

Abstract
Summary: Iterative similarity searches with PSI-BLAST position-specific score matrices (PSSMs) find many more homologs than single searches, but PSSMs can be contaminated when homologous alignments are extended into unrelated protein domains—homologous over-extension (HOE). PSI-Search combines an optimal Smith–Waterman local alignment sequence search, using SSEARCH, with the PSI-BLAST profile construction strategy. An optional sequence boundary-masking procedure, which prevents alignments from being extended after they are initially included, can reduce HOE errors in the PSSM profile. Preventing HOE improves selectivity for both PSI-BLAST and PSI-Search, but PSI-Search has ~4-fold better selectivity than PSI-BLAST and similar sensitivity at 50% and 60% family coverage. PSI-Search is also produces 2- for 4-fold fewer false-positives than JackHMMER, but is ~5% less sensitive.
Availability and implementation: PSI-Search is available from the authors as a standalone implementation written in Perl for Linux-compatible platforms. It is also available through a web interface (www.ebi.ac.uk/Tools/sss/psisearch) and SOAP and REST Web Services (www.ebi.ac.uk/Tools/webservices).
Contact: pearson@virginia.edu; rodrigo.lopez@ebi.ac.uk
1 INTRODUCTION
PSI-BLAST (Altschul et al., 1997) uses an iterative strategy to construct a protein profile, in the form of a position-specific score matrix (PSSM), which dramatically improves homology detection in diverse protein families. Improved versions of PSI-BLAST have more accurate statistics and more sensitive consensus profiles (Agrawal et al., 2009; Altschul et al., 2005, 2009; Bhadra et al., 2006; Li et al., 2011; Przybylski and Rost, 2008; Stojmirović et al., 2008), but the most common cause of PSI-BLAST errors is contamination of the PSSM by extension of an homologous domain into a non-homologous region (homologous over-extension, HOE) (Gonzalez and Pearson, 2010a). Even searches with a single well-defined domain do not guarantee uncontaminated profiles (Kim et al., 2010). Some HOE errors can be reduced by ‘profile cleaning’; HangOut (Kim et al., 2010) focuses on long insertions, but requires insertion boundaries to be specified by the user, thus assuming a priori knowledge of the domain structure of the query protein.
Here we present PSI-Search, an iterated profile search application for identifying distantly related protein sequences. PSI-Search is similar to PSI-BLAST, but substitutes a rigorous Smith–Waterman local alignment (Smith and Waterman, 1981) search strategy (SSEARCH, Pearson, 1991) to produce optimal local alignment scores from the profile PSSM. PSI-Search includes an optional alignment boundary-masking procedure that reduces HOE errors in the PSSM profile. SCANPS (Walsh et al., 2008) implements a similar iterative search strategy using Smith–Waterman alignments; however, it does not currently scale to large protein databases and does not include boundary masking.
2 METHODS
In PSI-Search, library searches are performed with ssearch, selected hit sequences from the result are processed with an automated sequence boundary-masking procedure, and PSSM profiles are built using blastpgp. The PSI-Search iteration workflow (Fig. 1a) iterates through search and alignment/PSSM construction steps:
The initial iteration is a normal ssearch run with a sequence input.
During the second iteration, aligned sequences with statistically significant scores from the previous search are retrieved using fastacmd; details of the alignment boundaries are stored; sequence regions outside the boundaries are masked with ‘X’s to remove potential HOE regions; masked sequences are formatted into BLAST indexes using formatdb with an additional 10 000 random protein sequences created by makeprotseq (Rice et al., 2000); and a PSSM checkpoint constructed with a blastpgp search; finally ssearch is run with the input sequence, using the generated PSSM, to complete the second iteration and output alignments.
Further iterations repeat Step (2). To avoid HOEs, PSI-Search always uses the alignment boundary information from the first significant alignment in which a library sequence appears. Thus, if the first significant alignment with a library sequence aligns residues 25–125 at iteration i, later alignment boundaries at iteration i+1 and beyond are ignored; only the initially aligned region (25–125) is used to form the PSSM.
Fig. 1.
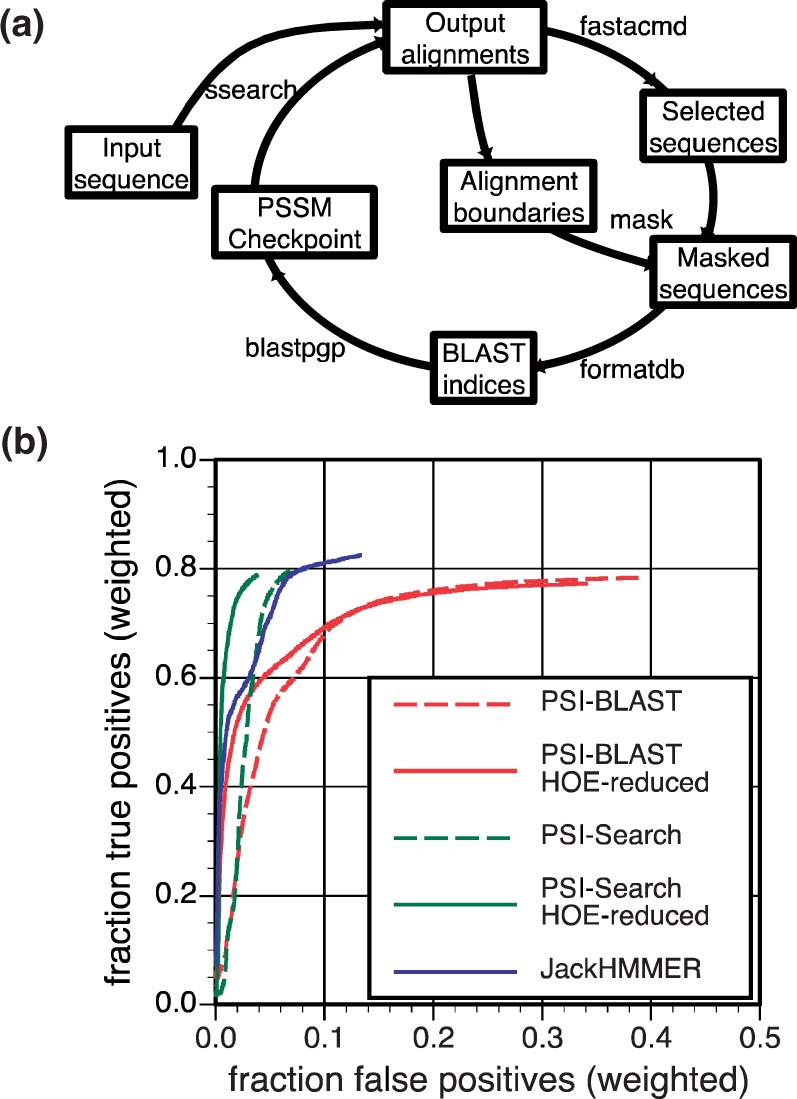
(a) HOE-reduced PSI-Search iteration workflow. (b) Fraction of true-positives versus false-positives found by PSI-BLAST, PSI-BLAST HOE-reduced, PSI-Search, PSI-Search HOE-reduced, and JackHMMER. Weighted true-positives and false-positives are calculated as 1/500∑5001 tpf (or fpf)/totalf where tpf (or fpf) is the number of true positives (or false positives) at iteration 5 and totalf is the total number of homologs for query f in the RefProtDom benchmark database. Alignments containing HOEs with >50% of the alignment outside the homologous boundary are counted as both true and false positives
3 RESULTS
Five iterative search strategies—PSI-BLAST (standard and HOE-reduced), PSI-Search (standard and HOE-reduced) and JackHMMER (Eddy, 2011)—were evaluated on the RefProtDom (Gonzalez and Pearson, 2010b) benchmark queries (500 sampled domain-embedded sequences) against the RefProtDom benchmark database using an E-value threshold of 0.001. JackHMMER is another iterative search tool that uses Hidden Markov Models (HMMs) (Johnson et al., 2010) rather than a PSSM. The output alignments from the fifth iteration were classified into true positives (TPs) and false positives (FPs, Fig. 1b). At 50% family coverage, PSI-Search reduces the weighted fraction of errors from 4.5% (PSI-BLAST) to 2.9% (PSI-Search). Reducing HOE improves sensitivity even more, to 1.7% for HOE-reduced PSI-BLAST and 0.5% for HOE-reduced PSI-Search. At 50% coverage, JackHMMER performs very well using its statistical alignment envelope, producing only 1% weighted FPs, but its selectivity is worse than PSI-Search or HOE-reduced PSI-Search at 60% and 75% coverage. Overall, HOE-reduced PSI-Search is 9-fold more selective than PSI-BLAST. At the end of iteration 5, 78.3, 79.5, 77.3, 78.8 and 82.5% of weighted homologs are found by PSI-BLAST, PSI-Search, HOE-reduced PSI-BLAST, HOE-reduced PSI-Search and JackHMMER respectively. Thus, (i) HOE-reduction greatly improves search selectivity with a small cost in sensitivity in both PSI-BLAST and PSI-Search; (ii) Both PSI-Search and JackHMMER are more sensitive and selective than PSI-BLAST; (iii) HOE-reduced PSI-Search is more selective, but slightly less sensitive, than JackHMMER. JackHMMER is the most sensitive tool, but HOE-reduced PSI-Search is the most selective iterative tool.
ACKNOWLEDGEMENTS
Funding: This research was supported by the National Library of Medicine (NIH grant LM04969 to W.R.P.); European Molecular Biology Laboratory; and European Commission Research Infrastructures of the FP7 [grant agreement number 226073 SLING (Integrating Activity)].
Conflict of Interest: none declared.
REFERENCES
- Agrawal A., Huang X. PSIBLAST_PairwiseStatSig: reordering PSI-BLAST hits using pairwise statistical significance. Bioinformatics. 2009;25:1082–1083. doi: 10.1093/bioinformatics/btp089. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., et al. PSI-BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res. 2009;37:815–824. doi: 10.1093/nar/gkn981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra R., et al. Cascade PSI-BLAST web server: a remote homology search tool for relating protein domains. Nucleic Acids Res. 2006;34:W143–W146. doi: 10.1093/nar/gkl157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M.W., Pearson W.R. Homologous over-extension: a challenge for iterative similarity searches. Nucleic Acids Res. 2010a;38:2177–2189. doi: 10.1093/nar/gkp1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez M.W., Pearson W.R. RefProtDom: a protein database with improved domain boundaries and homology relationships. Bioinformatics. 2010b;26:2361–2362. doi: 10.1093/bioinformatics/btq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L.S., et al. Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics. 2010;11:431. doi: 10.1186/1471-2105-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.H., et al. HangOut: generating clean PSI-BLAST profiles for domains with long insertions. Bioinformatics. 2010;26:1564–1565. doi: 10.1093/bioinformatics/btq208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., et al. A performance enhanced PSI-BLAST based on hybrid alignment. Bioinformatics. 2011;27:31–37. doi: 10.1093/bioinformatics/btq621. [DOI] [PubMed] [Google Scholar]
- Pearson W.R. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith–Waterman and FASTA algorithms. Genomics. 1991;11:635–650. doi: 10.1016/0888-7543(91)90071-l. [DOI] [PubMed] [Google Scholar]
- Przybylski D., Rost B. Powerful fusion: PSI-BLAST and consensus sequences. Bioinformatics. 2008;24:1987–1993. doi: 10.1093/bioinformatics/btn384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., et al. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Stojmirović A., et al. The effectiveness of position- and composition-specific gap costs for protein similarity searches. Bioinformatics. 2008;24:i15–i23. doi: 10.1093/bioinformatics/btn171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T.P., et al. SCANPS: a web server for iterative protein sequence database searching by dynamic programing, with display in a hierarchical SCOP browser. Nucleic Acids Res. 2008;36:W25–W29. doi: 10.1093/nar/gkn320. [DOI] [PMC free article] [PubMed] [Google Scholar]