

The diverse members of the Amaryllidaceae family of natural products (Figure 1) have long been recognized for their medicinal properties.[1] In particular, many members of this small subgroup of isocarbostyrils exhibit unusually high levels of in vitro and in vivo antitumor and antiviral activity.[2] Despite their potential for clinical use many of these compounds are found in low natural abundance; while narciclasine (1) is available in practical quantities[1a,3], limited availability of lycoricidine (2), pancratistatin (3) and other derivatives poses one limitation to their development as viable therapeutic agents. Not surprisingly, the limited availability of these isocarbostyrils, as well as their interesting frameworks, have made these attractive and relevant targets for organic synthesis, and many successful strategies for the construction of 1, 2, and 3 have been described.[4] In contrast, though the natural products transdihydronarciclasine (4)[5] and trans-dihydrolycoricidine (5)[6] also demonstrate potent cytotoxicities, their syntheses have not been explored as extensively.[7] To date, three syntheses of (+)-5 have been reported in the literature[8] and a third report describes the synthesis of the non-natural enantiomer (−)-5.[9] Given their promising biological activity, and their interesting structures, we have been attracted to the isocarbostyrils as synthetic targets and herein describe the preparation of trans-dihydrolycoricidine by a route that employs enantioselective conjugate allylation to control the absolute configuration. We also describe the first examples of diastereoselective diboration as a method for control of the oxygenation pattern in the target.
Figure 1.
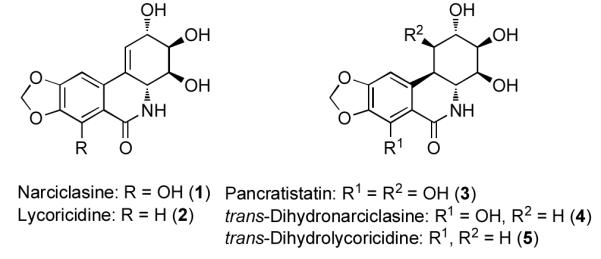
Representative Amaryllidaceae isocarbostyrils.
We envisioned an approach to the isocarbostyrils that centers on the diastereoselective 1,4-dioxygenation of cyclohexadiene 6, (Scheme 1), a compound that is readily available in enantiomerically pure form by a sequence employing an asymmetric conjugate allylation that was developed in our laboratory (vide infra).[10] A preliminary study of the stereoselective dioxygenation of 6 employed singlet oxygen cycloaddition as a method to establish the reuired functional group pattern.[11] While this transformation occurred in acceptable yield, the complete lack of stereocontrol in this reaction (1:1 diastereomer ratio) suggested that other methods for this dioxygenation would be required.[12]
Scheme 1.
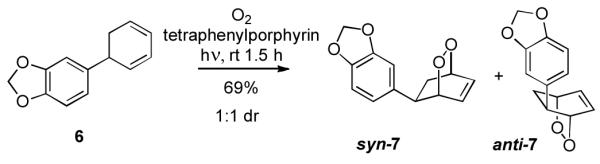
Singlet oxygen cycloaddition of chiral cyclohexadiene 6.
Recent studies in our laboratory have focused on the development of a platinum-catalyzed enantioselective 1,4-diboration of 1,3-dienes as a tool for 1,4-dioxygenation of these substrates.[13] In a related vein, we considered that a diastereoselective version of this reaction might be useful for the elaboration of complex diene substrates and enable the synthesis of the highly hydroxylated core of the isocarbostyrils such as 1–5. To initiate studies, the diboration of 5-phenyl-1,3-cyclohexadiene was examined with bis(pinacolato)diboron [B2(pin)2] and catalytic amounts of Pt(dba)3 and PCy3 (Table 1, entry 1). The reaction was allowed to proceed for 14 hours at 60 °C and was subjected to oxidative work-up with NaOH and H2O2. As depicted in Table 1, the 1,4-dihydroxylation in this manner occurred with excellent yield and >20:1 diastereoselectivity. Examination of other substrates revealed that high levels of stereocontrol and good reaction yields are a general feature of these reactions. Similar to examples with aryl substituted substrates, alkyl substituents are also sufficiently biased to provide for high levels of stereocontrol; X-ray analysis of both aryl- and alkyl-substituted reaction products indicated that the diboration occurs with anti stereoselection as might be anticipated from the steric influence of the aryl group on the substrate.[14] Lastly, diboration of α-phellandrene (entry 5) was examined. The [4+2] singlet-oxygen cycloaddition with this substrate has been the subject of numerous studies and is notoriously non-selective.[15] However, diene diboration occurs with excellent regio- and diastereoselectively, cleanly delivering the 1,4-dihydroxylation product in excellent yield. The outcome with this substrate suggests that substituents on the diene do not necessarily interfere with the reaction and can provide products with enhanced substitution.
Table 1.
Dioxygenation of 1,3-dienes via diboration/oxidation.[a]
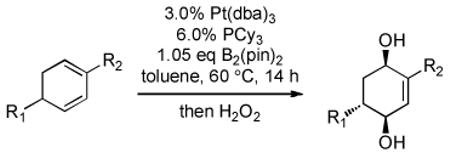
| entry | substrate | product | yield [%][b] | dr[c] |
|---|---|---|---|---|
| 1 |  |
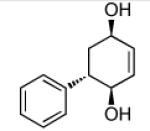 |
86 | >20:1 |
| 2 | 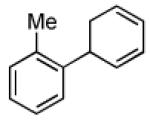 |
 |
71 | >20:1 |
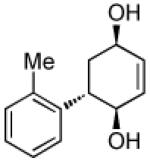
| 3 | 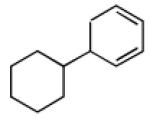 |
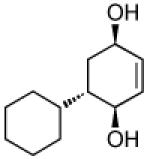 |
93 | >20:1 |
| 4 | 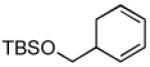 |
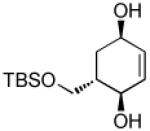 |
69 | >20:1 |
| 5[d] | 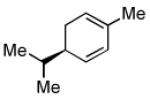 |
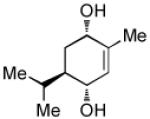 |
87 | >20:1 |
Reactions were conducted with exclusion of moisture and under a nitrogen atmosphere.
The yield represents the isolated yield of purified material and is an average of at least two experiments.
Diastereoselectivity was determined by 400 MHz 1H NMR analysis; (for >20:1, the minor diastereomer was not detected).
To achieve optimal yield with this substrate, the Pt(dba)3 and PCy3 were pre-mixed at rt for 1 h.
With an effective strategy for the diastereoselective dihydroxylation of cyclohexadiene substrates in place, efforts toward the construction of trans-dihydrolycoricidine were initiated. As depicted in Scheme 2, enantioselective conjugate allylation of dialkylidene ketone 7 (derived from the corresponding cinnamic acid in two steps) furnished allylated product 8 in 74% yield and 92% ee, as previously described for the enantiomer of 8.[10a] Ring-closing metathesis employing the second-generation Hoveyda-Grubbs catalyst[16] provided enone 9 (81% yield) which was subjected to Luche reduction to afford allylic alcohol 10 in 98% yield. Treatment of 10 with 2,4-dinitrobenzenesulfenyl chloride effected allylic transposition and subsequent sulfoxide elimination to provide enantioenriched diene 6.[17] As anticipated, the diboration/oxidation of 6 occurred with excellent levels of diastereoselection to provide diol 11 in 83% yield. Notably, in comparison to the simple phenyl-derived substrate (entry 1, Table 1), this example shows that diene 6 is processed efficiently and without interference from Lewis basic functionality in the substrate.
Scheme 2.

Synthesis of 13.
From 11, substitution at position C-4a with retention of configuration was required to transform the existing hydroxyl group into an amine. We anticipated that the steric hindrance provided by the aryl group should allow for selective protection of the C-2 hydroxyl group, allowing subsequent activation of the C-4a hydroxyl for allylic substitution. After surveying reaction conditions, it was found that this was best accomplished by treatment of 11 with TIPSOTf (1.1 equiv) at rt, providing 12 in 68% yield (Scheme 2). The remaining allylic alcohol at position C-4a was then derivatized as the ethyl carbonate, giving 13.
Palladium-catalyzed allylic substitution of 13 was enlisted as a strategy for amination of C4a. The identity of the nucleophile was critical with basic nucleophiles resulting in elimination of the intermediate π-allyl. The use of azide anion, however, was found to provide an optimal solution and allowed conversion of the methyl carbonate to the allylic azide 14. Azide 14 was converted to the corresponding carbamate 15 in 73% yield by a Staudinger reaction followed by the addition of ethyl chloroformate (Scheme 5). In an effort to convert 15 to the derived cyclic lactam 15, the substrate was subjected to Banwell-modified Bischler-Napieralski reaction conditions,[18] however, only decomposition products were observed. This problem was remedied by a strategy similar to that used by Kadas[7]; first 15 was subjected to an OsO4-catalyzed dihydroxylation to afford a single diastereomer of the diol, which was acylated with Ac2O. This alternative substrate for the ring-closure (16) was treated with Tf2O and DMAP (5:3) to afford the desired lactam 17 in 54% yield. Deprotection of the acetate and TIPS protecting groups with methanolic NaOH and TBAF, respectively, provided (+)-trans-dihydrolycoricidine (5). By NMR analysis, the synthetic material was found to be identical with the natural product. Moreover, HPLC analysis of the enantiomerically enriched material, in comparison to racemic material showed the synthetic material to have retained the 92% ee from the initial conjugate allylation step.
In conclusion, we have completed the synthesis of the natural product (+)-trans-dihydrolycoricidine (5) by a route that relies on enantioselective conjugate allylation and diastereoselective diboration to establish the absolute and relative stereochemistry. This represents the first reported application of both of these methodologies in natural product synthesis. Modification of the dialkylidine ketone substrate in the conjugate allylation should allow for a similar synthesis of trans-dihydronarciclasine (4). Furthermore, the diastereoselective diboration described in this paper promises convenient access to highly versatile 1,4-diols for a variety of other applications and further examination of this reaction in the synthesis of natural products will be the subject of future studies.
Experimental Section
Diastereoselective Diene Diboration
In the dry box, an oven-dried 6-dram scintillation vial equipped with a magnetic stir bar was charged with Pt(dba)3 (11.2 mg, 0.013 mmol, 0.03 equiv), tricyclohexylphosphine (6.9 mg, 0.025 mmol, 0.06 equiv), and toluene (4.1 mL). After stirring in the dry box for 5 min, diene 6 (82.2 mg, 0.41 mmol, 1 equiv) and B2(pin)2 (109.5 mg, 0.43 mmol, 1.05 equiv) were added. The vial was sealed with a polypropylene cap, removed from the dry box, and stirred at 60 °C for 15 h. The reaction mixture was cooled to 0 °C and charged with THF (4.5 mL), 3 M aqueous sodium hydroxide (3.0 mL) and 30% aqueous hydrogen peroxide (1.5 mL). The reaction was stirred under ambient atmosphere while slowly warming to rt over 4 h. The vial was cooled to 0 °C and saturated aqueous sodium thiosulfate (10 mL). The reaction mixture was diluted with EtOAc and the layers were separated. The aqueous layer was extracted with EtOAc (4 × 20 ml). The combined organic layers were dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude material was purified by silica gel chromatography (50-70% EtOAc in hexanes) to afford (1S,4S,5R)-5-(benzo[d][1,3]dioxol-5-yl)cyclohex-2-ene-1,4-diol as a white solid (80.0 mg, 83%).
Scheme 3.
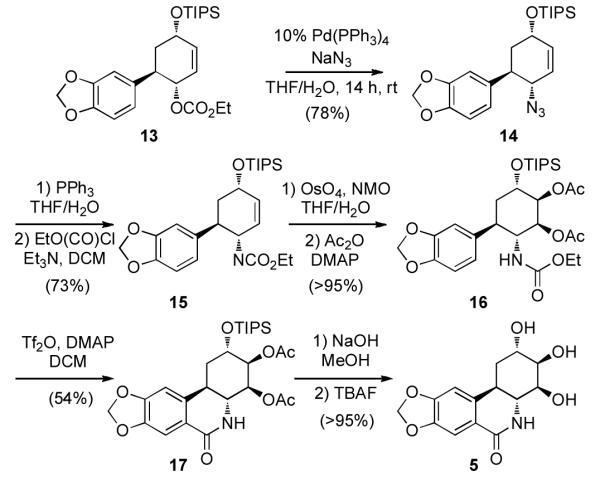
Completion of the synthesis of (+)-trans-dihydrolycoricidine.
Acknowledgments
We are grateful to the NIH (NIGMS GM-59417 and GM-64451) for support of this work.
References
- [1]a).For representative reviews of Amaryllidaceae alkaloids, see: Kornienko A, Evidente A. Chem. Rev. 2008;108:1982–2014. doi: 10.1021/cr078198u.; Ingrassia L, Lefranc F, Mathieu V, Darro F, Kiss R. Transl. Oncol. 2008;1:1–13. doi: 10.1593/tlo.08100.; Rinner U, Hudlicky T. Synlett. 2005;3:365–387.; Polt R. In: Organic Synthesis: Theory and Applications. Hudlicky T, editor. Vol 3. JAI Press; Greenwich, CT: 1997. p. 109.
- [2]a).Pettit GR, Eastham SA, Melody N, Orr B, Herald DL, McGregor J, Knight JC, Doubek DL, Pettit GR, III, Garner LC, Bell JA. J. Nat. Prod. 2006;69:7–13. doi: 10.1021/np058068l. [DOI] [PubMed] [Google Scholar]; b) Gabrielsen B, Monath TP, Huggins JW, Kefauver DF, Pettit GR, Groszek G, Hollingshead M, Kirsi JJ, Shannon WM, Schubert EM, Dare J, Ugarkar B, Ussery MA, Phelan MJ. J. Nat. Prod. 1992;55:1569–1581. doi: 10.1021/np50089a003. [DOI] [PubMed] [Google Scholar]
- [3]a).Ceriotti G. Nature. 1967;213:595–596. doi: 10.1038/213595a0. [DOI] [PubMed] [Google Scholar]; b) Okamoto T, Torii Y, Isogai Y. Chem. Pharm. Bull. 1968;24:1119–1131. doi: 10.1248/cpb.16.1860. [DOI] [PubMed] [Google Scholar]; c) Evidente A. Planta Med. 1991;57:293–295. doi: 10.1055/s-2006-960098. [DOI] [PubMed] [Google Scholar]; d) Pettit G, Melody N, Herald DL. J. Org. Chem. 2001;66:2583–2587. doi: 10.1021/jo000710n. [DOI] [PubMed] [Google Scholar]
- [4]a).For recent reviews on the synthesis of Amaryllidaceae alkaloids, see Manpadi M, Kornienko A. Org. Prep. Proc. Intl. 2008;40:107–161. doi: 10.1080/00304940809458083.; Chapleur Y, Chretien F, Ibn Ahmed S, Khaldi M. Curr. Org. Synth. 2006;3:341–378.; See also, reference 1c.
- [5].Pettit GR, Cragg GM, Singh SB, Duke JA, Doubek DI. J. Nat. Prod. 1990;53:176–178. doi: 10.1021/np50067a026. [DOI] [PubMed] [Google Scholar]
- [6](a).Pettit GR, Pettit GR, III, Backhaus RA, Boyd MR, Meerow AW. J. Nat. Prod. 1993;56:1682–1687. doi: 10.1021/np50100a004. [DOI] [PubMed] [Google Scholar]; (b) Pettit GR, Melody N. J. Nat. Prod. 2005;68:207–211. doi: 10.1021/np0304518. [DOI] [PubMed] [Google Scholar]
- [7]a).For the synthesis of racemic 5, see: Isobe K, Taga J-I. Hetereocycles. 1978;9:625–630.; Szántó G, Hegedüs L, Mattyasovszky L, Simon A, Simon A, Kádas I. Tetrahedron Lett. 2009;50:2857–2859.
- [8]a).Chida N, Jitsuoka M, Yamamoto Y, Ohtsuka M, Ogawa S. Heterocycles. 1996;43:1385–1390. [Google Scholar]; b) Fujimura T, Shibuya M, Ogasawara K, Iwabuchi Y. Heterocycles. 2005;66:167–173. [Google Scholar]; c) Collins J, Rinner U, Moser M, Hudlicky T, Ghiviriga I, Romero AE, Kornienko A, Ma D, Griffin C, Pandey S. J. Org. Chem. 2010;75:3069–3084. doi: 10.1021/jo1003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Szántó G, Hegedüs L, Mattyasovszky L, Simon A, Simon A, Bitter I, Tóth G, Töke L, Kádas I. Tetratrahedron. 2009;65:8412–8417. [Google Scholar]
- [10]a).Sieber JD, Morken JP. J. Am. Chem. Soc. 2008;130:4978–4983. doi: 10.1021/ja710922h. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sieber JD, Liu S, Morken JP. J. Am. Chem. Soc. 2007;129:2214–2215. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]
- [11].Clenan EL. For an excellent review on singlet oxygen cycloaddition. Tetrahedron. 1991;47:1343–1382. [Google Scholar]
- [12].For a similarly non-selective singlet oxygen [4+2], see: Robertson J, Stafford PM, Bell SJ. J. Org. Chem. 2005;70:7133–7148. doi: 10.1021/jo050644v.
- [13].Burks HE, Kliman LT, Morken JP. J. Am. Chem. Soc. 2009;131:9134–9135. doi: 10.1021/ja809610h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Coordinates have been deposited with the Cambridge Crystallographic Data Center. CDCC 799995; 799996.
- [15]a).For original studies on the product selectivity in this reaction, see: Matusch R, Schmidt G. Angew. Chem. 1988;100:729–730.; Angew. Chem. Int. Ed. Engl. 1988;27:717–718. Atkins R, Carless HAJ. Tetrahedron. 1987;28:6093–6096.; Schenck GO, Kinkel KG, Mertens H-J. Liebigs Ann. Chem. 1953;584:125.
- [16].Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J. Am. Chem. Soc. 2000;122:8168–8179. [Google Scholar]
- [17].Reich HJ, Wollowitz S. J. Am. Chem. Soc. 1982;104:7051–7059. [Google Scholar]
- [18]a).Banwell MG, Bissett BD, Busato S, Cowden CJ, Hockless DCR, Holman JW, Read RW, Wu AW. J. Chem. Soc., Chem. Commun. 1995:2551–2553. [Google Scholar]; b) Banwell MG, Cowden CJ, Gable RW. J. Chem. Soc., Perkin Trans. 1. 1994:3515–3518. [Google Scholar]