

Abstract
In many microorganisms, the first step of arginine biosynthesis is catalyzed by the classical N-acetylglutamate synthase (NAGS), an enzyme composed of N-terminal amino acid kinase (AAK) and C-terminal histone acetyltransferase (GNAT) domains that bind the feedback inhibitor arginine and the substrates, respectively. In NAGS, three AAK domain dimers are interlinked by their N-terminal helices, conforming a hexameric ring, whereas each GNAT domain sits on the AAK domain of an adjacent dimer. The arginine inhibition of Pseudomonas aeruginosa NAGS was strongly hampered, abolished, or even reverted to modest activation by changes in the length/sequence of the short linker connecting both domains, supporting a crucial role of this linker in arginine regulation. Linker cleavage or recombinant domain production allowed the isolation of each NAGS domain. The AAK domain was hexameric and inactive, whereas the GNAT domain was monomeric/dimeric and catalytically active although with ∼50-fold-increased and ∼3-fold-decreased Kmglutamate and kcat values, respectively, with arginine not influencing its activity. The deletion of N-terminal residues 1 to 12 dissociated NAGS into active dimers, catalyzing the reaction with substrate kinetics and arginine insensitivity identical to those for the GNAT domain. Therefore, the interaction between the AAK and GNAT domains from different dimers modulates GNAT domain activity, whereas the hexameric architecture appears to be essential for arginine inhibition. We proved the closeness of the AAK domains of NAGS and N-acetylglutamate kinase (NAGK), the enzyme that catalyzes the next arginine biosynthesis step, shedding light on the origin of classical NAGS, by showing that a double mutation (M26K L240K) in the isolated NAGS AAK domain elicited NAGK activity.
INTRODUCTION
In most microorganisms N-acetyl-l-glutamate synthase (NAGS), the enzyme encoded by argA, catalyzes the first step of arginine biosynthesis (Fig. 1A), producing N-acetyl-l-glutamate (NAG) from glutamate and acetyl coenzyme A (acetyl-CoA) and being feedback inhibited by arginine (Fig. 1A) (8, 10). Plants also use NAGS for making arginine (35). Although NAG is not a precursor of arginine in animals (2), NAGS is present in animals that make urea (8), since NAG is an essential activator of the urea cycle enzyme carbamoyl phosphate synthetase I (31) (Fig. 1B). Consequently, human NAGS deficiency is an inborn error of the urea cycle that causes clinical hyperammonemia (7).
Fig 1.
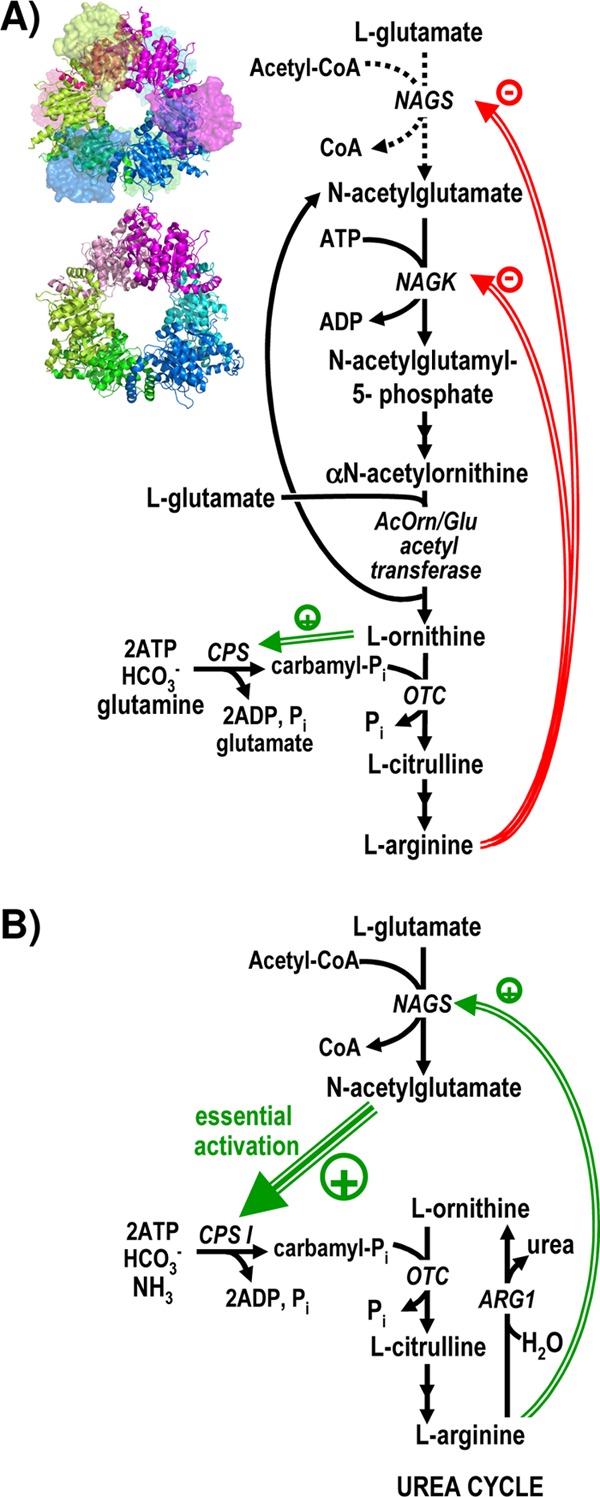
Roles of N-acetyl-l-glutamate synthase (NAGS) and of its homologous enzyme N-acetyl-l-glutamate kinase (NAGK) in arginine biosynthesis. Two arrows in succession indicate the existence of two steps that are not detailed. Double and triple green arrows and green-encircled plus signs denote activation, whereas double red arrows and red-encircled minus signs denote inhibition. OTC, ornithine transcarbamylase. ARG1, arginase 1. (A) Schematic representation of the arginine biosynthetic pathway of P. aeruginosa and of many other bacteria and plants. The dotted arrows for the NAGS reaction indicate an anaplerotic role of NAGS in those organisms, like P. aeruginosa, in which the N-acetyl group is recycled by transacetylation from acetylornithine to glutamate (10). However, some organisms, like E. coli, deacetylate acetylornithine hydrolytically, and in these cases, NAGS makes one NAG molecule per arginine molecule synthesized (10). The structures of the NAGS from N. gonorrhoeae (34) (PDB accession number 2R8V) and of the NAGK from P. aeruginosa (28) (PDB accession number 2BUF) are shown next to the steps catalyzed by them to grossly illustrate their structural similarity. They are viewed along their 3-fold axes, with each dimer colored differently and with both subunits of each dimer in different hues. NAGK and the AAK domain of NAGS are shown in a cartoon representation. In NAGS, to avoid occluding the view of the AAK domains, the GNAT domains are shown in a surface semitransparent representation, and those in the background are fainter. (B) Arginine and urea biosynthesis in urea-making terrestrial animals such as humans. Animals do not make ornithine through N-acetylated intermediates, and they lack NAGK and other enzymes of the route except NAGS (2). The large green triple arrow stresses the essentiality of the activation of carbamoyl phosphate synthetase (CPS I) by NAG.
Although the key role and widespread distribution of NAGS in all domains of life warrant studies of this enzyme, most detailed data concern the bacterial forms of this enzyme (17, 23, 27, 32, 33), including the crystal structure of NAGS from Neisseria gonorrhoeae (NgNAGS) in the substrate-bound and arginine-bound forms (24, 34). NgNAGS can be considered a typical example of classical bacterial NAGSs, as defined by the early-studied Escherichia coli and Pseudomonas aeruginosa enzymes (17, 23, 32, 33). These classical bacterial forms, encoded by argA (Fig. 2A), consist of a single polypeptide with a mass of ∼50 kDa that is composed of an N-terminal ∼260-residue domain and a C-terminal ∼150-residue domain, belonging to the amino acid kinase (AAK) and the histone acetyltransferase (GNAT) families, respectively (32). The NgNAGS structure proved that the GNAT domain binds both substrates (34), whereas the AAK domain was shown to host the site for the feedback inhibitor arginine (24, 34). As predicted from the homology of the classical bacterial NAGS with arginine-sensitive NAG kinase (NAGK) (28), the NgNAGS structure is nucleated by a ringlike hexameric trimer of dimers of AAK domains that closely resembles the NAGK hexamer (34) (Fig. 1A, and see Fig. S1 in the supplemental material). NAGK catalyzes the next step in the route of arginine synthesis in many microbes and in plants (10, 35) (Fig. 1A) and is considered a paradigm for the AAK domain family (28, 29). In both NAGS and arginine-sensitive NAGK, a kinked α helix emerging at the N-terminal end of each AAK domain is interlaced with the corresponding N-terminal helix of an adjacent dimer, linking the three AAK domain dimers to form the hexamer (28, 34) (see Fig. S1 in the supplemental material).
Fig 2.
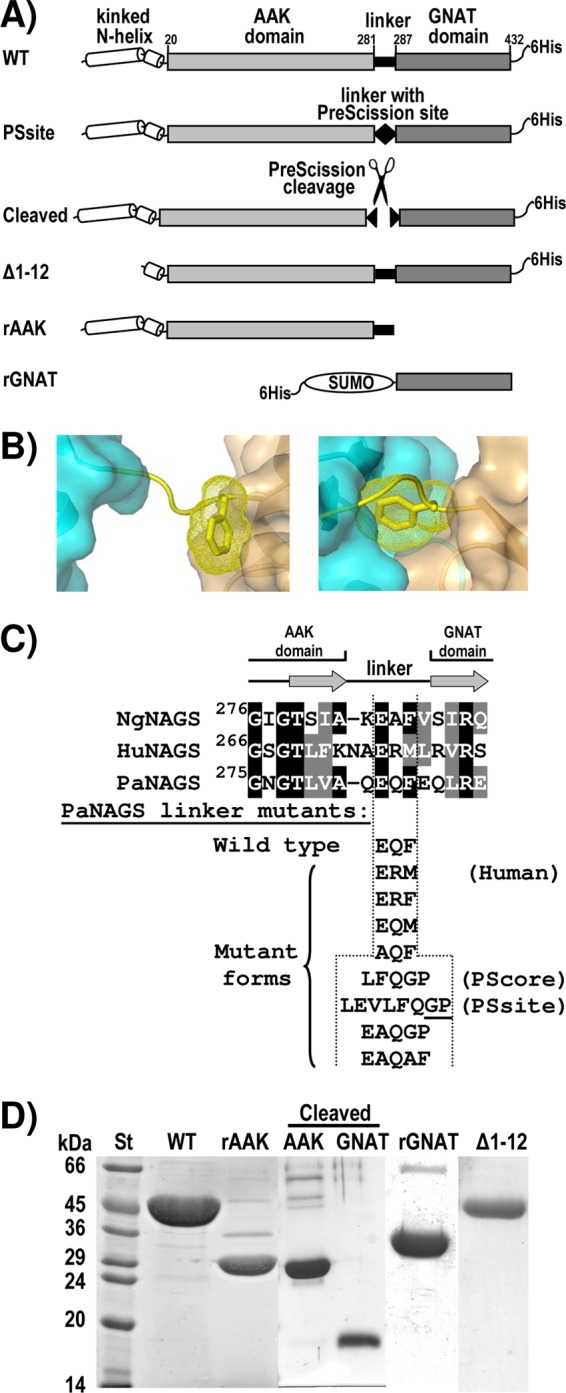
Engineered and mutant forms of PaNAGS. (A) Schematic representation of the PaNAGS polypeptide, illustrating the constructions prepared in the present work, giving the abbreviated denominations used here for each construction. The figures above the wild-type form correspond to the first and last residues of each domain. (B) Detail of the interdomain linker (in string representation and yellow) of a subunit of NgNAGS in the arginine-free form (left) (PDB accession number 2R8V) (34) and the arginine-bound form (right) (PDB accession number 3D2P) (24), showing the AAK and GNAT domains that are connected by this linker in a semitransparent surface representation and in blue and brown-orange, respectively. The side chain of the linker residue F286 (corresponding to F285 of PaNAGS) is shown in a stick representation, with its Van der Waals surface shown as yellow dots. (C) Linker mutants, shown below the alignment of the region encompassing the last and first β strands (shown as arrows) of the AAK and GNAT domains, respectively, of NgNAGS and the corresponding sequences of human NAGS (HuNAGS) and PaNAGS. Amino acid identities and conservative replacements are highlighted in black and gray backgrounds, respectively, with lettering in white. All the mutant sequences replaced the EQF sequence of the wild-type form. In the PSsite mutant, the cleavage by PreScission protease should take place immediately before the two underlined residues. (D) Coomassie-stained SDS-PAGE gel of the purified wild-type enzyme, of the rAAK and rGNAT domains, and of the Δ1–12 engineered form (see panel A for the composition of these forms) as well as of the gel-filtration-separated AAK and GNAT domains prepared by the PreScission cleavage of the PSsite form. St, molecular mass standard markers, with masses (in kilodaltons) given on the side.
In bacterial NAGS and arginine-sensitive NAGK, the feedback inhibitor arginine sits in the AAK domain (24, 28). The arginine sites flank the junctions between the AAK dimers, next to the kinked N-terminal helices, with participation in each arginine site of the short N-terminal helix segment following the kink. By binding at its site, arginine widens the hexameric ring of AAK domains (24, 28). In NAGK, this binding causes inhibition because it favors an open conformation of the AAK domain, where catalysis takes place, distorting and widening the active center (28). However, in NAGS, both substrates bind in the GNAT domain (34), and this inhibitory mechanism cannot apply. The globular GNAT domain of NgNAGS, connected to its cognate AAK domain by a 5-residue linker (Fig. 2B and C), sits on the AAK domain of an adjacent dimer (34) (Fig. 1A, and see Fig. S1A in the supplemental material). Judging from site-directed mutagenesis studies (32), the AAK domain may play a regulatory role in NAGS activity. In agreement with this view, the NgNAGS crystal structures revealed that arginine dramatically changes the spatial relations between the AAK and GNAT domains (24). Thus, in the arginine-bound form of NgNAGS, the GNAT domain interacts with the AAK domain of its own subunit and experiences a 109° rotation around its linker (Fig. 2B), drastically altering its interactions with the AAK domain on which it lies (24). Our previous studies (33) of the effects of linker shortening or lengthening by up to 2 residues on P. aeruginosa NAGS (PaNAGS) kinetic parameters support the importance of this rotation around the linker. NAGS inhibition by arginine involves a decrease in the Vmax as well as an increase in the apparent Km for glutamate (33). The latter effect fits the observation that in the arginine-bound form of NgNAGS, two loops of the glutamate site become disordered (24). It also fits the finding that two-residue linker shortening, expected to drag away the GNAT domain from its normal position, mimics arginine in increasing the Km for glutamate (33).
In the present work, we subject some key functional inferences that were based on the NgNAGS structure as well as our previous proposal (33) that the interdomain linker plays a paramount role in NAGS functionality to experimental corroboration by using PaNAGS. We engineered this linker, cleaved it, isolated and studied the properties of the individual domains, and also produced recombinantly the two domains in the isolated form (Fig. 2C), showing that the GNAT domain alone can catalyze the reaction but exhibits a low affinity for glutamate and that this isolated domain is insensitive to arginine. We also prove that the AAK domain is closely related to NAGK by restoring some NAGK activity by mutating two residues of this domain. The importance of the hexameric organization is demonstrated by rendering the enzyme dimeric by the deletion of the N-terminal helix up to the expected site for its kink (Fig. 2C). Our findings dissect functionally classical bacterial NAGS and help generate an integrated picture that may also contribute to an understanding of mammalian NAGS, which was reported to have the same domain organization as that of the bacterial enzyme (27, 32). In fact, by engineering the linker of PaNAGS, we managed to convert arginine from a potent inhibitor to a modest activator, rendering the effect of arginine on enzyme activity reminiscent of the more potent activation triggered by arginine on the NAGSs of terrestrial animals, including humans (3, 6, 36).
MATERIALS AND METHODS
Preparation of cloned DNA sequences encoding wild-type and engineered PaNAGS forms.
We previously reported the cloning in pET22b (from Novagene) of argA (PA5204 gene) (http://cmr.jcvi.org/tigr-scripts/CMR/CmrHomePage.cgi) from P. aeruginosa encoding wild-type (WT) NAGS with a GSLEH6 tail (Fig. 2A, top) (32). This plasmid is called here pETNAGS. Single and double point mutations (Fig. 2C) at adjacent positions in the interdomain linker were introduced by site-directed mutagenesis into pETNAGS, using the QuikChange kit (from Stratagene) and utilizing appropriate mutagenic forward and reverse oligonucleotides (see Table S1 in the supplemental material). The same approach was used to produce the isolated recombinant AAK (rAAK) domain (Fig. 2A) by replacing codon 287 of argA with a stop codon. Two further rounds of site-directed mutagenesis on the latter plasmid introduced the M26K L240K double mutation into rAAK. The preparation of PaNAGS having 283EAQAF replacing 283EQF was reported previously (33). Plasmid pET22b carrying this mutated argA form was used for the preparation of other engineered enzyme forms having a linker with an increased length (Fig. 2C). Thus, a single round of site-directed mutagenesis on this plasmid allowed the change of the mutant sequence from 283EAQAF to 283EAQGP. The resulting mutant plasmid was mutated further to the sequence 283LFQGP (PScore form), and this last plasmid was used for the insertion of three extra residues “en bloc” in a single mutagenic round to give the linker sequence 283LEVLFQGP (PSsite mutant), which was engineered to host the entire PreScission protease (an engineered derivative of human rhinovirus 3C protease [9], provided by GE Healthcare) cleavage site. All these changes were carried out with the QuikChange system as described above, using the primers indicated in Table S1 in the supplemental material.
The DNA sequence encoding the GNAT domain was cloned into pET26b (from Novagene) after the PCR amplification of codons 287 to 432 of argA from pETNAGS using a forward primer (see Table S1 in the supplemental material) that introduced an initial ATG codon as part of an NdeI site and a reverse primer that introduced an XhoI site (the same primer [32] used for the cloning of argA into pETNAGS, called “GNAT reverse” in Table S1 in the supplemental material), allowing directional insertion by ligation into the corresponding sites of pET26b digested with these enzymes. BL21(DE3) cells (from Novagene) transformed with this plasmid failed to express the GNAT domain. This plasmid was used as a template for the PCR amplification of the GNAT domain-encoding sequence in a way permitting its fusion to the C terminus of SUMO (the protein chimera encoded by this construction is called here rGNAT) (Fig. 2A). The primers used for this amplification (see Table S1 in the supplemental material) restored the stop codon at its normal site (thus, the C-terminal His6 tag was eliminated, although the SUMO moiety carries one such tag) and introduced BsaI and BamHI sites before and after the coding sequence, respectively, allowing directional insertion into the corresponding sites of plasmid pSUMO (from LifeSensors Inc.).
The same strategy used for the cloning of the GNAT domain into pET26b was used for the pET22b cloning of argA carrying a deletion of codons 1 to 12 (called here Δ1-12) (Fig. 2A). For this purpose, in the PCR amplification step, we used a forward primer corresponding to nucleotides 20 to 51 of the coding sequence in which 4 nucleotides were changed to include an NdeI site (see Table S1 in the supplemental material). In this way, the engineered gene encodes residues 13 to 432 of PaNAGS preceded by a methionine and followed by the C-terminal GSLEH6 tail.
PCR amplification was carried out with a high-fidelity thermostable DNA polymerase (Deep-Vent; New England BioLabs). The correctness of all the constructs and mutants prepared here was confirmed by DNA sequencing.
Expression and purification of the protein constructs.
We produced wild-type PaNAGS and its mutant forms, including Δ1–12, as reported previously (32) for the wild enzyme. The procedure used included growing transformed BL21(DE3) cells at 37°C to an optical density at 600 nm (OD600) of ∼0.5 in Luria-Bertani medium containing 0.1 mg/ml ampicillin and then keeping the culture standing for 45 min in ice, followed by the addition of 2% (vol/vol) ethanol and 0.02 mM isopropyl-β-δ-thiogalactoside (IPTG) and the continuing of the culture with aeration overnight at 15°C. However, for the production of rGNAT and rAAK, either the wild type or with the M26K L240K double mutation, induction was done for 3 h at 37°C with 1 mM IPTG.
Cells were harvested by centrifugation, and subsequent steps were performed at 4°C. The cells were disrupted by sonication in a solution containing 20 mM Na phosphate (pH 8), 1 mM dithiothreitol (DTT), 0.5 M NaCl, and 20 mM imidazole. Insoluble material was centrifuged away. There was an abundant production of the recombinant proteins in a soluble form, allowing purification to essential homogeneity (Fig. 2D; point and linker mutants are not shown) by Ni affinity chromatography. His-Spin Trap centrifugal columns (GE Healthcare) were used when small amounts of protein were required (32). For larger amounts, the cell pellet from a 0.5-liter culture was suspended in 15 ml of sonication buffer, and the centrifuged sonicate was applied onto a 1-ml His Trap-HP column mounted onto an Äkta fast protein liquid chromatography (FPLC) system (both from GE Healthcare), eluting the His6-tagged protein with a 30-ml linear gradient of 20 mM to 500 mM imidazole-containing buffer. The different proteins were placed into storage buffer (10 mM sodium phosphate [pH 7.0], 15% [vol/vol] glycerol, 1 mM EDTA, 1 mM DTT, 20 mM NaCl, and 10 mM NAG) (23) by either centrifugal desalting through PD SpinTrap G-25 columns (from GE Healthcare) or, for larger volumes, repeated cycles of centrifugal ultrafiltration (Amicon Ultra with a 10,000-molecular-weight [10K] cutoff; Millipore) and dilution using the same buffer.
For the purification of the rAAK domain and its double mutant, which had no His6 tag, 0.5-liter cultures were used, the sonication buffer used contained 15 ml of 20 mM Na phosphate (pH 8)–1 mM DTT, and the postcentrifugal supernatant was subjected to sequential precipitations with ammonium sulfate at 30% and 60% saturations, dissolving the final precipitate in 15 ml of sonication buffer, followed by desalting by repeated centrifugal ultrafiltration as described above by using the same buffer. The protein solution was then applied onto a 1-ml HiTrap Q HP column (GE Healthcare) mounted onto an Äkta FPLC system equilibrated with sonication buffer, followed by washing and by the elution of essentially pure rAAK (Fig. 2D) with a 35-ml linear gradient of 0 to 1 M NaCl in the same buffer. The protein was concentrated to 25 mg/ml by centrifugal ultrafiltration.
Cleavage with PreScission protease and separation of the two domains.
PreScission protease (from GE Healthcare) and the wild-type enzyme or the engineered PScore or PSsite form of PaNAGS (Fig. 2C), at the indicated concentrations, were incubated for 5 h at 15°C in a solution containing 50 mM Tris-HCl (pH 7.1), 0.15 M NaCl, 1 mM EDTA, and 1 mM DTT, monitoring cleavage by SDS-PAGE. At the end of the incubation, either the mixture was centrifuged through a His Spin Trap column or, for the separation of the two domains, 0.3 ml of the digestion mixture or of an equivalent nondigested mixture was applied onto a Superdex 200HR (10/300) column (GE Healthcare) mounted onto an Äkta fast protein liquid chromatography system. The column was equilibrated and run (flow rate, 0.50 ml/min) with a solution containing 50 mM Tris-HCl (pH 8.5)–0.1 M NaCl at 4°C, monitoring the absorbance of the effluent at 280 nm and collecting 0.5- to 1-ml fractions over ice. The fractions corresponding to the AAK domain peak or to the GNAT domain peak from two identical chromatographic experiments were pooled together and concentrated by centrifugal ultrafiltration to ∼1 mg protein/ml.
Enzyme activity assays.
NAGS activity was determined colorimetrically with Ellman's reagent as CoA release (12) at 37°C, as previously reported (32), using a solution of 0.2 M Tris-HCl (pH 9) containing, in the standard assay mixture, 30 mM l-glutamate (sodium salt), 4 mM acetyl-CoA, and, when used, the indicated concentrations of arginine hydrochloride. For estimations of the kinetic parameters for the substrates, the acetyl-CoA concentration was fixed at 4 mM while the glutamate concentration was varied, or the glutamate concentration was fixed at 30 mM (except where indicated) and the acetyl-CoA concentration was varied. For assays, the enzyme was appropriately diluted in storage buffer lacking both NAG and NaCl and supplemented with 30 mg/ml bovine serum albumin (albumin concentration in the assays, 1.5 to 3 mg/ml). Reactions were carried out at least in duplicate, and blanks in which the enzyme was replaced with dilution solution were run in parallel and were subtracted. One enzyme unit produces 1 μmol CoA min−1. Results (means ± standard errors [SE]) were fitted with GraphPadPrism (GraphPad Software, San Diego, CA) to either hyperbolic kinetics or substrate inhibition kinetics, as reported previously (33).
NAGK activity was determined at 37°C and at pH 7.5 as previously described (13), using a hydroxylamine-containing colorimetric assay mixture described previously by Haas and Leisinger (18). One enzyme unit is the amount of enzyme that generates 1 μmol of product in 1 min.
Other techniques.
SDS-PAGE (20) was carried out with 15% polyacrylamide gels. The protein concentration was determined by a Bradford assay (4), using bovine serum albumin as the standard. The structures of E. coli NAGK (Protein Databank [PDB] accession number 1GS5) (29) and of the AAK domain of NgNAGS (PDB accession number 2R8V) (34) were superposed with the program Coot (Crystallographic Object-Oriented Toolkit) (11), using default parameters. Figures representing protein structures were generated by using PyMOL (http://www.pymol.org/).
RESULTS AND DISCUSSION
Influence of the interdomain linker on the arginine modulation of NAGS activity.
To ascribe functions to the GNAT domain, we engineered the interdomain linker (Fig. 2A and C, and see Materials and Methods) to introduce a cleavage site for the highly specific PreScission protease by replacing the EQF sequence within the linker connecting both domains with LEVLFQGP (Fig. 2A and C). The enzyme with this replacement, called here the PSsite form, was expressed and purified similarly to the recombinant wild-type enzyme (not shown). This form was catalytically active, with kinetic parameters not too different from those of wild-type PaNAGS (Table 1), but it failed to be inhibited by arginine, whereas the wild type enzyme was nearly totally inhibited by 2 mM arginine (Fig. 3A and Table 1). This observation confirms our previous conclusion (33) that the interdomain linker is highly important for the arginine modulation of NAGS activity.
Table 1.
Influence of linker changes on PaNAGS kinetics
| Linker changea | Mean apparent kinetic parameter value ± SE forb: |
(%) | ||||
|---|---|---|---|---|---|---|
| Acetyl-CoA |
l-Glutamate |
|||||
| V[acetyl-CoA]=∞ (U mg−1) | Kmacetyl-CoA (μM) | V[glutamate]=∞ (U mg−1) | Kmglutamate (mM) | KIglutamate (mM) | ||
| None (EQF) (WT) | 80 ± 1 | 92 ± 9 | 136 ± 20 | 5.2 ± 1.4 | 72 ± 36 | 10.2 ± 3.6c |
| LEVLFQGP (PSsite) | 55 ± 1 | 85 ± 5 | 83 ± 8 | 10.8 ± 1.6 | 95 ± 36 | 105.1 ± 0.6 |
| LFQGP (PScore) | 49 ± 2 | 63 ± 14 | 85 ± 5 | 2.7 ± 0.4 | 28 ± 4 | 124.5 ± 2.8 |
| EAQGP | 47 ± 1 | 25 ± 2 | 67 ± 5 | 4.9 ± 0.8 | 69 ± 13 | 156.3 ± 4.4 |
| EAQAF | 42 ± 1 | 68 ± 15 | 70 ± 11 | 2.6 ± 1.0 | 67 ± 37 | 58.0 ± 3.8 |
| ERM | 21 ± 1 | 25 ± 5 | 53 ± 4 | 5.5 ± 0.6 | 20 ± 2 | 114.0 ± 1.1 |
| EQM | 84 ± 2 | 31 ± 4 | 125 ± 5 | 4.3 ± 0.4 | 58 ± 7 | 94.0 ± 0.6 |
| ERF | 54 ± 1 | 35 ± 4 | 84 ± 3 | 2.7 ± 0.3 | 30 ± 3 | 5.6 ± 0.3 |
| AQF | 63 ± 1 | 35 ± 4 | 82 ± 5 | 7.7 ± 1.0 | 150 ± 30 | 9.4 ± 0.5 |
Changes as defined in the legend of Fig. 2C.
See Materials and Methods for details.
[Arg] equals 10 mM.
Fig 3.
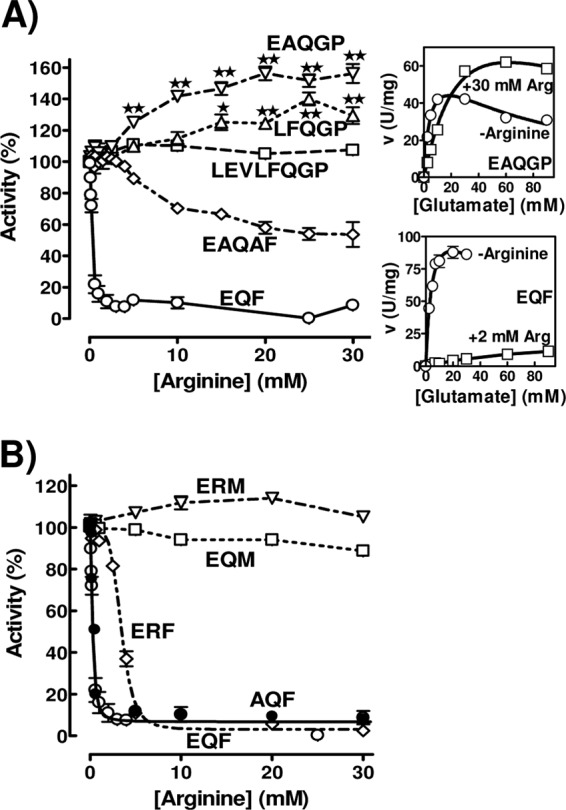
Influence of linker mutations on the effect of arginine on PaNAGS activity (expressed as a percentage of that for each form in the absence of arginine). See Fig. 2C for the key to each mutant form. Mutant forms having (A) or not having (B) a lengthened linker are compared with the wild-type form (EQF). The activation of the EAQGP and LFQGP forms by arginine is statistically significant for the points marked with double stars (P < 0.001) or single stars (P < 0.01) (tested by analysis of variance [ANOVA], followed by the Bonferroni test for individual points; n = 4 to 8). The insets to the right of panel A show the glutamate concentration dependency of the velocity for the form with the EAQGP linker sequence (top inset) and for the wild-type enzyme (bottom inset). The curves drawn in these insets are those for hyperbolic kinetics with substrate inhibition for KmGlu, KIGlu, and Vmax, as follows: 4.9 mM, 69 mM, and 67 U/mg, respectively, for EAQGP without arginine; 37 mM, 103 mM, and 136 U/mg, respectively, for EAQGP with 30 mM arginine; 5.2 mM, 72 mM, and 136 U/mg, respectively, for EQF (wild type) without arginine; and 85 mM, ∞ (no substrate inhibition), and 22 U/mg, respectively, for EQF with 2 mM arginine.
Surprisingly, when we engineered the linker sequence to replace the wild-type EQF linker sequence with LFQGP (called here the PScore form) and, particularly, with EAQGP (Fig. 2C), a form prepared as an intermediate in the stepwise engineering of the linker from the wild-type to the PSsite form, the enzyme was modestly but significantly activated by arginine rather than being inhibited (Fig. 3A and Table 1). This effect, which is somewhat reminiscent of the activation of the NAGSs from terrestrial animals by arginine (6, 19), suggests that the linker sequence might determine whether arginine is an inhibitor or an activator. The importance not only of the linker length (33) but also of the linker sequence in determining the effect of arginine is illustrated by the observation that the replacement of EAQGP with EAQAF in the modified linker (Fig. 2C) resulted in enzyme inhibition by arginine although with less potency and at higher arginine concentrations than in the case of the wild-type sequence, EQF (Fig. 3A and Table 1). Since all these linker changes reduced the Vmax somewhat (Table 1), the linker also influences the efficiency of the enzyme as a catalyst in the absence of arginine.
We attempted to replicate, without success, the arginine activation of the human enzyme by replacing the Q and F of the PaNAGS wild-type linker sequence (EQF) with R and M, respectively, the two residues found at these positions in human NAGS (Fig. 2C). This change prevented enzyme inhibition by arginine, with perhaps a slight trend toward activation (Fig. 3B and Table 1). When enzyme forms with only one or the other of these mutations were studied, it was found that the EQF-to-EQM change (the changed residue is underlined) virtually abolished arginine inhibition although without any evidence of arginine-triggered activation, whereas the EQF-to-ERF change failed to abolish inhibition by arginine, although the arginine concentration needed for inhibition was increased (Fig. 3B). In contrast, the replacement of the E residue, which is common to human and bacterial NAGSs, with A (change of EQF to AQF) (Fig. 2C) did not alter the sensitivity of the enzyme to arginine (Fig. 3B and Table 1). These results clearly show that the phenylalanine found in the bacterial linker is essential for arginine inhibition. The crystal structures of NgNAGS (24, 34) with and without arginine (Fig. 2B) support the importance of this phenylalanine, which would act as an end lever, stabilizing the orientations of the GNAT domain relative to the AAK domain across the linker in the arginine-free and arginine-bound enzyme conformations. Thus, in the arginine-free form, the benzene ring of phenylalanine makes extensive and close contacts with the GNAT domain (Fig. 2B, left), whereas in the arginine-bound form, it makes extensive contacts with the AAK domain (Fig. 2B, right). Our observation that the F285M mutation, although abolishing arginine inhibition, had virtually no effect on the kinetic parameters of the enzyme in the absence of arginine (Table 1) strongly supports the view that the methionine cannot fulfill this lever function, failing to stabilize the arginine-bound conformation. In any case, our results show that the mere replacement of this phenylalanine or of the Q preceding it with its human counterparts is not enough for rendering arginine an activator.
We examined the reasons for the activation of the enzyme with the EAQGP linker sequence by arginine. In the presence of 30 mM arginine, the glutamate dependency of the activity (Fig. 3A, top inset) revealed that only when glutamate concentrations exceeded 20 mM was the activity higher with arginine than without arginine. Below this glutamate concentration, arginine was an inhibitor, revealing that arginine can be an inhibitor or an activator depending on the glutamate concentration. The effective glutamate concentration at which the enzyme reaches its experimental activity maximum is higher with arginine than without arginine, and substrate inhibition also appears to occur at increased glutamate concentrations when arginine is present (Fig. 3A, top inset). These changes are qualitatively similar to those triggered by arginine on the kinetics of wild-type PaNAGS (Fig. 3A, bottom inset) (33), but their magnitude was smaller with the EAQGP form than with the wild-type enzyme. However, in the case of the modified enzyme, arginine did not appear to trigger a reduction in the apparent Vmax for glutamate, whereas kinetic results for the wild-type enzyme were consistent with a nearly 7-fold reduction in the Vmax triggered by as little as 2 mM arginine (Fig. 3A, insets) (33). This different effect on the Vmax appears to be the main reason for the observed activation of the enzyme with the modified linker by arginine.
Influence of linker cleavage on activity and arginine inhibition.
PreScission protease cleaved the PSsite enzyme form at its interdomain linker, but it failed to cleave wild-type PaNAGS (Fig. 4A) or the PScore form (not shown), as shown by SDS-PAGE, which revealed bands with the expected masses for the AAK (31 kDa) and GNAT (18 kDa) domains in the digestion of the PSsite form. Linker cleavage did not lead to the immediate dissociation of the two domains, since only a small fraction (∼25%) (Fig. 4B) of the cleaved AAK domain was not retained by centrifugation through an Ni-affinity column, whereas the majority was retained (Fig. 4B), being eluted together with the His6-tag-containing GNAT domain by the application to the centrifugal column of 0.5 M imidazole.
Fig 4.
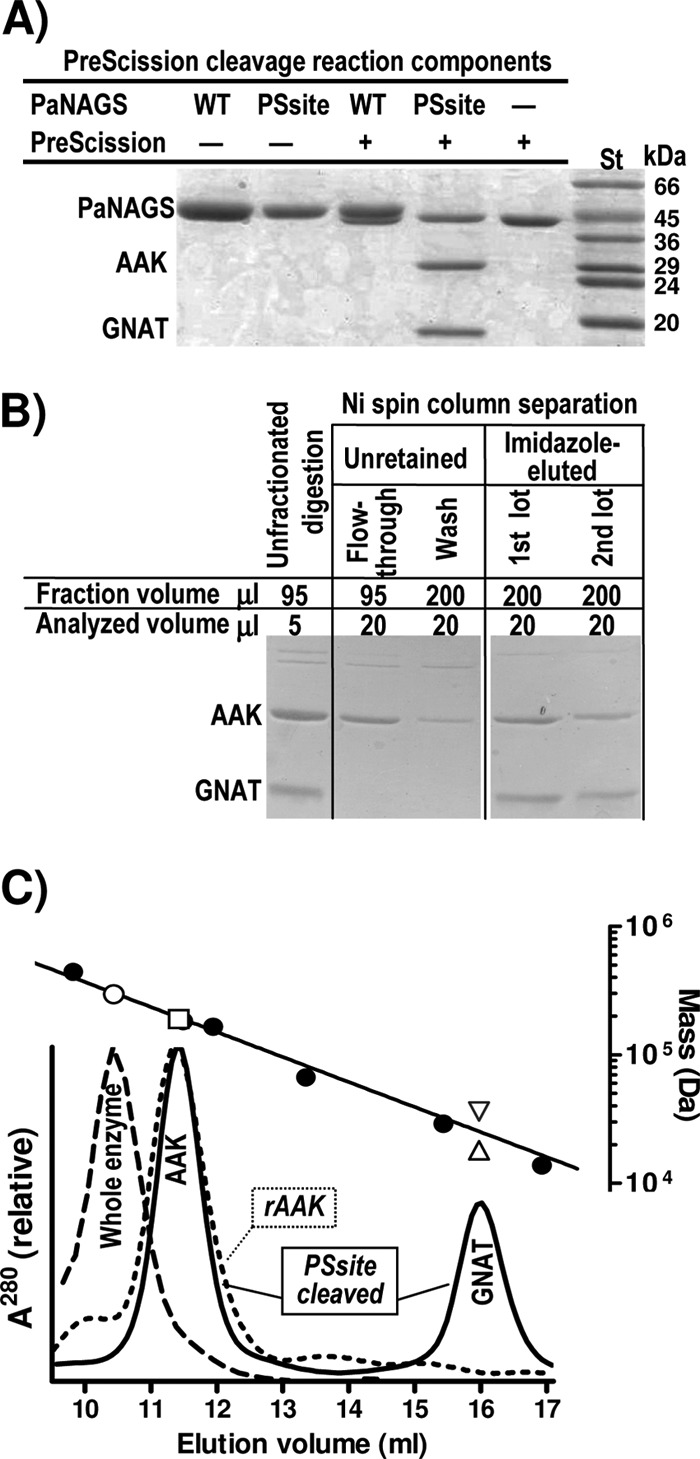
PreScission protease cleavage of the linker and size-exclusion chromatography of the digested enzyme and of the recombinant AAK domain. (A) SDS-PAGE analysis (Coomassie staining) revealing that wild-type PaNAGS (WT) is not cleaved, whereas the enzyme engineered to include the PreScission cleavage site (PSsite) (Fig. 2A and C) in the linker is cleaved. The PaNAGS and PreScission concentrations were 0.5 mg/ml and 167 U/ml, respectively. Note that a large excess of protease was added and that the polypeptide mass of this protease is very close to that of PaNAGS. St, protein markers of the indicated masses. (B) Ninety-five microliters of a digestion mixture of the PSsite form of PaNAGS (1 mg/ml) with 30 U/ml of PreScission protease was centrifuged through a 0.1-ml His Spin Trap column, followed by a 0.2-ml wash with 20 mM Na phosphate (pH 8)–1 mM DTT–0.5 M NaCl–20 mM imidazole and elution with two lots of the same buffer supplemented with 0.5 M imidazole, collecting separately each eluate from the sample, the washing, and the two-lot elutions. The figure shows the result of the SDS-PAGE analysis of the various fractions. AAK and GNAT denote the bands corresponding to these individual domains. (C) Size-exclusion chromatography of cleaved (continuous line) or noncleaved (broken line) PaNAGS and of recombinant AAK domain (dotted line). For details, see Materials and Methods. A total of 0.3 mg of each protein was injected. The digestion is that shown in panel B. The upper line is the semilogarithmic plot of the masses of marker proteins (closed circles) versus their elution volumes. The open symbols correspond to the protein peaks below them for the following sequence-deduced masses: 294.4 kDa for the whole enzyme, assuming that it is hexameric (○); 189.1 kDa for AAK domain, either recombinant or produced by cleavage, assuming that it is hexameric (□); and 18.1 kDa or 36.2 kDa for the GNAT domain, assuming that it is monomeric (▵) or dimeric (▽), respectively. The following protein standards were used: thyroglobulin (669 kDa) (not shown), ferritin (440 kDa), Thermotoga maritima acetylglutamate kinase (182 kDa) (28), E. coli UMP kinase (165 kDa) (5), bovine serum albumin (66.4 kDa), carbonic anhydrase (29 kDa), and RNase (13.7 kDa).
Linker cleavage did not inactivate the enzyme, but it caused a very large increase in the concentrations of glutamate required for activity (Fig. 5A, diamonds). Given the lack of an immediate dissociation of the two domains, this strongly suggests that the physical continuity across the linker is crucial to endow the enzyme with its normal affinity for glutamate. Similarly to the noncleaved form of the PSsite form, the cleaved form remained insensitive to arginine (data not shown), suggesting that the physical continuity between both domains provided by the linker is a requisite for the arginine modulation of enzyme activity.
Fig 5.
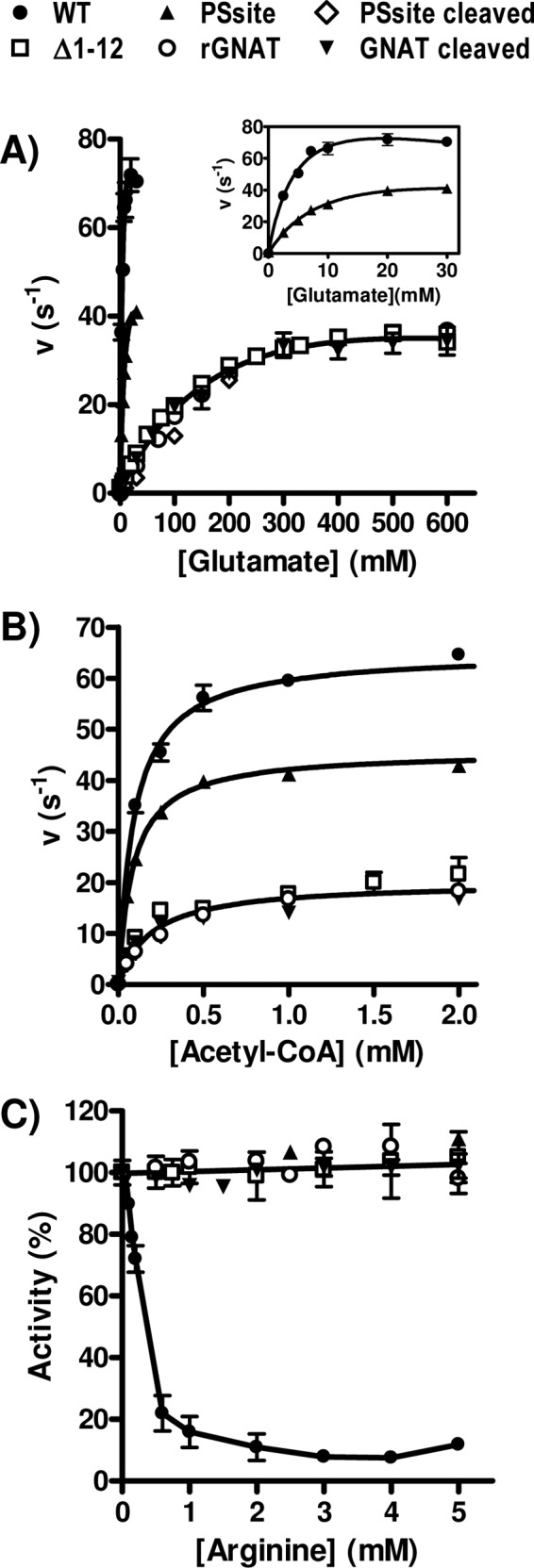
Dependency of NAGS activity on the concentration of both substrates (A and B) and of arginine (C) for the WT, the PSsite form (either noncleaved or PreScission protease cleaved), and the Δ1–12 form of PaNAGS and of the GNAT domain either isolated after cleavage (GNAT cleaved) or produced recombinantly (rGNAT). When the concentration of acetyl-CoA was varied, the concentration of glutamate was fixed at 100 mM, except for the WT and the noncleaved PSsite forms, with which it was kept at 30 mM. When the concentration of glutamate was varied, the concentration of acetyl-CoA was fixed at 4 mM. To allow a meaningful comparison of the activities of various enzyme forms having different masses, velocities are given as turnover numbers per polypeptide chain (units are s−1). A similar concentration dependency of the PSsite-cleaved form, the Δ1–12 form, and the isolated GNAT domains is evident for each substrate. (A and B) Therefore, single curves were fitted for the results for all these forms for glutamate (A) and for acetyl-CoA (B). The curve for glutamate corresponds to hyperbolic kinetics with substrate inhibition and with apparent values of KmGlu, KIGlu, and kcat at infinite glutamate of 240 ± 45 mM, 1,254 ± 480 mM, and 66 ± 8 s−1, respectively. The curve for acetyl-CoA (B) is a hyperbola, with apparent values of Kmacetyl-CoA and kcat at infinite acetyl-CoA of 190 ± 30 μM and 20.0 ± 0.7 s−1, respectively. The KmGlu, KIGlu, and Kmacetyl-CoA values for the WT and for the noncleaved PSsite forms of the enzyme are those shown in Table 1, and the apparent kcat values are 111 ± 16 s−1 for the WT and 69 ± 7 s−1 for the noncleaved PSsite form (see inset in panel A) for infinite glutamate and 65 ± 1 s−1 for the WT and 46 ± 1 s−1 for the PSsite form for infinite acetyl-CoA (see panel B). (C) Influence of arginine concentration on enzyme activity. Results are expressed as a percentage of the activity of the same enzyme form in the absence of arginine. A single line corresponding to no inhibition has been fitted to the results for all forms except the wild-type enzyme. Substrate concentrations in these assay mixtures were 4 mM acetyl-CoA and either 30 mM glutamate for the WT and noncleaved PSsite forms or 100 mM glutamate for all other forms.
Gel filtration separates the AAK and GNAT domains of the cleaved enzyme.
The application of the PreScission-cleaved PSsite enzyme form (Fig. 2A) onto a gel filtration column (Fig. 4C) resulted in the separation of the AAK and GNAT domains as individual peaks (Fig. 2D and 4C). We also succeeded in producing the AAK domain recombinantly by replacing codon 287 of the pET22b-encoded PaNAGS with a stop codon (Q287X mutation) (Fig. 2A and D). The AAK domain generated by PreScission cleavage or produced recombinantly was eluted identically from the column, at a volume corresponding to hexamers (Fig. 4C). Similarly, as reported previously (23, 32), the noncleaved enzyme was also eluted as expected for a hexamer (Fig. 4C). These findings are in agreement with observations made for the crystal structure of NgNAGS (24, 34) (Fig. 1A, and see Fig. S1A in the supplemental material) of a hexameric enzyme architecture nucleated by a hexameric ring of AAK domains that closely resembles the hexameric ring of AAK domains forming the structure of arginine-sensitive bacterial NAGK (28) (Fig. 1A, and see Fig. S1B in the supplemental material). In contrast to the hexameric oligomerization of the enzyme and the AAK domain, the GNAT domain produced by PreScission cleavage appeared late in the effluent from the gel filtration column, at a position intermediate between those expected for monomers and dimers (Fig. 4C) although somewhat closer to that of the monomer (mass estimate by interpolation of the calibration line of 25.6 kDa, corresponding to 1.4 protomers). A monomeric architecture would agree with the lack of interactions between GNAT domains in the crystal structure of the NgNAGS hexamer (24, 34).
The isolated GNAT domain catalyzes the NAGS reaction and is insensitive to arginine.
In agreement with the observation made for the NgNAGS structure that the sites for both substrates are located in the GNAT domain (34), only this domain exhibited NAGS activity (Fig. 5A and B, inverted closed triangles). However, the concentrations of glutamate required for activity with the isolated GNAT domain were much higher than those for the noncleaved PSsite form (∼25-fold higher) (Fig. 5A, compare main panel and inset), in agreement with the results obtained (see above) with the cleaved PSsite form in which the two domains had not been separated (Fig. 5A, diamonds). In addition, as expected from the binding of arginine in the AAK domain of PaNAGS (32) and NgNAGS (24), the isolated GNAT domain was insensitive to arginine (Fig. 5C). Identical results were obtained, within experimental errors (Fig. 5, open circles), for the substrate dependency of the activity and the lack of arginine sensitivity with the recombinant GNAT domain which we finally succeeded in producing as a chimera with SUMO (Fig. 2A and D).
The deletion of the N-terminal helix renders PaNAGS dimeric.
The present results indicate that the AAK domain, when connected covalently to the GNAT domain by the normal version or by mutated versions of the interdomain linker (including 5-residue-longer versions as in the engineered linker of the PSsite form), triggers a very important increase in the apparent affinity of the GNAT domain for glutamate. The structure of NgNAGS (34) reveals that each GNAT domain is connected to two or even three (in the arginine bound-form [24]) AAK domains: one belonging to the same subunit, to which the GNAT domain is covalently linked, and the others from an adjacent dimer on which the GNAT domain lies in the hexameric structure. Therefore, we sought to clarify whether it was the cognate AAK domain or the ones from an adjacent dimer that are responsible for the triggering of the increased affinity of the GNAT domain for glutamate.
We decided to dissociate the PaNAGS hexamer to dimers by deleting the N-terminal helix, which, by interlacing with the corresponding N-terminal helix from another dimer, links the three dimers into the hexamer (34) (see Fig. S1A in the supplemental material). In this way, the connection of each GNAT domain with its cognate AAK domain of the same subunit would be preserved, while the interaction with the AAK domains from another dimer would be lost if the hexamer was dissociated to its three composing dimers. We actually deleted only residues 1 to 12 (Fig. 2A), which should correspond to the larger and more N-terminal portion of the N-terminal helix, down to the kink before the second portion of the helix (34), which is shorter and is involved in arginine binding (24, 33), thus expectedly preserving the arginine binding site. The resulting protein (called the Δ1–12 form) was expressed as the wild-type enzyme, it was soluble and was purified easily (Fig. 2D), and it behaved in gel filtration experiments as dimers (Fig. 6), as expected, although with some skew toward larger elution volumes, which may indicate coexistence with a small fraction of monomers. Thus, these results reveal hexamer dissociation and, therefore, the abolition of interdimeric contacts. The peak of the dissociated enzyme exhibited enzyme activity (Fig. 6) with a constant ratio, within experimental errors, of activity versus the protein concentration (monitored as the OD280 [Fig. 6] and confirmed by SDS-PAGE [not shown]), indicating that the whole peak consists of a single species in terms of specific activity. The substrate kinetic parameters for this enzyme form (Fig. 5A and B, open squares) appeared to be identical, within experimental errors, to those of the isolated recombinant or PreScission-cleaved GNAT domain when velocities were expressed as the turnover number per polypeptide chain. Thus, single concentration-activity curves for glutamate and acetyl-CoA were fitted to the pooled values for these enzyme forms (Fig. 5A and B). The curve for glutamate, adjusted to hyperbolic kinetics with substrate inhibition, corresponds to a ∼50-fold increase and a ∼2-fold decrease in the Km and kcat, respectively, relative to the values for the wild-type enzyme (Fig. 5A, inset). The hyperbola fitted to the pooled data for acetyl-CoA (Fig. 5B) shows a modest (∼2-fold) increase in the Kmacetyl-CoA relative to that of the wild type. The identical kinetic properties of dimeric PaNAGS and of the isolated GNAT domain indicate that the modulating effect of the AAK domain on the activity of the GNAT domain results from the interactions that occur in the hexamer between the GNAT domain from one dimer and the AAK domains from another dimer. Furthermore, arginine had no effect on the activity of the enzyme with the helix deletion (Fig. 5C). Since the portion of the helix that is deleted is not involved in arginine binding (24), arginine would be expected to bind to the dimeric enzyme. Therefore, the lack of an influence of arginine on enzyme activity possibly reflects the requirement of a hexameric organization for inhibition in the classical NAGS.
Fig 6.
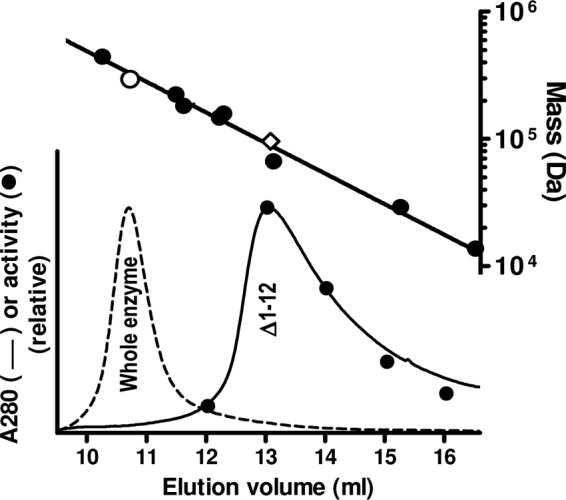
Size-exclusion chromatography of the Δ1–12 form of PaNAGS. The chromatographic profile (continuous line) is compared with that of the wild-type enzyme (broken line), both injected in 0.2-mg amounts. The procedure and system are described in the section on cleavage with PreScission protease and the separation of the two domains in Materials and Methods. In the case of Δ1–12, enzyme activity was measured in 1-ml collected fractions (closed circles) (bottom plot). The upper line is the semilogarithmic plot of the masses of marker proteins (closed circles) versus their elution volumes. The open symbols correspond to the protein peaks below them for the following sequence-deduced masses: 294.4 kDa for the whole enzyme, assuming that it is hexameric (○), and 95.5 kDa for Δ1–12 enzyme form, assuming that it is dimeric (♢). The protein standards used are ferritin (440 kDa), β-amylase (224 kDa), T. maritima acetylglutamate kinase (182 kDa) (28), aldolase (158 kDa), alcohol dehydrogenase (147 kDa), bovine serum albumin (66.4 kDa), carbonic anhydrase (29 kDa), and RNase (13.7 kDa).
The AAK domain of NAGS is derived from an ancestral NAGK domain, as shown by the triggering of NAGK activity by a double point mutation.
As indicated above, the isolated AAK domain did not exhibit NAGS activity, as expected. However, the similarity of this domain of NgNAGS to the enzyme NAGK (34), with the preservation of the active center crevice (Fig. 7A), led us to investigate whether this domain had any NAGK activity. In fact, a bifunctional NAGS/NAGK was identified previously in Xanthomonas campestris (27), although its NAGK activity was quite low compared with its own NAGS activity or with the NAGK activities of classical bacterial NAGKs (13, 22).
Fig 7.

The AAK domain of NAGS is an ancestral NAGK. (A) Stereo view of the superimposition of the structure of the active center of E. coli NAGK (in green) bound to NAG and the ATP inert analog AMPPNP (PDB accession number 1GS5) (29) with the corresponding region of NgNAGS (in orange-brown) (PDB accession number 2R8V). The main chain of the structural elements (labeled) that make the site is shown. A few important residues are illustrated in the same color as the main chain (except the N atoms of the side chains, which are in blue) and are labeled. NAG and AMPPNP are shown in a stick representation, with the C, P, O, and N atoms shown in yellow, green, red, and blue, respectively. (B and C) Dependency of the activity of the M26K L240K double mutant (squares) of rAAK on ATP and NAG concentrations. The circle illustrates the lack of activity of the wild-type enzyme (0.3 mg/ml) assayed under the same conditions at concentrations of 20 mM ATP and 100 mM NAG. The hyperbola fitted to the points for variable ATP yields KmATP and V[ATP]=∞ values of 1.6 ± 0.2 mM and 0.122 ± 0.003 U/mg, respectively. The nearly linear NAG concentration dependency of the activity indicates that the KmNAG and the V[NAG]=∞ values exceed 0.225 M and 0.5 U/mg, respectively. The curve fitted over the points would correspond to the hyperbola for KmNAG and V[NAG]=∞ values of 0.83 M and 1.1 U/mg, respectively.
We failed to detect any NAGK activity with PaNAGS, even at very high enzyme concentrations (up to 0.16 mg/ml in the assay mixture; at this concentration, the X. campestris enzyme would have consumed all the NAG present in the assay mixture). This lack of NAGK activity might have been expected, since P. aeruginosa has a separate gene (argB) that encodes a highly active arginine-sensitive NAGK (28). Similarly, we failed to detect any NAGK activity with the isolated recombinant AAK domain of PaNAGS, even at concentrations of 0.3 mg/ml in the assay mixture (Fig. 7B and C, closed circles), excluding that the lack of NAGK activity of the complete enzyme was due to some inhibitory effect of the GNAT domain on that activity. However, when we mutated two residues of the putative NAGK active center of the AAK domain of PaNAGS to lysine (M26K L240K double mutation), rAAK became active as an NAGK. These residues were chosen because they are the counterparts of two invariant NAGK active center lysines in PaNAGS (K8 and K217 of E. coli NAGK) (Fig. 7A) that play key catalytic roles in acetylglutamate phosphorylation (16, 29). Whereas no activity was observed when only one of the two mutations was introduced into rAAK (results not shown), the double mutant exhibited clear although low NAGK activity (Fig. 7B and C). The activity depended hyperbolically on the ATP concentration (Fig. 7B) and also presented a nonlinear dependency on the concentration of NAG (Fig. 7C), although the apparent affinity for this substrate was too low to approach saturation. The apparent KmATP of the double mutant (1.6 ± 0.2 mM) was in the range typical for a bacterial NAGK in the same type of hydroxylamine-based assay (13, 18). In contrast, the KmNAG could not be estimated accurately, given its high value, since the range of NAG concentrations used did not reach the levels needed for saturation. In any case, from the data shown in Fig. 7C, it appears evident that the KmNAG and the activity at saturation of this substrate are >0.225 M and >0.5 U/mg, respectively. Actually, the best hyperbolic fit for the NAG data gives apparent KmNAG and Vmax values of ∼0.8 M and ∼1 U/mg, respectively. The Vmax value is not too far from the activity exhibited by the bifunctional X. campestris enzyme (27). However, the KmNAG is much higher than that for genuine NAGKs (13, 18), possibly reflecting a tendency toward a closed conformation of the NAG site of the AAK domain of NAGS, as revealed by the lowered position of the lid for this site (the β3-β4 hairpin) (Fig. 7A) in the NgNAGS structure (24) despite the emptiness of this site, whereas in NAGK, this site closes down only when NAG is bound (15).
How and why did two-domain NAGSs emerge?
The closeness of the AAK domains of NAGS and NAGK goes beyond the overall domain fold, extending to the similarity of the NAGK active center crevice (Fig. 7A) and even to the conservation of a key NAG-binding arginine (R66 of E. coli NAGK, R85 of NgNAGS [Fig. 7A], and R82 of PaNAGS) (29, 34). Indeed, although with a low affinity, the AAK domain of NAGS binds NAG, as attested by the NAGK activity of its M26K L240K double mutant despite the fact that these mutations do no affect NAG site residues. An invariant aspartate (D162 in E. coli NAGK), which in NAGKs has a key active center-organizing role by coordinating the two catalytic lysines and by binding MgATP (16, 29), is also preserved in many NAGSs as a glutamate (E185 of PaNAGS) although not in NgNAGS (34). The NAGK activity of the M26K L240K double mutant indicates that classical NAGS lost its ancestral NAGK activity primarily because of the mutation of these two catalytic lysines (16, 22, 29). Indeed, as expected from its NAGK activity, the bifunctional NAGS/NAGK of X. campestris has these two lysines preserved (27).
Since Mycobacterium tuberculosis NAGS is merely a GNAT domain and yet is feedback inhibited by arginine (12), the AAK-GNAT domain organization of the classical bacterial NAGS is clearly unessential for arginine-regulated NAG synthesis from acetyl-CoA and glutamate. A previously reported proposal by Xu et al. (37) that the classical two-domain NAGSs arose from the fusion of a NAGS similar to that found in M. tuberculosis and an arginine-sensitive NAGK is supported very strongly by our finding that a mere two-residue mutation in the AAK domain of PaNAGS can render this domain an NAGK. This proposal is also supported by the closeness of the structures of NAGK (28, 29) and the AAK domain of NgNAGS (34) (Fig. 7A) and by the discovery of bifunctional NAGS/NAGK having the same domain organization as that of PaNAGS (27). A question remaining to be answered concerning the evolutionary process from single-domain to two-domain NAGS is whether the arginine sensitivity of M. tuberculosis NAGS (12) was a late evolutionary acquisition or whether this trait of the GNAT component was originally present and has been lost over the course of the evolution of two-domain NAGSs.
A major advantage of associating AAK and GNAT domains into a two-domain NAGS that has become patent in our studies is that because of the important modulating role of the AAK domain on the activity of the GNAT domain, the Km for glutamate is brought down from the very high values for the M. tuberculosis enzyme (12) to the millimolar range that is characteristic of the canonical microbial enzymes (17, 23, 24, 27, 32). This value for these enzymes is closer to the range of glutamate concentrations present in bacteria (see, for example, reference 25), thus increasing the efficiency of the enzyme in the catalysis of the reaction in vivo. Another potential reason for the shift from a single-domain to a two-domain NAGS may be related to NAG channeling between NAGS and NAGK, since such channeling would prevent unwanted NAG hydrolysis by aminoacylases such as those prevalent in animals (30) or existing (although nowadays in the periplasmic space) in P. aeruginosa (14). This channeling requires the direct interaction of the GNAT domain of one subunit with the AAK domain of another subunit, as observed for hexameric NAGS. Reminiscent of such channeling might be the association in Saccharomyces cerevisiae of NAGS and NAGK, both having the two-domain composition of PaNAGS (27), to form a metabolon when NAG is produced by NAGS (1, 26). It would be important to clarify whether such channeling occurs in this metabolon and/or in bifunctional NAGS/NAGK.
A third potential advantage of the two-domain organization of NAGS is the possibility of easily modulating the arginine regulation of NAGS activity. In two-domain NAGS, arginine regulation results from primary arginine-triggered changes in the architecture of the AAK domain hexamer (24). These changes are similar to those observed previously for hexameric NAGK upon arginine binding to this enzyme (28). Since with the latter enzyme, a signaling protein, PII, was shown previously to modulate the sensitivity of the enzyme to arginine inhibition (21), it cannot be excluded that analogous regulatory mechanisms may exist for modulating the arginine sensitivity of canonical hexameric NAGSs. Furthermore, the key role of the interdomain linker as a mediator of the effects of arginine that has been revealed by our previous (33) and present studies provides the basis for adapting arginine regulation by changes merely in the 5-residue linker sequence. This endows AAK-GNAT domain NAGSs with an enormous potential for adaptation to the specific physiological needs of different organisms. The best example of this ability to adapt is provided by the change in the effect of arginine on NAGS from inhibition to activation with the shift of animals from marine life to terrestrial ureotelism (19), a change that we have partially reproduced by linker manipulation in the present studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants BFU2008-05021 of the Spanish Ministry of Science (MEC and MICINN) and Prometeo/2009/051 of the Valencian Government. E.S.-V. and M.L.F.-M. were supported by contracts from the Instituto de Salud Carlos III and the JAE-DOC Program of the Consejo Superior de Investigaciones Científicas.
We thank N. Gougeard (IBV-CIBERER) and the late E. Cogollos (IBV-CSIC) for technical assistance and F. Gil-Ortiz (IBV-CSIC) for help with Fig. 2B and 7A.
Footnotes
Published ahead of print 23 March 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Abadjieva A, Pauwels K, Hilven P, Crabeel M. 2001. A new yeast metabolon involving at least the two first enzymes of arginine biosynthesis: acetylglutamate synthase activity requires complex formation with acetylglutamate kinase. J. Biol. Chem. 276:42869–42880 [DOI] [PubMed] [Google Scholar]
- 2. Alonso E, Rubio V. 1989. Participation of ornithine aminotransferase in the synthesis and catabolism of ornithine in mice. Studies using gabaculine and arginine deprivation. Biochem. J. 259:131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bachmann C, Krähenbühl S, Colombo JP. 1982. Purification and properties of acetyl-CoA:L-glutamate N-acetyltransferase from human liver. Biochem. J. 205:123–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 5. Briozzo P, et al. 2005. Structure of Escherichia coli UMP kinase differs from that of other nucleoside monophosphate kinases and sheds new light on enzyme regulation. J. Biol. Chem. 280:25533–25540 [DOI] [PubMed] [Google Scholar]
- 6. Caldovic L, et al. 2006. Biochemical properties of recombinant human and mouse N-acetylglutamate synthase. Mol. Genet. Metab. 87:226–232 [DOI] [PubMed] [Google Scholar]
- 7. Caldovic L, Morizono H, Tuchman M. 2007. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum. Mutat. 28:754–759 [DOI] [PubMed] [Google Scholar]
- 8. Caldovic L, Tuchman M. 2003. N-Acetylglutamate and its changing role through evolution. Biochem. J. 372:279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cordingley MG, Callahan PL, Sardana VV, Garsky VM, Colonno RJ. 1990. Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitro. J. Biol. Chem. 265:9062–9065 [PubMed] [Google Scholar]
- 10. Cunin R, Glansdorff N, Pierard A, Stalon V. 1986. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50:314–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 12. Errey JC, Blanchard JS. 2005. Functional characterization of a novel ArgA from Mycobacterium tuberculosis. J. Bacteriol. 187:3039–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernández-Murga ML, Gil-Ortiz F, Llácer JL, Rubio V. 2004. Arginine biosynthesis in Thermotoga maritima: characterization of the arginine-sensitive N-acetyl-L-glutamate kinase. J. Bacteriol. 186:6142–6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Früh H, Leisinger T. 1981. Properties and localization of N-acetylglutamate deacetylase from Pseudomonas aeruginosa. J. Gen. Microbiol. 125:1–10 [DOI] [PubMed] [Google Scholar]
- 15. Gil-Ortiz F, Ramón-Maiques S, Fernández-Murga ML, Fita I, Rubio V. 2010. Two crystal structures of Escherichia coli N-acetyl-L-glutamate kinase demonstrate the cycling between open and closed conformations. J. Mol. Biol. 399:476–490 [DOI] [PubMed] [Google Scholar]
- 16. Gil-Ortiz F, Ramón-Maiques S, Fita I, Rubio V. 2003. The course of phosphorus in the reaction of N-acetyl-L-glutamate kinase, determined from the structures of crystalline complexes, including a complex with an AlF4− transition state mimic. J. Mol. Biol. 331:231–244 [DOI] [PubMed] [Google Scholar]
- 17. Haas D, Kurer V, Leisinger T. 1972. N-Acetylglutamate synthetase of Pseudomonas aeruginosa. An assay in vitro and feedback inhibition by arginine. Eur. J. Biochem. 31:290–295 [DOI] [PubMed] [Google Scholar]
- 18. Haas D, Leisinger T. 1975. N-Acetylglutamate 5-phosphotransferase of Pseudomonas aeruginosa. Catalytic and regulatory properties. Eur. J. Biochem. 52:377–393 [PubMed] [Google Scholar]
- 19. Haskins N, et al. 2008. Inversion of allosteric effect of arginine on N-acetylglutamate synthase, a molecular marker for evolution of tetrapods. BMC Biochem. 9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 21. Llácer JL, et al. 2007. The crystal structure of the complex of PII and acetylglutamate kinase reveals how PII controls the storage of nitrogen as arginine. Proc. Natl. Acad. Sci. U. S. A. 104:17644–17649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marco-Marín C, Ramón-Maiques S, Tavárez S, Rubio V. 2003. Site-directed mutagenesis of Escherichia coli acetylglutamate kinase and aspartokinase III probes the catalytic and substrate-binding mechanisms of these amino acid kinase family enzymes and allows three-dimensional modelling of aspartokinase. J. Mol. Biol. 334:459–476 [DOI] [PubMed] [Google Scholar]
- 23. Marvil DK, Leisinger T. 1977. N-Acetylglutamate synthase of Escherichia coli: purification, characterization, and molecular properties. J. Biol. Chem. 252:3295–3303 [PubMed] [Google Scholar]
- 24. Min L, et al. 2009. Mechanism of allosteric inhibition of N-acetyl-L-glutamate synthase by L-arginine. J. Biol. Chem. 284:4873–4880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ogahara T, Ohno M, Takayama M, Igarashi K, Kobayashi H. 1995. Accumulation of glutamate by osmotically stressed Escherichia coli is dependent on pH. J. Bacteriol. 177:5987–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pauwels K, Abadjieva A, Hilven P, Stankiewicz A, Crabeel M. 2003. The N-acetylglutamate synthase/N-acetylglutamate kinase metabolon of Saccharomyces cerevisiae allows co-ordinated feedback regulation of the first two steps in arginine biosynthesis. Eur. J. Biochem. 270:1014–1024 [DOI] [PubMed] [Google Scholar]
- 27. Qu Q, Morizono H, Shi D, Tuchman M, Caldovic L. 2007. A novel bifunctional N-acetylglutamate synthase-kinase from Xanthomonas campestris that is closely related to mammalian N-acetylglutamate synthase. BMC Biochem. 8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramón-Maiques S, et al. 2006. Structural bases of feed-back control of arginine biosynthesis, revealed by the structures of two hexameric N-acetylglutamate kinases, from Thermotoga maritima and Pseudomonas aeruginosa. J. Mol. Biol. 356:695–713 [DOI] [PubMed] [Google Scholar]
- 29. Ramón-Maiques S, Marina A, Gil-Ortiz F, Fita I, Rubio V. 2002. Structure of acetylglutamate kinase, a key enzyme for arginine biosynthesis and a prototype for the amino acid kinase enzyme family, during catalysis. Structure 10:329–342 [DOI] [PubMed] [Google Scholar]
- 30. Reglero A, Rivas J, Mendelson J, Wallace R, Grisolia S. 1977. Deacylation and transacetylation of acetyl glutamate and acetyl ornithine in rat liver. FEBS Lett. 81:13–17 [DOI] [PubMed] [Google Scholar]
- 31. Rubio V, Ramponi G, Grisolia S. 1981. Carbamoyl phosphate synthetase I of human liver. Purification, some properties and immunological cross-reactivity with the rat liver enzyme. Biochim. Biophys. Acta 659:150–160 [DOI] [PubMed] [Google Scholar]
- 32. Sancho-Vaello E, Fernández-Murga ML, Rubio V. 2008. Site-directed mutagenesis studies of acetylglutamate synthase delineate the site for the arginine inhibitor. FEBS Lett. 582:1081–1086 [DOI] [PubMed] [Google Scholar]
- 33. Sancho-Vaello E, Fernández-Murga ML, Rubio V. 2009. Mechanism of arginine regulation of acetylglutamate synthase, the first enzyme of arginine synthesis. FEBS Lett. 583:202–206 [DOI] [PubMed] [Google Scholar]
- 34. Shi D, et al. 2008. The crystal structure of N-acetyl-L-glutamate synthase from Neisseria gonorrhoeae provides insights into mechanisms of catalysis and regulation. J. Biol. Chem. 283:7176–7184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Slocum RD. 2005. Genes, enzymes and regulation of arginine biosynthesis in plants. Plant Physiol. Biochem. 43:729–745 [DOI] [PubMed] [Google Scholar]
- 36. Sonoda T, Tatibana M. 1983. Purification of N-acetyl-L-glutamate synthetase from rat liver mitochondria and substrate and activator specificity of the enzyme. J. Biol. Chem. 258:9839–9844 [PubMed] [Google Scholar]
- 37. Xu Y, Glansdorff N, Labedan B. 2006. Bioinformatic analysis of an unusual gene-enzyme relationship in the arginine biosynthetic pathway among marine gamma proteobacteria: implications concerning the formation of N-acetylated intermediates in prokaryotes. BMC Genomics 7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.