino)methyl]piperidine-1-carboxylate, an Orally Bioavailable, Brain Penetrant NR2B Selective N-Methyl-d-Aspartate Receptor Antagonist. J. Med. Chem. 50, 807–819. [DOI] [PubMed] [Google Scholar]
- Liverton N. J., Butcher J. W., McIntyre C. J., Claiborne C. F., Claremon D. A., McCauley J. A., Romano J. J., Thompson W., and Munson P. M.. Preparation of N-substituted nonaryl heterocyclyl amides as NMDA/NR2B antagonists for relieving pain. PCT Int. Appl. WO 2002080928 A1, 17 October 2002.
- Thompson W., Young S. D., Phillips B. T., Munson P., Whitter W., Liverton N., Dieckhaus C., Butcher J., McCauley J. A., McIntyre C. J., Layton M. E., and Sanderson P. E.. Preparation of 4-Cycloalkylaminopyrazolopyrimidines as NMDA/NR2B Antagonists. PCT Int. Appl. WO 2005019221 A1, 3 March 2005.
- Yamazaki M; Neway W. E.; Ohe T; I-wu Chen; Rowe J. F.; Hochman J. H.; Chiba M.; Lin J. H. (2001) In vitro substrate identification studies for P-glycoprotein-mediated transport: Species difference and predictability of in vivo results. J. Pharmacol. Exp. Ther. 296, 723–735. [PubMed] [Google Scholar]
- Kiss L.; Cheng G.; Bednar B.; Bednar R.; Bennett P.; Kane S. A.; McIntyre C. J.; McCauley J. A.; Koblan K. S. (2005) In vitro characterization of novel NR2B selective NMDA receptor antagonists. Neurochem. Int. 46, 453–464. [DOI] [PubMed] [Google Scholar]
- Butcher J. W., Claremon D. A., Connolly T. M., Dean D. C., Karczewski J., Koblan K. S., Kostura M. J., Liverton N. J., and Melillo D. G.. Radioligand and Binding Assay. PCT Int. Appl. WO 0205860 A1, 24 January 2002.
- Mahar Doan K. M.; Humphreys J. E.; Webster L. O.; Wring S. A.; Sampine L. J.; Serabjit-Singh C. J.; Adkison K. K.; Polli J. W. (2002) Passive Permeability and P-Glycoprotein-Mediated Efflux Differentiate Central Nervous System (CNS) and Non-CNS Marketed Drugs. J. Pharm. Exp. Ther. 303, 1029–1037. [DOI] [PubMed] [Google Scholar]
- Greengrass P.; Bremner R. (1979) Binding Characteristics of Prazosin-H-3 to Rat-Brain Alpha-Adrenergic Receptors. Eur. J. Pharmacol. 55, 323–326. [DOI] [PubMed] [Google Scholar]
- All animal studies described in this report were approved by the Merck Research Laboratories Institutional Animal Care and Use Committee.
- Kim S. H.; Chung J. M. (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363. [DOI] [PubMed] [Google Scholar]
- %MPE = [(post-treatment value) – (pretreatment value)]/[(preoperation cutoff value) – pretreatment value)].
- Valenti O.; Marino M. J.; Wittmann M.; Lis E.; DiLella A. G.; Kinney G. G.; Conn P. J. (2003) Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J. Neurosci. 23(18), 7218–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamze A.; Hernandez J.-F.; Fulcrand P.; Martinez J. (2003) Synthesis of Various 3-Substituted 1,2,4-Oxadiazole-Containing Chiral β3- and α-Amino Acids from Fmoc-Protected Aspartic Acid. J. Org. Chem. 68(19), 7316–7321. [DOI] [PubMed] [Google Scholar]
- McCauley J. A., Theberge C. R., Liverton N. J., Claremon D. A., and Claiborne C. F.. 2-Benzyl and 2-Heteroaryl Benzimidazole NMDA/NR2B Antagonists. U.S. Patent 6,316,474 B1, Nov. 13, 2001.
- Bednar B.; Cunningham M. E.; Kiss L.; Cheng G.; McCauley J. A.; Liverton N. J.; Koblan K. S. (2004) Kinetic characterization of novel NR2B antagonists using fluorescence detection of calcium flux. J. Neurosci. Methods 137, 247–255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.