

Abstract
Ethanol abuse and chronic ethanol consumption remains a major public health problem and is responsible for a high rate of morbidity. Alcohol-induced fatty liver generally begins as hepatic steatosis, and if the cause persists, this invariably progresses to steatohepatitis and cirrhosis. The original biochemical explanation for an alcoholic fatty liver centered on the ability of ethanol metabolism to shift the redox state of the liver and inhibit fatty acid oxidation. Subsequent studies found repression of fatty acid oxidation and that the induction of lipogenesis can occur in alcoholic conditions. Ethanol activates sterol regulatory element binding protein 1, inducing a battery of lipogenic enzymes. These effects may be due in part to inhibition of AMP-dependent protein kinase, reduction in plasma adiponectin or increased levels of TNF-α the liver. They in turn activate lipogenic pathways and inhibit fatty acid oxidation. Besides the fatty acid synthesis and oxidation, ethanol also alters lipid droplet (LD, the storage form of triglycerides, TG) metabolism in hepatocytes and very low-density lipoprotein (VLDL) secretion from liver. Because steatosis is now regarded as a significant risk factor for advanced liver pathology, an understanding of the molecular mechanisms in its etiology provides new therapeutic targets to reverse the alcoholic fatty liver.
KEY WORDS: Adiponectin, alcohol, adenosine monophosphate-activated protein kinase, lipid droplets, steatosis/fatty liver
Introduction
Alcohol abuse and alcohol-induced liver disease (ALD) are a major public health problem both in the US and worldwide. ALD is probably the main cause of death among people with severe alcohol abuse and is responsible for about 3.8% of global mortality.[1] An early manifestation of alcohol liver disease (ALD) is the presence of fatty liver (hepatic steatosis) that, with continued insult, can progress to alcoholic liver disease (ALD).
The spectrum of disease ranges from fatty liver (steatosis) to alcoholic steatohepatitis (ASH), hepatic fibrosis and cirrhosis.[2,3] Steatosis is an earliest stage of alcoholic liver disease and the most common alcohol-induced liver disorder. Steatosis is characterized, in part, by the excessive buildup of fat inside liver cells. This condition can be reversed, however, when the alcohol intake stops.[4] ASH is defined by the presence of fatty liver, an inflammatory infiltrate (which mainly consists of polymorphonuclear leukocytes) and hepatocellular damage.[5,6] Patients with ASH can develop progressive fibrosis. In livers affected by ALD, the fibrotic tissue is typically located in pericentral and perisinusoidal areas, collagen deposits are evident and bridging fibrosis develops, which precedes the development of regeneration nodules and liver cirrhosis.[7] Cirrhosis involves replacement of the normal hepatic parenchyma with extensive thick bands of fibrous tissue and regenerative nodules, which results in the clinical manifestations of portal hypertension and liver failure.[8,9] The prevalence of alcoholic liver disease is influenced by many factors, including genetic factors (e.g., predilection to alcohol abuse, sex) and environmental factors (e.g., availability of alcohol, social acceptability of alcohol use, concomitant hepatotoxic insults), which make this disease, difficult to define.[10] In general, however, the risk of liver disease increases with the quantity and duration of an alcohol intake. Although necessary, excessive alcohol use is not sufficient to promote alcoholic liver disease. Only 1 in 5 heavy drinkers develops alcoholic hepatitis, and 1 in 4 develops cirrhosis.[11–13] The pathogenesis of ALD is multifactorial and includes several overlapping events. The “two-hit” hypothesis postulates that the steatotic liver is the first hit, and this steatotic liver is susceptible to secondary insults including a vulnerability to reactive oxygen species (ROS), gut-derived endotoxins, and adipocytokines such as, tumor necrosis factor (TNF-α) and other cytokines. All of these hits are widely believed to be major contributors to alcohol-induced liver injury and may compound an initial steatosis. An evolving concept that is gaining acceptance is that certain accumulated fatty acids are toxic to the liver. Thus, ethanol-elicited hepatic lipid accumulation, as well as that caused by dietary sources, has prompted renewed interest as a cornerstone of liver toxicity as it may not only initiate but enhance the progression of alcoholic liver disease.[14] This review will discuss the factors that contribute to ethanol-induced steatosis.
General Mechanism for Alcohol-induced Fatty Acid Synthesis
The liver and, to a lesser extent, the gastrointestinal tract, are the main sites of alcohol metabolism. Within the liver, there are 2 main pathways of alcohol metabolism, alcohol dehydrogenase and cytochrome P-450 2E1 (CYP2E1). Alcohol dehydrogenase (ADH) is a hepatocyte cytosolic enzyme that converts alcohol to acetaldehyde. Acetaldehyde subsequently is metabolized to acetate via the mitochondrial enzyme acetaldehyde dehydrogenase (ALDH). Both steps are coupled to the reduction of NAD to NADH.[15–17] An increased NADH/NAD ratio has profound effects on the metabolism of carbohydrates and lipids. Gluconeogenesis is impaired and substrate flow through the citric acid cycle is diminished with acetyl-coA diverted towards ketogenesis and fatty acid synthesis. Together with the inhibition of mitochondrial fatty acid β-oxidation, this latter effect of the altered redox state contributes to the pathogenesis of fatty liver (steatosis).[16,18] This metabolic explanation for fatty liver has been a prevailing concept for many years, but it was not sufficient to explain the rapid formation of fatty liver after an acute ethanol administration. In addition, the degree of change in liver redox potential that occurs in vivo after chronic ethanol administration to rodents is significant but rather modest. Thus, the metabolic explanation for alcohol-induced fatty liver became insufficient to account for all changes that occurred in hepatic lipids after ethanol consumption. Later discoveries in cell signaling and characterization of specific transcription factors prompted investigations of causes that are operative in the pathophysiology of ethanol-induced steatosis.
Updated Mechanisms in Ethanol-induced Hepatic Steatosis
The total amount of fat in the liver depends on fatty acid synthesis and its oxidation. As shown in Figure 1 the pathogenesis of alcoholic fatty liver is based upon the combination of an increased glycerolipid synthesis and decreased fatty acid oxidation in mitochondria. In addition to synthesis and oxidation, fatty acid export processes also influence the fat levels.
Figure 1.
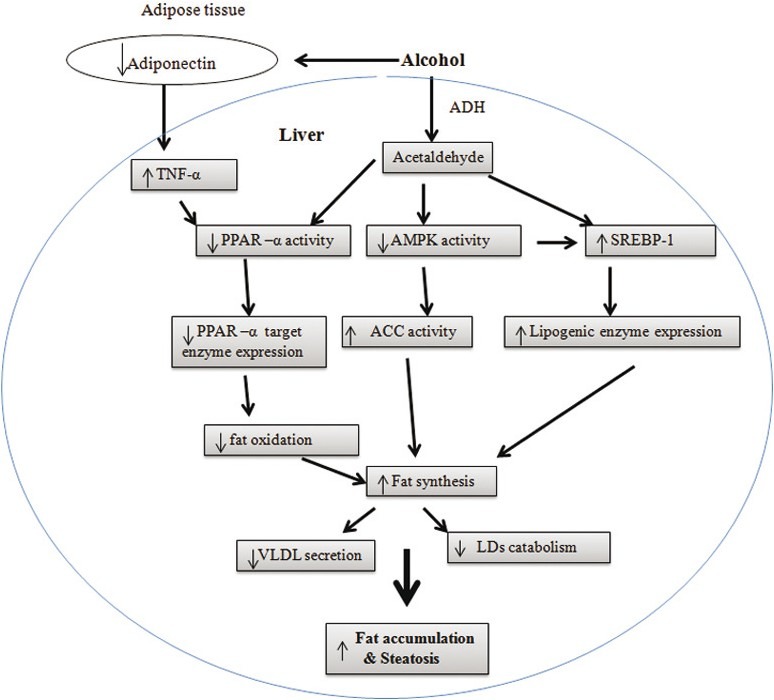
Potential mechanisms underlying the alcoholic fatty liver. In liver, sterol regulatory element binding protein 1 is responsible for fatty acid synthesis and peroxisome proliferator activated receptor-α, AMPdependent protein kinase (AMPK) and adiponectin are responsible for fatty acid oxidation. Ethanol may influence the activity of PPAR-α, SREBP-1, and AMPK directly or through adiponectin and tumor necrosis factor-α. These effects activate the lipogenic pathways and inhibit fatty acid oxidation. Besides the fatty acid synthesis and oxidation, ethanol also alters lipid droplets (LD, the storage form of TG) metabolism in hepatocytes and very low-density lipoproteins secretion from liver. All these alterations contribute to alcohol-induced fatty liver
Accelerated Synthesis
The enzymes involved in fatty acid synthesis are predominantly controlled by sterol regulatory element binding protein 1 (SREBP-1).[19] Sterol regulatory element-binding proteins (SREBPs) are a family of transcription factors that regulate the enzymes responsible for the synthesis of cholesterol, fatty acids, and triglycerides in liver and other tissues. SREBPs are synthesized as precursors (~125 kDa) bound to the endoplasmic reticulum and nuclear envelope. Upon an activation, SREBPs are released from the membrane into the nucleus as a mature protein (~68 kDa) by a sequential 2-step cleavage process.[20] Ethanol exposure has been shown to significantly increase the SREBP-regulated transcription via elevated levels of the mature SREBP-1 protein. Thus, ethanol increases the fatty acid synthesis through SREBP-1. The effect of ethanol on SREBP-1 appears to be mediated through its metabolism to acetaldehyde. Results from published work suggest that acetaldehyde can increase the synthesis of the mature SREBP-1 protein, which enhances hepatic lipogenesis and leads to the development of fatty liver [Figure 1].[21]
Impaired Fatty Acid Oxidation
Oxidation of fatty acids occurs in 3 subcellular organelles, with β-oxidation confined to mitochondria and peroxisomes and cytochrome P450 4A (CYP4A)-catalyzed ω-oxidation occurring in the endoplasmic reticulum. Some of the key enzymes of these 3 fatty acid oxidation systems in liver are regulated by peroxisome proliferator-activated receptor-alpha (PPAR-α), adiponectin and adenosine monophosphate-activated protein kinase (AMPK).
Beta-oxidation is the major degradative pathway for fatty acid esters in humans, and this is regulated by carnitine palmitoyltransferase-1 (CPT-1), the carnitine concentration, and malonyl-CoA, which inhibits CPT-1.[22] With the concomitant up-regulation of fatty acid biosynthesis by ethanol, the intracellular accumulation of intermediary products of fatty acid synthesis, such as malonyl-CoA, can negatively affect fatty acid transport into mitochondria and its oxidation by inhibiting CPT-1. Fatty acids, fatty acyl-CoAs, and several structurally different synthetic compounds, known as peroxisome proliferators, can activate PPAR-α, and thus regulate CPT-1 levels in the liver.[22]
PPAR-α in hepatic steatosis
Peroxisome proliferator-activated receptor-alpha (PPAR-α) is a member of the nuclear hormone receptor super family and functions as a lipid sensor in the liver. PPAR-α recognizes and responds to the influx of fatty acids by stimulating the transcription of PPAR-α-regulated genes, which are involved in oxidation, transport and export of free fatty acids. These include membrane transporters such as CPT-1, apolipoprotein genes, and several components of the mitochondrial and peroxisomal fatty acid β-oxidation pathways.[23,24] The PPAR-α regulated enzymes involved in fatty acid oxidation include acyl-CoA oxidase (AOX), 3-hydroxyacyl-CoA dehydrogenase, multifunctional β-oxidation protein (3-ketoacyl-CoA thiolase), acyl-CoA synthase, malonyl -CoA decarboxylase (MCD), CYP4A, and CPT-1. In addition, MCD, which controls the levels of malonyl-CoA, is positively regulated by PPAR-α.[25]
The effect of alcohol on PPAR-α in alcoholic fatty liver has been investigated in cultured hepatocytes as well as in ethanol-fed rodents. Alcohol has shown to inhibit PPAR-α activity both in vitro and in vivo conditions.[26,27] Acetaldehyde, the metabolite of alcohol, is a key factor in the effect of alcohol on PPAR-α. It is possible that acetaldehyde, because of its ability to covalently bind proteins, can form adducts with the PPAR-α transcription complex, thereby preventing its ability to bind the promoter element(s).[28,29]
Adiponectin in Alcohol Liver Disease
In addition to PPAR-α, hepatic lipid metabolism has been shown to be tightly regulated by adiponectin and adenosine monophosphate-activated protein kinase (AMPK). Adiponectin is an adipose-derived hormone with a variety of beneficial biological functions.[30] Increasing evidence suggests that altered adiponectin production in adipose tissue and impaired expression of hepatic adiponectin receptors (AdipoRs) are associated with the development of alcoholic liver steatosis in several rodent models.[31] It has been shown that the effect of adiponectin is largely mediated by an increase in fatty acid oxidation, associated with activation of AMPK and PPAR-α pathways and suppressing hepatic production of TNF-α.[32,33] It is well known that adiponectin and TNF-α regulate the mutual production and antagonize their biological effects on the target tissues. Although it is known that chronic ethanol consumption leads to increased circulating and local concentrations of TNF-α,[34,35] it remains unclear whether increased TNF-α resulting from ethanol feeding causes adiponectin reduction or whether suppression of adiponectin production by ethanol leads to TNF-α induction.
Role of Adenosine Monophosphate-activated Protein Kinase
AMPK is known to act as a key metabolic “master switch” by phosphorylating the target enzymes involved in lipid metabolism in many tissues including the liver. This enzyme, a heterotrimeric protein, is, itself activated by AMP as well as by phosphorylation by liver kinase B-1.[36] When AMP activates the AMPK, it down-regulates energy requiring pathways, generally lipid, RNA and protein synthesis. Conversely, AMPK activates ATP-generating catabolic pathways, such as fatty acid oxidation, the TCA cycle and glycolysis.[37] It phosphorylates and inhibits enzymes involved in lipid metabolism such as, 3-hydroxy-3-methyl glutamate-CoA reductase and acetyl-CoA carboxylase (ACC).[38] In addition to the direct regulation of the activity of the lipid metabolizing enzymes, AMPK also modulates SREBP-1 activity,[39] which plays an important role in fatty acid synthesis.
Dephosphorylation of AMPK by protein phosphatase 2A (PP2A) causes AMPK inactivation. This PP2A can be activated by ceramide, which is known to be increased after ethanol administration.[40–42] Thus, ethanol increases the ceramide levels thus increasing PP2A activity, which in turn inhibits the AMPK activity and increases the fatty acid synthesis.
Hepatic TGs: Export in VLDL and store in LDs
The increased synthesis of free fatty acids in the liver of alcoholics along with reduced ability of liver to oxidize these compounds can lead to increased synthesis of TG, the main storage form of fat in liver. Fatty acids, stored as TG, are then imported into very low-density lipoproteins (VLDL) particles, which are exported and transported in the serum to peripheral tissues. The amount of fat, that can be exported in VLDL, will depend on synthesis of the protein components, as well as the availability of TGs. Excess TGs are stored as lipid droplets (LDs) in the liver.[43]
Ethanol Impairs Secretion of Very Low-density Lipoproteins from Liver
Triacylglycerols are generally exported from the liver by VLDL particles. These are assembled through a complex process and made up of triglycerides, cholesterol, phosphatidylcholine, and apolipoproteins. These VLDLs are released into circulation for delivery to the various tissues (primarily muscle and adipose tissue) for storage or production of energy through oxidation. Inhibition of this process at any of several levels may result in accumulation of triglycerides in hepatocytes and consequently development of fatty liver. Indeed, in vivo studies[44,45] and in vitro studies,[46] VLDL secretion has been shown to be impaired after ethanol administration. Alcohol may impair triglyceride export by inhibiting the synthesis of phosphatidylcholine (via inactivating phosphatidylethanolamine methyl transferase activity), which is an important component of VLDL formation.[47] Another proposed mechanism for reduced VLDL secretion is inhibition of microtubular assembly by acetaldehyde produced during ethanol metabolism. This is supported by studies in which inhibition of alcohol dehydogenase prevented the alcohol-induced reduction of triacylglycerol.[47] In addition, alcohol may impair transport by inhibiting apolipoprotein synthesis through inhibition of PPAR-α activity. Studies are required to understand the mechanisms which impair the formation, intracellular transport through the cytoskeleton, and secretion of VLDL.
Impaired Lipid Droplet Metabolism
Organisms store lipid when they take in more energy that can be immediately used. This excess energy is packaged and stored for later use when the need for energy outstrips available nutrient supply. In mammals, excess lipid intracellularly is stored in structures, most commonly referred to as lipid droplets (LDs). LDs consist of a core of neutral lipids, surrounded by a monolayer of phospholipids with attached or embedded proteins. The LDs proteome contains structural proteins (e.g. proteins of the perilipin family), lipid-synthesis enzymes, lipases and membrane-trafficking proteins.[48–50] Under normal or fasting conditions, healthy hepatocytes will metabolize and degrade LDs by several mechanism that include the action of a series of associated lipases that directly metabolize stored lipids. Members of the perilipin (PAT) family of LD-associated proteins, including perilipin, adipophilin (ADRP) and TIP 47. These proteins regulate TG accumulation, either through promotion of processes that enhance TG synthesis and packing in LDs or by controlling TG lipolysis.[51–53] ADRP has been shown to be one of the major LD-associated proteins that can act to attenuate lipolysis.[53] In animal studies, mice lacking ADRP have been shown to be protected against fatty liver disease[54,55] and that ethanol feeding in rats is found to enhance ADRP expression in association with hepatosteatosis.[56] In our studies, hepatic cells treated with alcohol have showed an elevated LD accumulation and expression of the LD-associated protein, adipophilin.[46] Considered together, these reports underline that the ethanol-amplified LD accumulation in hepatic cells involves impairments in the LD metabolism as a consequence of altered LD associated proteins.
Conclusion
Alcoholic liver disease is associated with a state of hepatic fatty acid overload. In this review, we have summarized the accepted pathways for alcohol-induced hepatic steatosis. In summary, ethanol metabolism leads to excessive generation of reducing equivalents, thereby inhibiting fatty acid oxidation. Recent studies indicate that additional effects of ethanol impair fat oxidation as well as stimulate lipogenesis. The effects of ethanol on lipid metabolism result from inhibition of PPAR-α and stimulation of SREBP-1, resulting in metabolic remodeling of the liver toward a fat-storing, rather than fat oxidizing organ. These effects may, in turn, result from effects of ethanol on AMPK and adiponectin. Inhibition of AMPK by ethanol feeding results in an increase in SREBP-1 activity. As a result, target genes for SREBP-1 are up-regulated, contributing to an increased hepatic lipid synthesis and AMPK inhibition also results in decreased fatty acid oxidation. AMPK may also affect the activity of PPAR-α. Reduced PPAR-α activity will lead to reduced capacity for fatty acid oxidation. In addition to fatty acid synthesis and oxidation, ethanol also alters the LDs (TG storage form) metabolism in hepatocytes and inhibits the VLDL secretion from liver. All these reports help us to understand the intricacies of ethanol-induced fatty liver.
Acknowledgments
The authors acknowledge the support provided by the Department of Veterans’ Affairs, and the NIH (5RC1 AA019032).
Footnotes
Source of Support: Department of Veteransߣ Affairs, and the NIH (5RC1 AA019032)
Conflict of Interest: None declared.
References
- 1.World Health Organization. The world health report -2002. Reducing risks, promoting healthy life. 2002. Available from: http://www.who.int/whr/2002/en/ [DOI] [PubMed]
- 2.Sozio M, Crabb DW. Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295:E10–6. doi: 10.1152/ajpendo.00011.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.French SW. Ethanol and hepatocellular injury. Clin Lab Med. 1996;16:289–306. [PubMed] [Google Scholar]
- 4.Zhou Z, Wang L, Song Z, Lambert JC, McClain CJ, Kang YJ. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol. 2003;163:1137–46. doi: 10.1016/s0002-9440(10)63473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crabb DW, Galli A, Fischer M, You M. Molecular mechanisms of alcohol fatty liver: Role of peroxisome proliferator-activated receptor a. Alcohol. 2004;34:35–8. doi: 10.1016/j.alcohol.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Bataller R, Rombouts K, Altamirano J, Marra F. Fibrosis in alcoholic and nonalcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 2011;25:231–44. doi: 10.1016/j.bpg.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest. 2004;114:147–52. doi: 10.1172/JCI22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yip WW, Burt AD. Alcoholic liver disease. Semin Diagn Pathol. 2006;23:149–60. doi: 10.1053/j.semdp.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Fridman S. Stellate cell activation in alcoholic fibrosis-an overview. Alcohol Clin Exp Res. 1999;23:904–10. [PubMed] [Google Scholar]
- 10.Gramenzi A, Caputo F, BisellI M, Kuria F, Loggi E, Andreone P, et al. Review article: Alcoholic liver disease-pathophysiological aspects and risk factors. Aliment Pharmacol Ther. 2006;24:1151–61. doi: 10.1111/j.1365-2036.2006.03110.x. [DOI] [PubMed] [Google Scholar]
- 11.Savolainen VT, Liesto K, Mannikko A, Penttila A, Karhunen PJ. Alcohol consumption and alcoholic liver disease: Evidence of a threshold level of effects of ethanol. Alcohol Clin Exp Res. 1993;17:1112–7. doi: 10.1111/j.1530-0277.1993.tb05673.x. [DOI] [PubMed] [Google Scholar]
- 12.Lelbach WK. Cirrhosis in the alcoholic and its relation to the volume of alcohol abuse. Ann N Y Acad Sci. 1975;252:85–105. doi: 10.1111/j.1749-6632.1975.tb19146.x. [DOI] [PubMed] [Google Scholar]
- 13.Grant BF, Dufour MC, Harford TC. Epidemiology of alcoholic liver disease. Semin Liver Dis. 1988;8:12–25. doi: 10.1055/s-2008-1040525. [DOI] [PubMed] [Google Scholar]
- 14.Day CP, James OF. Steatohepatitis: A tale of two “hits”? Gastroenterology. 1998;114:842–5. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 15.Lieber CS. New pathway of ethanol metabolism in the liver. Gastroenterology. 1970;59:930–7. [PubMed] [Google Scholar]
- 16.Lieber CS. Hepatic, metabolic and toxic effects of ethanol: 1991 update. Alcohol Clin Exp Res. 1991;15:573–92. doi: 10.1111/j.1530-0277.1991.tb00563.x. [DOI] [PubMed] [Google Scholar]
- 17.Lieber CS, De Carli LM. Hepatotoxicity of ethanol. J Hepatol. 1991;12:394–401. doi: 10.1016/0168-8278(91)90846-4. [DOI] [PubMed] [Google Scholar]
- 18.Grunnet N, Kondrup J. The effect of ethanol on the beta-oxidation of fatty acids. Alcohol Clin Exp Res. 1986;10:64S–8. doi: 10.1111/j.1530-0277.1986.tb05182.x. [DOI] [PubMed] [Google Scholar]
- 19.Min Y, David WC. Recent advances in alcoholic liver disease II.Minireview: Molecular mechanisms of alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol. 2004;287:G1–6. doi: 10.1152/ajpgi.00056.2004. [DOI] [PubMed] [Google Scholar]
- 20.Brown MS, Goldstein JL. A proteolytic pathways that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A. 1999;96:11041–8. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid pathways by activation of steroid regulatory element binding protein (SREBP) J Biol Chem. 2002;277:29342–7. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 22.Reddy JK, Hashimoto T. Peroxisomal β-oxidation and peroxisome proliferator-activated receptor a: An adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- 23.Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10:1–7. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 24.Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. 2003;3:561–72. doi: 10.2174/1566524033479537. [DOI] [PubMed] [Google Scholar]
- 25.Lee GY, Kim NH, Zhao ZS, Cha BS, Kim YS. Peroxisomal proliferator activated receptor- a activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: A key regulation of malonyl-CoA level. Biochem J. 2004;378:983–90. doi: 10.1042/BJ20031565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanji AA, Dannenberg AJ, Jokelainen K, Bass NM. Alcoholic liver injury in the rat is associated with reduced expression of peroxisome proliferator-a (PPAR-a)-regulated genes and is ameliorated by PPAR-a activation. J Pharmacol Exp Ther. 2004;310:417–24. doi: 10.1124/jpet.103.064717. [DOI] [PubMed] [Google Scholar]
- 27.Galli A, Pinaire J, Fischer M, Dorris R, Crabb DW. The transcriptional and DNA binding activity of peroxisome proliferator activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J Biol Chem. 2001;276:68–75. doi: 10.1074/jbc.M008791200. [DOI] [PubMed] [Google Scholar]
- 28.Tuma DJ, Hoffman T, Sorrell MF. The chemistry of acetaldehyde-protein adducts. Alcohol Alcohol Suppl. 1991;1:271–6. [PubMed] [Google Scholar]
- 29.Tuma DJ, Casey CA. Dangerous by products of alcohol breakdown-focus on adducts. Alcohol Res Health. 2003;27:285–90. [PMC free article] [PubMed] [Google Scholar]
- 30.Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84–9. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 31.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–95. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 33.Yamauchi T, Kamon J, Waki H, Imai Y, Shimozawa N, Hioki K, et al. Globular adiponectin protected ob ⁄ ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278:2461–8. doi: 10.1074/jbc.M209033200. [DOI] [PubMed] [Google Scholar]
- 34.Bird GL, Sheron N, Goka AK, Alexander GJ, Williams RS. Increased plasma tumor necrosis factor in severe alcoholic hepatitis. Ann Intern Med. 1990;112:917–20. doi: 10.7326/0003-4819-112-12-917. [DOI] [PubMed] [Google Scholar]
- 35.Jordi C, Ramo B, Pau S, Pablo B, Rosa M, Montserrat M, et al. Hepatic expression of candidate genes in patients with alcoholic hepatitis: Correlation with disease severity. Gastroenterology. 2009;136:687–97. doi: 10.1053/j.gastro.2006.12.036. [DOI] [PubMed] [Google Scholar]
- 36.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–83. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am J Physiol. 1999;277:E1–10. doi: 10.1152/ajpendo.1999.277.1.E1. [DOI] [PubMed] [Google Scholar]
- 38.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–6. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
- 39.Shklyaev S, Aslanidi G, Tennant M, Prima V, Kohlbrenner E, Kroutov V, et al. Sustained peripheral expression of transgene adiponectin offsets the development of diet-induced obesity in rats. Proc Natl Acad Sci U S A. 2003;100:14217–22. doi: 10.1073/pnas.2333912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–48. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liangpunsakul S, Wou SE, Zeng Y, Ross RA, Jayaram HN, Crabb DW. Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am J Physiol Gastrointest Liver Physiol. 2008;295:G1173–81. doi: 10.1152/ajpgi.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Ruiz C, Colell A, Mari M, Albert M, Maria C, Carlos E, et al. Defective TNF-α mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. J Clin Invest. 2003;111:197–208. doi: 10.1172/JCI16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michael WB. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: Possible role in steatosis. Am J Physiol Gastrointest Liver Physiol. 2006;290:194–8. doi: 10.1152/ajpgi.00413.2005. [DOI] [PubMed] [Google Scholar]
- 44.Naoumova RP, Kim KD, Neuwirth C, Niththyananthan S, Rendell NB, Taylor GW, et al. Effect of chronic ethanol feeding on the hepatic secretion of very-low-density lipoproteins. Biochim Biophys Acta. 1988;960:61–6. doi: 10.1016/0005-2760(88)90009-4. [DOI] [PubMed] [Google Scholar]
- 45.Kusum KK, Sandra LT, Brian WW, John JC, Dean JT. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol Cell Biochem. 2009;327:75–8. doi: 10.1007/s11010-009-0044-2. [DOI] [PubMed] [Google Scholar]
- 46.Benita LM, Karuna R, Dean JT, Mark AM, Carol AC. Lipid Droplet Accumulation and Impaired Fat Efflux in Polarized Hepatic Cells: Consequences of Ethanol Metabolism. Int J Hepatol. 2012;2012:ID 978136. doi: 10.1155/2012/978136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wehr H, Rodo M, Lieber CS, Baraona E. Acetaldehyde adducts and autoantibodies against VLDL and LDL in alcoholics. J Lipid Res. 1993;34:1237–44. [PubMed] [Google Scholar]
- 48.Brown DA. Lipid droplets: Proteins floating on a pool of fat. Curr Biol. 2001;11:R446–9. doi: 10.1016/s0960-9822(01)00257-3. [DOI] [PubMed] [Google Scholar]
- 49.Bartz R, Zehmer JK, Zhu M, Chen Y, Serrero G, Zhao Y, et al. Dynamic activity of lipid droplets: Protein phosphorylation and GTP-mediated protein translocation. J Proteome Res. 2007;6:3256–65. doi: 10.1021/pr070158j. [DOI] [PubMed] [Google Scholar]
- 50.Brasaemle DL. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J Lipid Res. 2007;48:2547–59. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Robenek H, Hofnagel O, Buers I, Robenek MJ, Troyer D, Severs NJ. Adipophilin-enriched domains in the ER membrane are sites of lipid droplet biogenesis. J Cell Sci. 2006;119:4215–24. doi: 10.1242/jcs.03191. [DOI] [PubMed] [Google Scholar]
- 52.Wolins NE, Brasaemle DL, Bickel PE. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett. 2006;580:5484–91. doi: 10.1016/j.febslet.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 53.Listenberger LL, Ostermeyer-Fay AG, Goldberg EB, Brown WJ, Brown DA. Adipocyte differentiation-related protein reduces lipid droplet association of adipose triglyceride lipase and slows triglycerol turnover. J Lipid Res. 2007;48:2751–61. doi: 10.1194/jlr.M700359-JLR200. [DOI] [PubMed] [Google Scholar]
- 54.Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, et al. absence of perilipin results in leanness and reverse obesity in Lepr(db/db) mice. Nat Genet. 2000;26:474–9. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- 55.Tansy JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, et al. Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci U S A. 2001;98:6494–9. doi: 10.1073/pnas.101042998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mak KM, Ren C, Ponomarenko A, Cao Q, Lieber CS. Adipose differentiation-related protein is a reliable lipid droplet marker in alcoholic fatty liver of rats. Alcohol Clin Exp Res. 2008;32:683–9. doi: 10.1111/j.1530-0277.2008.00624.x. [DOI] [PubMed] [Google Scholar]