

Abstract
An orthogonally protected disaccharide (GlcN(α1→4)Glc) with a β-linked 2′-aminoethyl linker was used to generate a series of sulfated derivatives (sulfoforms), with a 6-O-sulfate on the glucose residue and one or more sulfate esters on the terminal glucosamine. Deprotection and sulfonation steps were performed in solution and in variable order, with isolated yields of 36–54% (85–90% per operation) after HPLC purification. The modular deprotection–sulfonation sequences can be performed with efficient recovery of the polysulfate products, and avoids complications associated with heterogeneous reactivity in solid-phase synthesis.
1. Introduction
Proteoglycans and sulfomucins provide a number of protective and recognition functions, the latter being involved in cell adhesion, migration, proliferation, and infection.1,2 Heparan sulfate (HS) proteoglycans and other members of this diverse family of cell-surface carbohydrates are implicated in the recruitment of growth factors and chemokines known to promote or inhibit angiogenesis,3,4 and in the attraction of leukocytes that regulate the assembly and degradation of the extracellular matrix.5,6 Many studies have shown that the specificity of HS-binding proteins is defined in large part by their affinity to specific sulfate patterns (sulfoforms) encoded within proteoglycans, 7, 8 and the identification of such structures can be applied toward the development of new anti-inflammatory agents and immunotherapies based on glycan recognition.9,10 Even relatively simple sulfoforms can exhibit significant influence over protein recognition and signaling pathways, 11, 12 but progress in the structure–activity relationships of HS-like compounds is hampered by the difficulty of obtaining well-defined sulfoforms in sufficient quantities for further study.
In order to create libraries of sulfoforms with structures similar to those found in cell-surface glycans, we have investigated the modular deblocking and sulfonation of orthogonally protected carbohydrate derivatives. This approach enables us to generate both natural and unnatural (or unidentified) sulfoforms, as opposed to the more conventional strategy of developing precursors with pre-designated sites for sulfonation. A set of six orthogonal protecting groups for five hydroxyls and the C2 amine of a heparan disaccharide was previously identified and validated, and their cleavage was also shown to be compatible with neighboring sulfate esters.13 To obtain fully deprotected sulfoforms, we have conducted modular deblocking–sulfonation conditions on orthogonally protected glucosamines (GlcNs) immobilized on tritylated polystyrene (PS) resins.14,15 The solid-phase method can generate entire sets of mono- and disaccharide sulfoforms from a common precursor with up to three sulfate esters per GlcN, followed by mild cleavage and ion-exchange conditions to yield the final sulfoforms as sodium salts. We recently optimized this procedure to generate free GlcN sulfoforms in 8 operations or less with overall yields ranging from 41% (average yield per step: 90%) to 76% (average yield per step: >96%).15 This strategy can also produce sulfoforms with 2-aminoethyl linkers, to enable their conjugation onto activated substrates or for the preparation of neoglycolipids or neoglycoproteins.
The solid-phase method of generating sulfoforms does have some limitations, however. The mild condition for cleaving the aminoethyl group from the trityl–PS resin (30% hexafluoroisopropanol in CH2Cl2) precludes the use of stronger acids in the orthogonal deprotection scheme. Furthermore, the efficiency of sulfonation can vary depending on the number of sulfate esters per glycan: as the charge density increases the environment within the resin becomes heterogeneous, encouraging the formation of ionic bridges within a nonpolar environment. This can have adverse effects on resin swelling and raise the barrier to chemical diffusion and exchange. Resins are also mechanically fragile and may be less well suited for reactions involving large temperature increases (due to thermal expansion of the resin), or aggressive chemical reagents that may cause polymer degradation.
In light of these factors, we considered whether modular deblocking–sulfonation sequences might generate fully deprotected sulfoforms in solution with comparable yields and purity as those obtained using solid-phase methods. Working with charged intermediates in solution can be daunting: Most synthetic routes to sulfated oligosaccharides are designed so that sulfonation is the last step, which minimizes the number of operations needed to isolate these highly polar products. If the order of deprotection and sulfonation is variable, then some charged intermediates will be subjected to multiple processing steps. On the other hand, solution-based methods are clearly compatible with the formation of multiple sulfate esters, so long as the products are sufficiently stable to subsequent deprotection steps.
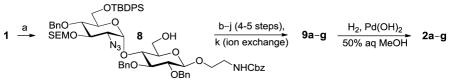
In this article we describe seven sulfoforms generated from a single, orthogonally protected disaccharide with a β-aminoethyl linker (Figure 1), using modular deblocking–sulfonation sequences performed in solution. We focus on derivatives with a 6-O-sulfate ester on the reducing-end glucoside and variable sulfate patterns on the terminal glucosamine, with the latter representing the structural diversity of HS and sulfomucins. Low molecular-weight disaccharide sulfoforms (unnatural derivatives as well as natural) have been found to exhibit significant binding affinity for mucin- and HS-binding proteins such as L-selectin16,17 and FGF-2.11 We compare the efficiency of generating sulfoforms using solution- and solid-phase methods, and the relative benefits of reaction optimization in solution versus the expedient purification of synthetic intermediates immobilized on resins.
Figure 1.

GlcN(α1→4)Glc sulfoforms with 2-aminoethyl linker (2a–g), generated from an orthogonally protected disaccharide (1). R1,R2 = H or SO3−; R3 = Ac or SO3− (see Table 1).
2. Results and Discussion
2.1 Synthesis of orthogonally protected GlcN(α1→4)Glc disaccharide
2′-Azidoethyl β-glucoside tetraacetate 3, which was prepared according to literature procedures,15,18 was converted into glycosyl acceptor 4 in 4 steps and 61% overall yield. The azide was then converted into carboxybenzyl (Cbz) carbamate 5 in 92% yield (Scheme 1). For the reductive cleavage of the 4,6-anisylidene acetal using BH3/Bu2BOTf, we note that the regioselectivity is temperature-dependent, and that performing the reaction at 78 °C yields exclusively the 6-O-PMB ether.19 Orthogonally protected 2-azido-2-deoxyglucose 6, which was also prepared according to literature procedures,14,15 was converted into 3-O-SEM ether 7 in 91% yield, then coupled with 5 using N-iodosuccinimide (NIS)/TfOH activation to yield α-1,4-linked disaccharide 1 as the sole product in 68% yield. We also examined activation conditions using benzenesulfinyl piperidine (BSP)/Tf2O13,20 and phenylsulfenyl chloride (PhSCl)/AgOTf,21 but these were found to be less effective than NIS/TfOH activation.
Scheme 1.

Synthesis of orthogonally protected GlcN(α1→4)Glc derivative 1
Reaction conditions: (a) (i) NaOMe, MeOH, rt; (ii) p-MeO(C6H4)CH(OMe)2, CSA, THF, reflux; (iii) BnBr, NaH, DMF, 0 °C to rt (62% over 3 steps). (b) BH3, Bu2BOTf, THF, −78°C (99%). (c) (i) Bu3P, CH2Cl2, then 1:1 CH2Cl2:H2O, rt; (ii) Cbz-Cl, NaHCO3, 5:1 THF:H2O, 0 °C to rt (92% over 2 steps); (d) (i) NaOMe, MeOH, rt; (ii) SEM-Cl, iPr2NEt, TBAI, CH2Cl2, rt (91% over 2 steps). (e) NIS, TfOH, 2:1 Et2O:(CH2Cl)2, −30°C (68%). Selected abbreviations: Cbz = carboxybenzyl; PMB = p-methoxybenzyl; SEM = 2-(trimethylsilyl)-ethoxymethyl; TBDPS = tert-butyldiphenylsilyl; Tol = p-tolyl.
2.2 Generation of GlcN sulfoforms
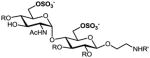
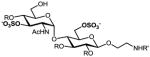
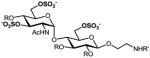
We have previously developed deblocking conditions for the azide, PMB, SEM, and TBDPS groups that are fully orthogonal and also compatible with sulfate esters.13 In this study, all derivatives have a sulfate ester at the C6 position, so the first step is to remove the PMB ether from 1 using ceric ammonium nitrate (CAN) in aqueous CH3CN at 0 °C to afford C6 alcohol 8 in 88% yield (step a). To obtain variable sulfate patterns on the 1,4-α-linked GlcN moiety, the following operations were performed in variable order: (i) TBDPS deprotection at the C6′ position using tetrabutylammonium fluoride (TBAF, step b or c), (ii) conversion of the azide at the C2′ position to –NHAc (step d)22 or a free amine (step i),23 (iii) removal of the SEM ether at the C3′ position using MgBr2 and CH3NO2 (step g or h),24 and (iv) sulfonation of the free hydroxyls and amines (step e, f, or j). These are followed by ion-exchange and HPLC chromatography to obtain partially benzylated sulfoforms 9a–g as sodium salts (step k). Finally, global deprotection of the benzyl (Bn) and Cbz groups was performed under standard hydrogenation conditions to obtain aminoethyl sulfoforms 2a–g in high yields. The sequence of operations and isolated yields for each sulfoform are summarized in Table 1.
Table 1.
Generation of partially and fully deprotected GlcN(α1→4)Glc sulfoforms
 | ||
|---|---|---|
| sulfoforma | 9 (yield from 1, ops)b | 2 (yield from 9)c |
 a |
52%, 6 ops (a, b, d, e, h, k) | 87% |
 b |
39%, 6 ops (a, g, d, e, c, k) | 83% |
 c |
36%, 7 ops (a, e, h, c, i, j, k) | 84% |
 d |
48%, 6 ops (a, b, g, d, f, k) | 97% |
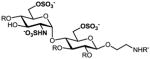 e |
42%, 6 ops (a, b, i, f, h, k) | 98% |
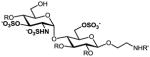 f |
40%, 6 ops (a, g, i, f, c, k) | 97% |
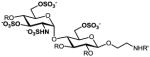 g |
54%, 6 ops (a, b, g, i, f, k) | 99% |
9a–g: R=Bn, R′=Cbz; 2a–g: R, R′=H. All products are isolated as sodium salts.
Isolated yield after HPLC purification. Each step was conducted at rt unless otherwise noted. Reagents and conditions: (a) CAN (3 equiv), 90% aq CH3CN, 0 °C, 10 h; (b) TBAF (6 equiv), THF, 6 h; (c) TBAF (8–10 equiv), 1:1 THF:DMF, 24–40 h; (d) 1:5 AcSH:Py, 48 h; (e) SO3·Me3N (20–30 equiv), DMF, 55 °C, 12 h; (f) SO3·Py (20–30 equiv), 1:5 Et3N:Py, 70 °C, microwave, 1 h; (g) MgBr2·Et2O (10 equiv.), CH3NO2 (20 equiv), Et2O, 10 h; (h) MgBr2·Et2O (10 equiv.), 1:2 CH3NO2:Et2O, 24–48 h; (i) HS(CH2)3SH (35 equiv), Et3N (35 equiv), MeOH, 24 h; (j) SO3·Py (10 equiv), 5:1 H2O:DMF, pH 9.5, 8 h; (k) Na+ ion exchange chromatography.
10% Pd(OH)2, H2 (1 atm), 1:1 MeOH:H2O, 20 h.
With respect to TBDPS and SEM deprotections, there exist significant differences in deblocking conditions when performed before or after sulfonation. Deprotections prior to sulfonation (steps b and g) took less time (6–10 hours) than those performed after sulfonation (steps c and h: 1–2 days); the latter also required more polar solvents. TBDPS deprotections after sulfonation using TBAF were also somewhat lower yielding, possibly due to complications caused by counterion exchange with nearby sulfate esters.
With respect to the efficiency of sulfonation, we observed this reaction to become increasingly sluggish when attempting to install several sulfate esters under thermal conditions. We thus investigated conditions involving microwave-assisted heating, which has been used to generate carbohydrates with multiple sulfate esters.25 A systematic variation in reaction solvent, time, and temperature revealed that microwave-assisted sulfonation is indeed more effective, but also highly temperature-sensitive. Reactions could be performed at 70 °C with minimal decomposition and generation of by-products, but reactions over 80 °C led to the rapid formation of a black tar. Sulfonation was most efficient when performed in pyridine buffered with Et3N, which also enabled simultaneous N- and O-sulfate generation (2e–g). In comparison, the synthesis of 2′,6-di-N,O-sulfate (2c) under conventional thermal conditions required separate sulfonation steps.
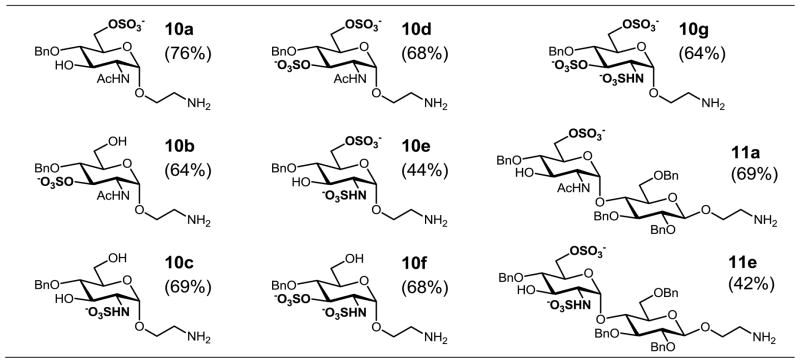
It is worthwhile to compare the synthetic efficiency of the modular deprotection–sulfonation sequence in solution with that performed on resin supports. We recently reported the solid-phase synthesis of a similar series of α-glucosamine sulfoforms (mono- and disaccharides 10 and 11), with overall yields ranging from 42% to 76% over 7–8 operations (Table 2).15 These yields are higher on average than those reported in Table 1, but not uniformly so; for example, the solid-phase syntheses of 2,6-di-N,O-sulfate derivatives 10e and 11e are comparable to the solution synthesis of the 2′,6′,6-tri-N,O,O-sulfate derivative 9e. In addition, we have encountered several practical limits with solid-phase methodologies. These limits included scalability and compound loading, the range of solvents that permit appreciable resin swelling, and the long reaction times required for the completion of each step, particularly sulfonation. We considered using microwave-assisted heating to increase the efficiency of solid-phase sulfonation; however, attempts to do so with SO3·Me3N in DMF at 70 °C resulted in extensive degradation of the supporting resin.
Table 2.
Mono- and disaccharide α-GlcN sulfoforms prepared by solid-phase synthesis15 (overall yields)
 |
In closing, we find that orthogonal deprotection–sulfonation sequences can be performed in solution without a serious attrition in synthetic efficiency, relative to that performed by solid-phase synthesis. This process can produce aminoethyl-linked disaccharide sulfoforms in good overall yield, for subsequent conjugation onto substrates for affinity screening against heparin-binding proteins and pathogens.
3. Experimental details
3.1 General methods
All starting materials and reagents were obtained from commercial sources and used as received unless otherwise noted. All solvents were freshly distilled prior to use. IR spectra were acquired from NaCl plate, using a Thermo-Nicolet Nexus 670 FT-IR spectrometer. 1H and 13C NMR chemical shifts were referenced to the solvent used (δ 7.27 and 77.00 for CDCl3, δ 3.31 and 49.15 for CD3OD, and δ 4.80 for D2O at 295 K). Mass spectra were acquired using either a Hewlett-Packard 5989B or a Finnigan 40000 mass spectrometer. Optical rotations were measured by polarimetry at rt. Silica gel chromatography was performed with ICN SiliTech 32–63D. Ion-exchange chromatography was performed by dissolving sulfoforms in 50% aq MeOH (up to 100 mg/mL) and passing them through a Dowex Marathon MSC column (14 × 1.5 cm), which was activated with 0.1 M NaOH and washed with water prior to use. HPLC purifications was performed by dissolving sulfoforms in CH3CN (up to 20 mg/mL) and injecting them onto a C18 reverse-phase column (250 × 10 mm, 4 μm beads, 80 Å pores) with UV detection at 214 nm, using a binary solvent gradient (up to 40% aq CH3CN) with a flow rate of 5 mL/min.
3.2 2′-Azidoethyl 2,3-di-O-benzyl-6-O-p-methoxybenzyl-β-D-glucopyranoside (4)
2′-azidoethyl tetra-O-acetyl-β-D-glucopyranoside 315,18 (14.2 g, 34.2 mmol) was dissolved in anhydrous MeOH (200 mL) and treated at rt with 1 M NaOMe solution in MeOH (10.3 mL). The mixture was stirred at rt for 5 h, neutralized with acid-washed Dowex 50W-X8 ion-exchange resin, filtered, concentrated, and dried under reduced pressure. The crude tetraol was redissolved in THF (80 mL) and treated with p-methoxybenzaldehyde dimethyl acetal (9.8 mL, 57.4 mmol) and camphorsulfonic acid (CSA, 476 mg, 2.1 mmol), then heated to reflux for 10 h. The reaction mixture was quenched with saturated aq NaHCO3 (50 mL), extracted with CH2Cl2 (3 × 150 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (50% EtOAc in hexanes) to afford the 4,6-anisylidene acetal as an amorphous white solid (9.3 g). The resulting 2,3-diol was redissolved in anhydrous DMF (160 mL) and treated at 0 °C with BnBr (5.8 mL, 49.1 mmol) and NaH (1.47 g, 36.8 mmol). The mixture was warmed to rt over 2 h and stirred for another 10 h, then quenched with saturated aq NH4Cl (100 mL), extracted with CH2Cl2 (3 × 200 mL), washed with brine (50 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (20% EtOAc in hexanes) to afford the 2,3-di-O-benzyl ether as an amorphous white solid (7.7 g, 62% over three steps).
The 4,6-anisylidene acetal (7.6 g, 13.8 mmol) was dissolved in THF (274 mL) and treated at −78 °C with a 1 M solution of BH3 in THF (76.0 mmol) and a 1 M solution of Bu2BOTf in THF (34.8 mL). The mixture was stirred at −78 °C for 10 h, then quenched with Et3N (6.8 mL, 48.6 mmol) and MeOH (200 mL), and warmed slowly to rt over 1 h. The product was extracted with CH2Cl2 (3 × 200 mL), washed with brine (40 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (25% EtOAc in hexanes) to afford 6-O-PMB ether 4 as an amorphous white solid (7.5 g, 99%). 1H NMR (400 MHz, CDCl3): δ 7.35 (m, 12H), 6.89 (d, 2H, J = 8.6 Hz), 4.99 (t, 2H, J = 11.5 Hz), 4.75 (d, 2H, J = 11.3 Hz), 4.52 (m, 2H), 4.46 (d, 1H, J = 7.3 Hz), 4.07 (ddd, 1H, J = 3.8, 5.4, 9.9 Hz), 3.81 (s, 3H), 3.60 (m, 9H), 2.60 (br, 1H); 13C NMR (100 MHz, CDCl3): δ 159.23, 138.50, 138.31, 129.82, 129.35, 128.49, 128.34, 128.07, 127.90, 127.80, 127.67, 113.76, 103.62, 83.85, 81.63, 75.22, 74.77, 74.03, 73.25, 71.39, 69.77, 68.14, 55.21, 50.96; IR (NaCl): 3460, 2916, 2872, 2104, 1612, 1513, 1248, 1064 cm−1; [α]20D = −13.2 (c 1.21, CH2Cl2); HRESI–MS: m/z calcd for C30H35N3NaO7 [M+Na]+: 572.2373; found: 572.2375.
3.3 2′-(N-carboxybenzyl)aminoethyl 2,3-di-O-benzyl-6-O-p-methoxybenzyl-β-D-glucopyranoside (5)
A solution of 2′-azidoethyl glucoside 4 (478 mg, 0.87 mmol) in CH2Cl2 (20 mL) was treated with Bu3P (225 μL, 0.91 mmol), and stirred at rt for 3 h, then treated with H2O (10 mL) and stirred at rt for 1 day. The crude amine was concentrated and dried under reduced pressure with azeotropic distillation with toluene (3 × 10 mL), to afford a yellowish oil. This was redispersed with NaHCO3 (263 mg, 3.10 mmol) in 5:1 THF:H2O (10 mL), then treated at 0 °C with Cbz-Cl (135 μL, 0.96 mmol). The reaction mixture was warmed to rt over 2 h and stirred for another 10 h, then extracted with CH2Cl2 (3 × 50 mL), washed with brine (20 mL), dried over Na2SO4 and concentrated, and purified by silica gel chromatography (33% EtOAc in hexanes) to afford Cbz-protected 2′-aminoethyl glucoside 5 as an amorphous white solid (526 mg, 92% over 2 steps). 1H NMR (400 MHz, CDCl3): δ 7.36 (m, 15H), 7.24 (d, 2H, J = 8.2 Hz), 6.88 (d, 2H, J = 8.2 Hz), 5.62 (br, 1H), 5.11 (s, 2H), 4.96 (d, 1H, J = 11.4 Hz), 4.90 (d, 1H, J = 11.1 Hz),4.78 (t, 2H, J = 13.0 Hz), 4.99 (m, 2H), 4.41 (d, 1H, J = 7.1 Hz), 3.87 (m, 6H), 3.63 (m, 2H), 3.46 (m, 5H), 2.84 (s, 1H); 13C NMR (100 MHz, CDCl3): δ 159.23, 156.53, 138.54, 138.24, 136.56, 129.81, 129.36, 128.49, 128.47, 128.41, 128.11, 128.00, 127.91, 127.80, 127.74, 113.78, 103.83, 84.00, 81.66, 75.23, 74.83, 74.09, 73.16, 71.33, 69.87, 69.61, 66.64, 55.20, 41.42; IR (NaCl): 3360, 2872, 1711, 1513, 1248, 1062 cm−1; [α]20D = −16.7 (c 0.65, CH2Cl2); HRESI MS: m/z calcd for C38H43NNaO9 [M+Na]+: 680.2836; found: 680.2835.
3.4 Thiotolyl 2-azido-4-O-benzyl-6-O-(tert-butyldiphenylsilyl)-2-deoxy-3-O-[2-(trimethyl-silyl)ethoxymethyl]-β-D-glucopyranoside (7)
3-O-Acetyl derivative 6 (1.0 g, 1.47 mmol) was prepared as previously described,15 then dissolved in anhydrous MeOH (60 mL) and treated at rt with 1 M NaOMe solution in MeOH (440 μL). The mixture was stirred for 5 h at rt, neutralized with activated Dowex 50X-W-H+ ion-exchange resin, filtered, concentrated, and dried under reduced pressure. The crude C3 alcohol was redissolved in CH2Cl2 (40 mL) and treated at rt with TBAI (813 mg, 2.20 mmol) and iPr2NEt (2.56 mL, 14.70 mmol), then SEM-Cl (1.82 mL, 10.30 mmol). The mixture was stirred for 48 h at rt, then extracted with CH2Cl2 (2 × 100 mL), washed with brine (40 mL), dried over Na2SO4, then concentrated under reduced pressure. Purification by silica gel chromatography (4.8% EtOAc in hexanes) yielded 3-O-SEM ether 7 as an amorphous white solid (1.03 g, 91% over 2 steps). 1H NMR (400 MHz, CDCl3): δ 7.81 (dd, 2H, J = 1.2, 7.7 Hz), 7.71 (dd, 2H, J = 1.3, 9.1 Hz), 7.55 (d, 2H, J = 8.1 Hz), 7.36 (m, 9H), 7.15 (dd, 2H, J = 3.6, 7.3 Hz), 7.05 (d, 2H, J = 8.0 Hz), 4.98 (d, 1H, J = 6.4 Hz), 4.88 (d, 1H, J = 6.4 Hz), 4.79 (d, 1H, J = 10.8 Hz), 4.78 (d, 1H, J = 8.2 Hz), 4.69 (d, 1H, J = 10.6 Hz), 4.41 (d, 1H, J = 10.1 Hz), 4.01 (dd, 1H, J = 1.4, 11.5 Hz), 3.92 (dd, 1H, J = 3.1, 11.4 Hz), 3.70 (m, 5H), 3.30 (m, 2H), 2.33 (s, 3H), 1.11 (s, 9H), 0.97 (t, 2H, J = 8.3 Hz), 0.00 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 138.50, 137.72, 135.83, 135.59, 134.05, 133.25, 132.73, 129.76, 128.42, 127.78, 127.71, 127.67, 127.37, 96.51, 86.33, 81.65, 79.91, 74.81, 66.40, 64.57, 62.23, 26.84, 21.15, 19.27, 18.08, −1.51; IR (NaCl): 2952, 2857, 2111, 1428, 1248, 1112, 1049 cm−1; [α]20D = −21.8 (c 1.35, CH2Cl2); HRESI-MS: m/z calcd for C43H55N3NaO5SSi2 [M+Na]+: 792.3299; found: 792.3313.
3.5 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-azido-4-O-benzyl-6-O-(tert-butyldiphenyl-silyl)-2-deoxy-3-O-[2-(trimethylsilyl)ethoxymethyl]-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-p-methoxybenzyl-β-D-glucopyranoside (1)
Thioglycoside donor 7 (200 mg, 0.26 mmol) and glucoside acceptor 5 (248 mg, 0.38 mmol) were dissolved in 1:2 (CH2Cl)2:Et2O (6 mL) and treated with freshly activated 4A molecular sieves (800 mg) for 1 h at rt under argon. The mixture was then cooled to −30 °C and treated with NIS (64 mg, 0.29 mmol) and TfOH (3.5 μL, 0.04 mmol). The mixture was stirred for 5 h at −30 °C, then quenched with Et3N (22 μL, 0.16 mmol) and filtered through Celite, which was rinsed with CH2Cl2 (3 × 70 mL). The filtrate was washed with brine (40 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (20% EtOAc in hexanes) to yield orthogonally protected GlcN(α1→4)Glc disaccharide 1 as an amorphous white solid (229 mg, 68%). 1H NMR (400 MHz, CDCl3): δ 7.69 (d, 2H, J = 6.7 Hz), 7.64 (d, 2H, J = 7.0 Hz), 7.30 (m, 26H), 7.00 (d, 2H, J = 8.2 Hz), 6.60 (d, 2H, J = 8.3 Hz), 5.70 (d, 1H, J = 3.7 Hz), 5.60 (br, 1H), 5.04 (m, 5H), 4.89 (m, 5H), 4.72 (dd, 2H, J = 5.7, 11.0 Hz), 4.42 (d, 1H, J = 7.9 Hz), 4.29 (m, 2H), 3.73 (m, 18H), 3.06 (dd, 1H, J = 3.8, 7.9 Hz), 1.07 (s, 9H), 1.01 (t, 2H, J = 8.6 Hz), 0.04 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 158.92, 156.48, 138.43, 138.08, 137.99, 136.54, 135.84, 135.50, 133.55, 132.91, 129.60, 128.95, 128.42, 128.38, 128.11, 128.00, 127.92, 127.67, 127.53, 127.44, 113.53, 103.70, 97.66, 96.47, 84.71, 82.54, 78.11, 74.87, 74.72, 74.56, 74.00, 72.93, 72.18, 70.04, 68.88, 66.60, 66.19, 62.55, 61.86, 55.03, 41.51, 26.83, 19.28, 18.11, −1.51; IR (NaCl): 3032, 2952, 2108, 1724, 1513, 1454, 1360, 1248, 1112, 1040 cm−1; [α]20D = +55.0 (c 1.02, CH2Cl2); HRMALDI–MS: m/z calcd for C73H90N4NaO14Si2 [M+Na]+: 1325.5889; found: 1325.5785.
3.6 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-azido-4-O-benzyl-6-O-(tert-butyldiphenyl-silyl)-2-deoxy-3-O-[2-(trimethylsilyl)ethoxymethyl]-α-D-glucopyranosyl]-2,3-di-O-benzyl-β-D-glucopyranoside (8)
6-O-PMB ether 1 (800 mg, 0.61 mmol) was dissolved in 90% aqueous CH3CN (8 mL), treated at 0 °C with CAN (1 g, 1.84 mmol), then stirred for 10 h. The mixture was quenched with Et3N (342 μL, 2.46 mmol), concentrated, and purified by silica gel chromatography (20% EtOAc in hexanes) to afford C6 alcohol 8 as a colorless oil (630 mg, 87%). 1H NMR (400 MHz, CDCl3): δ 7.79 (dd, 4H, J = 7.1, 8.4 Hz), 7.39 (m, 26H), 5.81 (d, 1H, J = 3.5 Hz), 5.55 (t, 1H, J = 6.0 Hz), 5.16 (m, 4H), 4.99 (m, 3H), 4.92 (d, 1H, J = 10.8 Hz), 4.79 (d, 2H, J = 11.0 Hz), 4.52 (d, 1H, J = 7.7 Hz), 4.12 (t, 1H, J = 8.7 Hz), 3.13 (m, 13H), 3.47 (m, 5H), 3.20 (dd, 1H, J = 3.6, 10.4 Hz), 2.44 (t, 1H, J = 6.0 Hz), 1.18 (s, 9H), 1.10 (t, 3H, J = 8.4 Hz), 0.11 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 156.18, 138.30, 137.90, 137.62, 136.22, 135.68, 135.42, 133.23, 132.73, 129.51, 128.25, 127.98, 127.76, 127.60, 127.44, 127.32, 103.58, 97.82, 96.30, 84.43, 82.43, 78.08, 76.46, 74.79, 74.58, 72.42, 72.32, 69.36, 66.55, 66.06, 62.39, 62.18, 61.67, 41.05, 26.72, 19.13, 17.96, −1.60; IR (NaCl): 2952, 2108, 1717, 1456, 1249, 1113, 1041 cm−1; [α]20D = +53.1 (c 1.02, CH2Cl2); ESI–MS: m/z calcd for C65H82N4NaO13Si2 [M+Na]+: 1205.5; found: 1205.4.
3.7 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-acetamido-4-O-benzyl-2-deoxy-6-O-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9a)
Op. 1: Orthogonally protected disaccharide 8 (70 mg, 0.06 mmol) was dissolved in THF (0.85 mL), then treated with a 1 M solution of TBAF in THF (0.35 mL) and stirred at rt for 6 h. The mixture was concentrated then purified by silica gel chromatography (40% EtOAc in hexanes) to yield the C6′ alcohol as a white solid (49 mg, 87%). Op. 2: The intermediate azide (40 mg, 0.04 mmol) was dissolved in dry pyridine (0.6 mL), then treated with AcSH (121 μL, 1.69 mmol) and stirred at rt for 48 h. The mixture was concentrated and purified by silica gel chromatography (3% MeOH in CHCl3) to yield the C2′ N-acetamide as an amorphous white solid (38 mg, 95%). Op. 3: The intermediate diol (34 mg, 0.04 mmol) was dissolved in dry DMF (1.1 mL), then treated with SO3•Me3N (98 mg, 0.70 mmol) and stirred at 55 °C for 10 h. The reaction mixture was concentrated and purified by silica gel chromatography (5% MeOH in CHCl3 with 2% Et3N) to yield the 6′,6-di-O-sulfate Et3NH salt as a yellow solid (44 mg, 93%). Op. 4: The intermediate SEM ether (20 mg, 0.02 mmol) was dissolved in Et2O (1 mL), then treated with a solution of MgBr2·Et2O (46 mg, 0.18 mmol) in CH3NO2 (0.9 mL) and Et2O (0.8 mL) and stirred at rt for 48 h. The reaction mixture was quenched with saturated NaHCO3 (2 mL), extracted with CH2Cl2 (3 × 2 mL), washed with brine (2 mL), dried over Na2SO4, and concentrated. Op. 5: The crude disulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then concentrated and purified by reverse-phase HPLC to yield the disodium salt as an amorphous white solid (14 mg, 79%). The overall yield of 6′,6-di-O-sulfate 9a (disodium salt) from 1 was 52% over 6 operations. 1H NMR (400 MHz, CD3OD): δ 7.44 (d, 3H, J = 7.4 Hz), 7.27 (m, 17H), 5.44 (d, 1H, J = 3.9 Hz), 4.98 (d, 1H, J = 5.5 Hz), 4.91 (s, 2H), 4.81 (d, 2H, J = 2.1 Hz), 4.56 (t, 2H, J = 10.4 Hz), 4.50 (d, 1H, J = 7.7 Hz), 4.34 (m, 3H), 4.13 (dd, 1H, J = 5.6, 11.2 Hz), 4.04 (dd, 1H, J = 3.7, 10.7 Hz), 3.92 (m, 2H), 3.70 (m, 6H), 3.56 (t, 1H, J = 9.5 Hz), 3.43 (m, 1H), 1.80 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 173.82, 158.86, 139.83, 138.25, 129.87, 129.54, 129.39, 129.21, 129.13, 129.08, 128.88, 128.78, 128.66, 128.50, 128.34, 104.46, 99.22, 85.02, 83.56, 79.49, 77.76, 75.82, 75.72, 75.40, 75.27, 74.49, 73.99, 69.37, 67.86, 67.39, 62.01, 54.77, 41.97, 22.97; [α]20D = +73.6 (c 1.0, MeOH); HRESI–MS: m/z calcd for C45H52N2Na3O19S2 [M+3 Na]+: 1057.2299; found: 1057.2282.
3.8 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-acetamido-4-O-benzyl-2-deoxy-3-O-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9b)
Op. 1: Orthogonally protected disaccharide 8 (85 mg, 0.07 mmol) was dissolved in Et2O (3 mL), then treated with a solution of MgBr2·Et2O (185 mg, 0.72 mmol) in CH3NO2 (3 mL) and Et2O (3 mL) and stirred at rt for 48 h. The reaction mixture was quenched with saturated aq NaHCO3 (10 mL), extracted with EtOAc (3 × 15 mL), washed with brine (10 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (40% EtOAc in hexanes) to yield the C3′ alcohol as a white solid (70 mg, 93%). Op. 2: The intermediate azide (40 mg, 0.04 mmol) was dissolved in dry pyridine (0.58 mL), then treated with AcSH (0.11 mL) and stirred at rt for 48 h. The mixture was concentrated and purified by silica gel chromatography (3% MeOH in CHCl3) to yield the C2′ N-acetamide as an amorphous white solid (38 mg, 93%). Op. 3: The intermediate diol (38 mg, 0.04 mmol) was dissolved in dry DMF (1.2 mL), then treated with SO3·Me3N (99 mg, 0.71 mmol) and stirred at 55 °C for 10 h. The reaction mixture was concentrated and purified by silica gel chromatography (3% MeOH in CHCl3 with 2% Et3N) to yield the 3′,6-di-O-sulfate Et3NH salt as a yellow solid (44 mg, 93%). Op. 4: The intermediate TBDPS ether (45 mg, 0.04 mmol) was dissolved in DMF (0.4 mL), then treated with a 1 M solution of TBAF in THF (0.4 mL) for 48 h and concentrated to obtain a yellow oil. Op. 5: The crude disulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then concentrated and purified by reverse-phase HPLC to yield the disodium salt as an amorphous white solid (20 mg, 55%). The overall yield of 3′,6-di-O-sulfate 9b (disodium salt) from 1 was 39% over 6 operations. 1H NMR (400 MHz, CD3OD): δ 7.45 (m, 2H), 7.23 (m, 18H), 5.49 (d, 1H, J = 3.4 Hz), 5.15 (d, 2H, J = 10.3 Hz), 4.99 (d, 2H, J = 7.4 Hz), 4.93 (d, 2H, J = 11.2 Hz), 4.71 (m, 2H), 4.54 (m, 3H), 4.35 (dd, 1H, J = 2.3, 10.7 Hz), 4.23 (dd, 1H, J = 4.7, 10.9 Hz), 4.07 (dd, 1H, J = 3.5, 10.8 Hz), 3.94 (m, 1H), 3.95 (m, 3H), 3.76 (t, 1H, J = 8.6 Hz), 3.69 (dd, 3H, J = 4.1, 12.1 Hz), 3.59 (t, 1H, J = 9.4 Hz), 3.42 (t, 1H, J = 8.3 Hz), 3.36 (dd, 2H, J = 5.3, 11.0 Hz), 1.76 (s, 3H); 13C NMR (100 MHz, CD3OD): δ 173.40, 158.86, 140.07, 139.84, 139.29, 138.24, 129.39, 129.28, 129.17, 129.07, 128.96, 128.87, 128.77, 128.64, 128.57, 128.39, 104.53, 98.67, 85.17, 83.54, 79.33, 75.86, 75.48, 75.26, 74.48, 74.38, 73.36, 72.00, 69.40, 68.09, 67.58, 67.39, 55.11, 41.95, 22.64; [α]20D = +78.9 (c 1.0, MeOH); HRESI–MS: m/z calcd for C45H52N2Na3O19S2 [M+3 Na]+: 1057.2299; found: 1057.2288.
3.9 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-amino-4-O-benzyl-2-deoxy-2-N-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9c)
Op. 1: Orthogonally protected disaccharide 8 (40 mg, 0.03 mmol) was dissolved in dry DMF (1.13 mL), then treated with SO3·Me3N (98 mg, 0.34 mmol) at 55 °C for 10 h. The mixture was concentrated and purified by silica gel chromatography (2% MeOH in CHCl3 with 2% Et3N) to yield the 6-O-sulfate Et3NH salt as a yellow solid (40 mg, 90%). Op. 2: The intermediate sulfoform (40 mg, 0.03 mmol) was dissolve in Et2O (2 mL), then treated with MgBr2·Et2O (80 mg, 0.30 mol) in CH3NO2 (1.5 mL) and Et2O (1 mL) and stirred at rt for 24 h. The mixture was quenched with saturated NaHCO3 (10 mL), extracted with CH2Cl2 (3 × 15 mL), washed with brine (10 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (5% MeOH in CHCl3 with 2% Et3N) to yield the C3′ alcohol as a yellow solid (40 mg, 90%). Op. 3: The intermediate TBDPS ether (30 mg, 0.02 mmol) was dissolved in DMF (1 mL), then treated with a 1 M solution of TBAF in THF (0.19 mL) and stirred at rt for 24 h. The mixture was concentrated and purified by silica gel chromatography (9% MeOH in CHCl3 with 2% Et3N) to yield the C6′ alcohol as a yellow solid (19 mg, 78%). Op. 4: The intermediate azide (28 mg, 0.03 mmol) was dissolved in MeOH (1 mL), then treated with 1,3-propanedithiol (130 μL, 0.93 mmol) and Et3N (94 μL, 0.93 mmol) and stirred at rt for 24 h. The mixture was concentrated and purified by silica gel chromatography (10% MeOH in CHCl3 with 2% Et3N) to yield the C2′ amine as a yellow solid (25 mg, 93%). Op. 5: The amine (48 mg, 0.56 mmol) was dissolved in DMF (1 mL) and mixed with aq NaOH adjusted to pH 9.5 (5 mL), then treated with SO3·Py (162 mg, 5.60 mmol), stirred at rt for 8 h, and concentrated. Op. 6: The crude di-N,O-sulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then concentrated and purified by reverse-phase HPLC to yield the disodium salt as an amorphous white solid (33 mg, 63%). The overall yield of 2′,6-di-N,O-sulfate 9c (disodium salt) from 1 was 36% over 7 operations. 1H NMR (400 MHz, CD3OD): δ 7.07–7.44 (m, 20H), 5.52 (d, 1H, J = 3.5 Hz), 4.99 (d, 1H, J = 3.9 Hz), 4.95 (d, 1H, J = 4.6 Hz), 4.82–4.93 (m, 2H), 4.64 (d, 1H, J = 11.0 Hz), 4.56 (d, 1H, J = 11.2 Hz), 4.49 (d, 1H, J = 7.5 Hz), 4.38 (dd, 1H, J = 2.6, 10.9 Hz), 4.16 (dd, 1H, J = 5.3, 10.7 Hz), 3.81–3.98 (m, 3H), 3.58–3.80 (m, 7H), 3.42 (m, 2H), 3.32–3.37 (m, 3H); 13C NMR (100 MHz, CD3OD) δ 158.84, 140.23, 139.87, 139.55, 138.25, 129.64, 129.53, 129.40, 129.25, 129.14, 129.03, 128.88, 128.78, 128.50, 128.41, 104.50, 99.48, 83.41, 79.42, 75.74, 75.19, 75.15, 75.04, 74.94, 74.43, 73.28, 69.41, 68.03, 67.40, 62.26, 60.15, 41.97; [α]20D = +53.2 (c 0.2, MeOH); HRESI–MS: m/z calcd for C43H50N2Na3O18S2 [M+3 Na]+: 1015.2193; found: 1057.2209.
3.10 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-acetamido-4-O-benzyl-2-deoxy-3,6-di-O-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9d)
Op. 1: Orthogonally protected disaccharide 8 was treated with TBAF in THF to yield the C6′ alcohol, as described in the synthesis of 9a. Op. 2: The intermediate SEM ether (160 mg, 0.17 mmol) was dissolved in Et2O (7 mL), then treated with a solution of MgBr2·Et2O (437 mg, 1.69 mmol) and CH3NO2 (182 μL, 3.38 mmol) in Et2O (5 mL) and stirred at rt for 10 h. The mixture was quenched with saturated aq NaHCO3 (70 mL), extracted with EtOAc (3 × 100 mL), washed with brine (20 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (66% EtOAc in hexanes) to yield the C3′ alcohol as an amorphous white solid (134 mg, 97%). Op. 3: The intermediate azide (67 mg, 0.08 mmol) was dissolved in pyridine (2 mL), then treated with AcSH (234 μL, 3.29 mmol) and stirred at rt for 40 h. The mixture was concentrated and purified by silica gel chromatography (4.75% MeOH in CH2Cl2) to yield the C2′ N-acetamide as an amorphous white solid (60 mg, 88%). Op. 4: The intermediate amino alcohol (44 mg, 0.05 mmol) was dissolved in 5:1 Py:Et3N (3 mL) and treated with SO3·Py (228 mg, 1.43 mmol), then heated to 70 °C under microwave conditions for 1 h. The mixture was concentrated and purified by silica gel chromatography (9% MeOH in CH2Cl2 with 2% Et3N) to yield the 3′,6′,6-tri-O-sulfate Et3NH salt as a yellow solid. Op. 5: The crude trisulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then concentrated and purified by reverse-phase HPLC to yield the trisodium salt as an amorphous white solid (45 mg, 74%). The overall yield of 3′,6′,6-tri-O-sulfate 9d (trisodium salt) from 1 was 48% over 6 operations. 1H NMR (500 MHz, CD3OD): δ 7.56 (d, 2H, J = 7.2 Hz), 7.16–7.34 (m, 18H), 5.54 (d, 1H, J = 2.0 Hz), 5.12 (d, 1H, J = 9.6 Hz), 4.99 (m, 2H), 4.92 (d, 1H, J = 11.2 Hz), 4.68 (m, 3H), 4.54 (d, 1H, J = 12.0 Hz), 4.52 (d, 1H, J = 8.3 Hz), 4.40 (d, 1H, J = 10.0 Hz), 4.35 (m, 2H), 4.22 (dd, 1H, J = 4.0, 10.6 Hz), 4.11 (dd, 1H, J = 2.4, 8.4 Hz), 3.91–4.02 (m, 2H), 3.85 (t, 1H, J = 8.8 Hz), 3.78 (t, 1H, J = 8.4 Hz), 3.69 (m, 7H), 3.56 (t, 1H, J = 4.4 Hz), 3.42 (t, 1H, J = 7.9 Hz), 3.37 (br, 1H), 1.94 (s, 3H); 13C NMR (125 MHz, CD3OD): δ 173.98, 159.04, 139.99, 139.94, 139.77, 138.40, 130.70, 129.57, 129.40, 129.30, 129.19, 129.07, 128.95, 128.74, 128.51, 104.63, 99.03, 85.21, 83.71, 79.60, 77.58, 76.19, 75.59, 75.28, 75.24, 74.45, 73.59, 72.25, 69.57, 68.05, 67.56, 67.49, 62.32, 55.26, 54.68, 42.12, 23.18; [α]20D = +25.0 (c 0.79, MeOH); ESI–MS: m/z calcd for C45H51N2Na4O22S3 [M+4 Na]+: 1159.2; found: 1159.1.
3.11 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-amino-4-O-benzyl-2-deoxy-2,6-di-N,O-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9e)
Op. 1: Orthogonally protected disaccharide 8 was treated with TBAF in THF to yield the C6′ alcohol, as described in the synthesis of 9a. Op. 2: The intermediate azide (80 mg, 0.09 mmol) was dissolved in MeOH (3 mL), treated with 1,3-propanedithiol (84 μL, 0.85 mmol) and Et3N (118 μL, 0.85 mmol), then stirred at rt for 40 h. The mixture was concentrated and purified by silica gel chromatography (3.25% MeOH in CH2Cl2) to afford the C2 amine as an amorphous white solid (68 mg, 87%). Op. 3: The intermediate amino diol (60 mg, 0.07 mmol) was dissolved in 5:1 Py:Et3N (3 mL) and treated with SO3·Py (281 mg, 1.76 mmol), then heated to 70 °C under microwave conditions for 1 h. The mixture was concentrated and purified by silica gel chromatography (9% MeOH in CH2Cl2 with 2% Et3N) to yield the 2′,6′,6-tri-N,O,O-sulfate Et3NH salt as a light yellow oil. Op. 4: The intermediate SEM ether was dispersed in Et2O (4 mL), treated with a solution of MgBr2·Et2O (169 mg, 0.65 mmol) in CH3NO2 (3 mL, 55.80 mmol) in Et2O (2 mL), then stirred at rt for 40 h. The mixture was concentrated and used directly in next operation. Op. 5: The crude trisulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then purified by reverse-phase HPLC to yield the trisodium salt as an amorphous white solid (46 mg, 64% over 2 steps). The overall yield of 2′,6′,6-tri-N,O,O-sulfate 9e (trisodium salt) from 1 was 42% over 6 operations. 1H NMR (500 MHz, CD3OD): δ 7.45 (d, 2H, J = 7.1 Hz), 7.36 (d, 2H, J = 7.2 Hz), 7.15–7.32 (m, 16H), 5.57 (d, 1H, J = 3.1 Hz), 4.91–5.03 (m, 4H), 4.83 (d, 1H, J = 11.2 Hz), 4.77 (d, 1H, J = 10.5 Hz), 4.58 (d, 1H, J = 13.2 Hz), 4.56 (d, 1H, J = 11.2 Hz), 4.50 (d, 1H, J = 7.3 Hz), 4.27–4.40 (m, 3H), 4.20 (dd, 1H, J = 5.0, 10.8 Hz), 3.84–3.98 (m, 3H), 3.73–3.83 (m, 2H), 3.67 (m, 2H), 3.57 (m, 2H), 3.44 (t, 1H, J = 7.7 Hz), 3.33–3.40 (m, 3H), 1.94 (s, 1H); 13C NMR (125 MHz, CD3OD): δ 158.99, 140.29, 139.99, 139.82, 138.36, 129.62, 129.58, 129.38, 129.29, 129.24, 129.06, 128.94, 128.67, 128.57, 128.50, 104.64, 99.27, 83.78, 83.43, 79.24, 75.76, 75.35, 75.29, 75.00, 74.60, 71.47, 69.59, 68.15, 67.72, 67.55, 60.06, 47.99, 42.10; [α]20D = +33.6 (c 0.93, MeOH); ESI–MS: m/z calcd for C43H49N2Na4O21S3 [M+4 Na]+: 1117.2; found: 1117.0.
3.12 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-amino-4-O-benzyl-2-deoxy-2,3-di-N,O-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9f)
Op. 1: Orthogonally protected disaccharide 8 (158 mg, 0.13 mmol) was dissolved in Et2O (20 mL), then treated with a solution of MgBr2·Et2O (441 mg, 1.34 mmol) and CH3NO2 (143 μL, 2.68 mmol) in Et2O (6 mL) and stirred for 10 h. The mixture was quenched with saturated aq NaHCO3 (70 mL), extracted with EtOAc (3 × 100 mL), washed with brine (20 mL), dried over Na2SO4 and concentrated, then purified by silica gel chromatography (66% EtOAc in hexanes) to afford an amorphous white solid (136 mg, 96%). Op. 2: The intermediate azide (136 mg, 0.13 mmol) was dissolved in MeOH (3 mL), then treated with 1,3-propanedithiol (133 μL, 1.30 mmol) and Et3N (186 μL, 1.30 mmol) and stirred at rt for 40 h. The mixture was concentrated and purified by silica gel chromatography (4.75% MeOH in CH2Cl2) to afford the C2′ amine as an amorphous white solid (101 mg, 77%). Op. 3: A solution of amino diol (93 mg, 0.09 mmol) was dissolved in 5:1 Py:Et3N (3 mL) and treated with SO3·Py (385 mg, 2.42 mmol), then heated to 70 °C under microwave conditions for 1 h. The mixture was concentrated and purified by silica gel chromatography (9% MeOH in CH2Cl2 with 2% Et3N) to yield 2′,3′,6-tri-N,O,O-sulfate Et3NH salt as a light yellow oil. Op. 4: The intermediate TBDPS ether was redissolved in DMF (2 mL), then treated with 1 M TBAF solution in THF (0.9 mL) and stirred at rt for 40 h. The mixture was concentrated and used directly in next operation. Op. 5: The crude trisulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then purified by reverse-phase HPLC to yield the trisodium salt as an amorphous white solid (61 mg, 62%). The overall yield of 2′,3′,6-tri-N,O,O-sulfate 9f (trisodium salt) from 1 was 40% over 6 operations. 1H NMR (400 MHz, CD3OD): δ 7.46 (dd, 2H, J = 1.4, 8.3 Hz), 7.11–7.42 (m, 18H), 5.75 (d, 1H, J = 3.4 Hz), 5.14 (d, 1H, J = 10.8 Hz), 5.10 (d, 1H, J = 11.1 Hz), 4.99 (m, 2H), 4.82 (d, 1H, J = 11.2 Hz), 4.65 (dd, 1H, J = 8.9, 10.5 Hz), 4.58 (d, 1H, J = 10.6 Hz), 4.54 (d, 1H, J = 11.2 Hz), 4.52 (d, 1H, J = 7.3 Hz), 4.37 (dd, 1H, J = 2.0, 10.8 Hz), 4.24 (dd, 1H, J = 4.8, 10.7 Hz), 3.53–4.05 (m, 10H), 3.49 (dd, 1H, J = 3.4, 10.6 Hz), 3.33–3.44 (m, 3H), 1.94 (s, 2H); 13C NMR (100 MHz, CD3OD): δ 158.86, 140.36, 139.98, 139.74, 138.26, 129.93, 129.40, 129.33, 129.17, 129.07, 128.97, 128.90, 128.75, 128.59, 128.40, 128.06, 104.35, 98.36, 84.57, 83.58, 79.47, 78.25, 75.64, 75.19, 74.95, 74.51, 74.15, 73.66, 69.30, 68.13, 67.38, 62.18, 58.86, 41.97; [α]20D = +54.4 (c 0.62, MeOH); ESI–MS: m/z calcd for C43H49N2Na4O21S3 [M+4 Na]+: 1117.2; found: 1117.0.
3.13 2′-(N-carboxybenzyl)aminoethyl 4-O-[2-amino-4-O-benzyl-2-deoxy-2,3,6-tri-N,O,O-sulfonato-α-D-glucopyranosyl]-2,3-di-O-benzyl-6-O-sulfonato-β-D-glucopyranoside (9g)
Op. 1: Orthogonally protected disaccharide 8 was treated with TBAF in THF to yield the C6′ alcohol, as described in the synthesis of 9a. Op. 2: The intermediate SEM ether was treated with MgBr2·Et2O and CH3NO2 in Et2O to yield the C3′ alcohol, as described in the synthesis of 9d. Op. 3: The intermediate azide (68 mg, 0.08 mmol) was dissolved in MeOH (3 mL), then treated with 1,3-propanedithiol (101 μL, 1.67 mmol) and Et3N (232 μL, 1.67 mmol) and stirred at rt for 20 h. The mixture was concentrated and purified by silica gel chromatography (4.75% MeOH in CH2Cl2) to yield the C2′ amine as an amorphous white solid (62 mg, 94%). Op. 4: A solution of amino triol (76 mg, 0.10 mmol) was dissolved in 5:1 Py:Et3N (3 mL) and treated with SO3·Py (552 mg, 3.47 mmol), then heated to 70 °C under microwave conditions for 1 h. The mixture was concentrated and purified by silica gel chromatography (9% MeOH in CH2Cl2 with 2% Et3N) to yield 2′,3′,6′,6-tetra-N,O,O,O-sulfate Et3NH salt as a light yellow oil. Op. 4: The tetrasulfate ester was dissolved in 50% aq MeOH and passed through an ion-exchange column, then purified by reverse-phase HPLC to yield the tetrasodium salt as an amorphous white solid (90 mg, 78% over 2 steps). The overall yield of 2′,3′,6′,6-tetra-N,O,O,O-sulfate 9g (tetrasodium salt) from 1 was 54% over 6 operations.1H NMR (400 MHz, CD3OD): δ 7.57 (d, 2H, J = 7.3 Hz), 7.06–7.49 (m, 18H), 5.51 (d, 1H, J = 2.6 Hz), 5.13 (d, 1H, J = 9.5 Hz), 5.06 (d, 1H, J = 11.2 Hz), 4.98 (m, 2H), 4.89 (d, 1H, J = 11.7 Hz), 4.70 (d, 1H, J = 9.5 Hz), 4.63 (t, 1H, J = 9.8 Hz), 4.46–4.60 (m, 3H), 4.23–4.42 (m, 4H), 3.84–4.06 (m, 4H), 3.49–3.84 (m, 6H), 3.41 (t, 1H, J = 8.0 Hz), 3.33 (br, 1H); 13C NMR (125 MHz, CD3OD): δ 157.34, 138.84, 138.38, 137.93, 136.68, 129.12, 127.94, 127.73, 127.68, 127.61, 127.55, 127.53, 127.45, 127.31, 127.24, 127.00, 126.58, 102.88, 96.62, 82.75, 82.05, 78.36, 76.26, 74.55, 73.84, 73.07, 72.37, 71.79, 70.22, 67.93, 66.74, 65.95, 57.07, 40.42; [α]20D = +71.9 (c 0.98, MeOH); ESI–MS: m/z calcd for C43H48N2Na5O24S4 [M+5 Na]+: 1219.1; found: 1219.1.
3.14 2′-Aminoethyl 4-O-[2-acetamido-2-deoxy-6-O-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2a)
Partially protected sulfoform 9a (20 mg, 0.02 mmol) was dissolved in 50% aq MeOH (1 mL), then treated with 10% Pd(OH)2 on charcoal (8 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 6′,6-di-O-sulfate 2a (disodium salt) as an amorphous white solid (10.5 mg, 87%). 1H NMR (400 MHz, D2O): δ 5.38 (d, 1H, J = 3.6 Hz) 4.42 (d, 1H, J = 8.0 Hz), 4.26 (m, 4H), 4.15 (dd, 1H, J = 5.1, 11.4 Hz), 3.88 (d, 2H, J = 3.6 Hz), 3.84 (m, 3H), 3.73 (m, 1H) 3.68 (m, 1H) 3.63 (t, 2H, J = 8.8 Hz), 3.54 (t, 1H, J = 9.5 Hz), 3.24 (m, 1H), 2.98 (t, 1H, J = 5.2 Hz), 1.96 (s, 3H); 13C NMR (100 MHz, D2O): δ 174.98, 102.66, 98.02, 76.91, 75.37, 73.93, 73.03, 71.31, 71.17, 69.71, 69.47, 67.74, 67.16, 54.20, 40.42, 22.53; [α]20D = +56.0 (c 0.5, H2O); HRESI–MS: m/z calcd for C16H28N2NaO17S2 [M+Na]−: 607.0727; found: 607.0733.
3.15 2′-Aminoethyl 4-O-[2-acetamido-2-deoxy-3-O-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2b)
Partially protected sulfoform 9b (20 mg, 0.02 mmol) was dissolved in 50% aq MeOH (1 mL), then treated with 10% Pd(OH)2 on charcoal (8 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 3′,6-di-O-sulfate 2b (disodium salt) as an amorphous white solid (10 mg, 83%). 1H NMR (400 MHz, D2O): δ 5.30 (d, 1H, J = 3.7 Hz), 4.44 (m, 1H), 4.36 (m, 1H), 4.29 (dd, 1H, J = 2.1, 11.3 Hz), 4.18 (dd, 1H, J = 4.5, 11.2 Hz), 4.01 (dd, 1H, J = 3.6, 10.8 Hz), 3.90 (dd, 1H, J = 5.4, 11.0 Hz), 3.78 (dd, 2H, J = 2.9, 7.4 Hz), 3.70 (m, 3H), 3.62 (m, 3H), 3.22 (m, 1H) 2.93 (t, 1H, J = 5.3 Hz), 1.93 (s, 3H); 13C NMR (100 MHz, D2O): δ 174.91, 102.69, 99.28, 80.18, 76.80, 76.58, 73.93, 73.23, 73.08, 70.09, 68.64, 67.52, 60.64, 53.10, 40.49, 22.73; [α]20D = +31.1 (c 0.45, H2O); HRESI–MS: m/z calcd for C16H28N2NaO17S2 [M+Na]−: 607.0727; found: 607.0722.
3.16 2′-Aminoethyl 4-O-[2-amino-2-deoxy-2-N-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2c)
Partially protected sulfoform 9c (20 mg, 0.02 mmol) was dissolved in 50% aq MeOH (1 mL), then treated with 10% Pd(OH)2 on charcoal (8 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 2′,6-di-N,O-sulfate 2c (disodium salt) as an amorphous white solid (10 mg, 84%). 1H NMR (400 MHz, D2O): δ 5.50 (d, 1H, J = 3.5 Hz), 4.44 (d, 1H, J = 8.1 Hz), 4.28 (dd, 1H, J = 1.9, 11.2 Hz), 4.13 (dd, 1H, J = 4.9, 11.5 Hz), 3.97 (m, 1H), 3.89 (m, 1H), 3.78 (dd, 1H, J = 2.4, 12.6 Hz), 3.71 (m, 3H), 3.66 (m, 2H), 3.52 (t, 1H, J = 9.5 Hz), 3.43 (t, 1H, J = 9.5 Hz), 3.27 (m, 1H), 3.14 (m, 3H); 13C NMR (100 MHz, D2O): δ 102.57, 98.98, 76.54, 76.15, 73.35, 73.10, 73.04, 71.74, 70.10, 67.60, 67.11, 60.80, 58.72, 40.13; [α]20D = +26.2 (c 0.8, H2O); HRESI–MS: m/z calcd for C14H26N2NaO16S2 [M+Na]−: 565.0622; found: 565.0613.
3.17 2′-Aminoethyl 4-O-[2-acetamido-2-deoxy-3,6-di-O-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2d)
Partially protected sulfoform 9d (20.3 mg, 0.02 mmol) was dissolved in 50% aq MeOH (3 mL), then treated with 10% Pd(OH)2 on charcoal (20 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 3′,6′,6-tri-O-sulfate 2d (trisodium salt) as an amorphous white solid (12.7 mg, 97%). 1H NMR (400 MHz, D2O): δ 5.32 (d, 1H, J = 3.6 Hz), 4.43 (d, 1H, J = 8.0 Hz), 4.36 (t, 1H, J = 10.4 Hz), 4.15–4.30 (m, 5H), 4.02 (dd, 1H, J = 3.6, 10.7 Hz), 3.86–3.98 (m, 3H), 3.70–3.77 (m, 1H), 3.59–3.69 (m, 3H), 3.25 (t, 1H, J = 8.4 Hz), 3.08–3.21 (m, 2H), 1.92 (s, 3H); 13C NMR (125 MHz, D2O): δ 174.21, 101.86, 98.17, 79.24, 75.83, 75.58, 73.11, 72.36, 70.69, 67.59, 66.84, 66.42, 66.12, 52.27, 39.42, 22.04; [α]20D = +39.2 (c 0.85, H2O); HRESI–MS: m/z calcd for C16H27N2Na4O20S3 [M+4 Na]+: 754.9910; found: 754.9918.
3.18 2′-Aminoethyl 4-O-[2-acetamido-2-deoxy-2,6-di-O-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2e)
Partially protected sulfoform 9e (24.4 mg, 0.02 mmol) was dissolved in 50% aq MeOH (3 mL), then treated with 10% Pd(OH)2 on charcoal (24 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 2′,6′,6-tri-N,O,O-sulfate 2e (trisodium salt) as an amorphous white solid (15.1 mg, 98%). 1H NMR (400 MHz, D2O): δ 5.65 (d, 1H, J = 3.6 Hz), 4.56 (t, 1H, J = 7.7 Hz), 4.31–4.38 (m, 3H), 4.25 (dd, 1H, J = 4.9, 11.4 Hz), 4.00–4.19 (m, 2H), 3.80–3.90 (m, 3H), 3.74 (t, 1H, J = 9.1 Hz), 3.61 (m, 2H), 3.34–3.48 (m, 2H), 3.30 (m, 2H), 2.91 (s, 2H), 2.76 (s, 2H); 13C NMR (125 MHz, D2O): δ 101.90, 97.94, 75.87, 75.06, 72.62, 72.40, 70.98, 70.44, 69.01, 66.96, 66.53, 64.89, 57.89, 42.80; [α]20D = +48.4 (c 1.0, H2O); HRESI–MS: m/z calcd for C14H25N2Na4O19S3 [M+4 Na]+: 712.9804; found: 712.9809.
3.19 2′-Aminoethyl 4-O-[2-amino-2-deoxy-2,3-di-N,O-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2f)
Partially protected sulfoform 9f (8.8 mg, 0.01 mmol) was dissolved in 50% aq MeOH (3 mL), then treated with 10% Pd(OH)2 on charcoal (9 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 2′,3′,6-tri-N,O,O-sulfate 2f (trisodium salt) as an amorphous white solid (5.4 mg, 97%). 1H NMR (400 MHz, D2O): δ 5.46 (d, 1H, J = 2.9 Hz), 4.46 (t, 1H, J = 8.0 Hz), 4.28 (m, 2H), 4.13 (dd, 1H, J = 4.8, 11.3 Hz), 3.90–4.07 (m, 2H), 3.66–3.86 (m, 6H), 3.61 (t, 2H, J = 9.2 Hz), 3.26–3.37 (m, 3H), 3.11–3.21 (m, 1H), 2.86 (s, 2H), 2.66 (s, 1H); 13C NMR (125 MHz, D2O): δ 101.93, 99.19, 78.22, 75.77, 72.45, 72.28, 68.08, 66.88, 64.86, 63.40, 59.94, 57.00, 42.73; [α]20D = +56.7 (c 0.36, H2O); HRESI–MS: m/z calcd for C14H25N2Na4O19S3 [M+4 Na]+: 712.9804; found: 712.9794.
3.20 2′-Aminoethyl 4-O-[2-amino-2-deoxy-2,3,6-tri-N,O,O-sulfonato-α-D-glucopyranosyl]-6-O-sulfonato-β-D-glucopyranoside (2g)
Partially protected sulfoform 9g (47.9 mg, 0.04 mmol) was dissolved in 50% aq MeOH (3 mL), then treated with 10% Pd(OH)2 on charcoal (24 mg) and stirred under positive H2 pressure at rt for 20 h. The product was filtered, concentrated, and purified by reverse-phase HPLC to yield 2′,3′,6′,6-tetra-N,O,O,O-sulfate 2g (tetrasodium salt) as an amorphous white solid (31.4 mg, 99%). 1H NMR (400 MHz, D2O): δ 5.55 (d, 1H, J = 3.0 Hz), 4.59 (d, 1H, J = 8.0 Hz), 4.25–4.44 (m, 5H), 3.97–4.18 (m, 3H), 3.68–3.96 (m, 5H), 3.16–3.50 (m, 4H); 13C NMR (125 MHz, D2O): δ 101.94, 98.75, 81.70, 78.10, 76.52, 75.79, 72.49, 72.29, 70.56, 67.73, 66.90, 66.32, 55.87, 39.50; [α]20D = +34.6 (c 1.0, H2O); HRESI–MS: m/z calcd for C14H24N2Na5O22S4 [M+5 Na]+: 814.9192; found: 814.9194.
Highlights.
Synthesis of an orthogonally protected disaccharide (GlcN(α1→4)Glc) with 2′-aminoethyl linker
generation of sulfoforms with a 6-O-sulfate on glucose and multiple sulfate esters on glucosamine
Deprotection and sulfonation steps performed in variable order with 85–90% per operation
Microwave-assisted sulfonation provides significant improvement to overall yields
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM-06982) and the Department of Defense (W911SR-08-C-0001). The authors also gratefully acknowledge NMR and MS support from the Purdue University Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 2.Hattrup CL, Gendler SJ. Annu Rev Physiol. 2008;70:431–457. doi: 10.1146/annurev.physiol.70.113006.100659. [DOI] [PubMed] [Google Scholar]
- 3.Bussolino F, Albini A, Camussi G, Presta M, Viglietto G, Ziche M, Persico G. Eur J Cancer. 1996;32A:2401–2412. doi: 10.1016/s0959-8049(96)00390-5. [DOI] [PubMed] [Google Scholar]
- 4.Vladovsky I, Christofori C. In: Antiangiogenic Agents in Cancer Therapies. Teicher BA, editor. Humana Press; Totowa: 1999. pp. 93–118. [Google Scholar]
- 5.Arenberg DA, Polverini PJ, Kunkel SL, Shanafelt A, Strieter RM. In: Methods in Enzymology. Horuk R, editor. Vol. 288. Academic Press; San Diego: 1997. pp. 190–220. [DOI] [PubMed] [Google Scholar]
- 6.Parish CR. Nat Rev Immunol. 2006;6:633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 7.(a) Raman R, Sasisekharan V, Sasisekharan R. Chem Biol. 2005;12:267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]; (b) Sasisekharan R, Raman R, Prabhakar V. Annu Rev Biomed Eng. 2006;8:181–231. doi: 10.1146/annurev.bioeng.8.061505.095745. [DOI] [PubMed] [Google Scholar]; (c) Raman R, Sasisekharan R. Chem Biol. 2007;14:873–874. doi: 10.1016/j.chembiol.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 8.(a) Capila I, Linhardt RJ. Angew Chem Int Ed. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (b) Tumova S, Woods A, Couchman JR. Int J Biochem Cell Biol. 2000;32:269–288. doi: 10.1016/s1357-2725(99)00116-8. [DOI] [PubMed] [Google Scholar]; (c) Powell AK, Yates EA, Fernig DG, Turnbull JE. Glycobiology. 2004;14:17R–30R. doi: 10.1093/glycob/cwh051. [DOI] [PubMed] [Google Scholar]
- 9.Schumacher B, Pecher P, von Specht BU, Stegmann T. Circulation. 1998;97:645–50. doi: 10.1161/01.cir.97.7.645. [DOI] [PubMed] [Google Scholar]
- 10.Isner JM, Losordo DW. Nat Med. 1999;5:491–2. doi: 10.1038/8374. [DOI] [PubMed] [Google Scholar]
- 11.(a) Noti C, de Paz JL, Polito L, Seeberger PH. Chem-Eur J. 2006;12:8664–8686. doi: 10.1002/chem.200601103. [DOI] [PubMed] [Google Scholar]; (b) Shipp EL, Hsieh-Wilson LC. Chem Biol. 2007;14:195–208. doi: 10.1016/j.chembiol.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Shaunak S, Thomas S, Gianasi E, Godwin A, Jones E, Teo I, Mireskandari K, Luthert P, Duncan R, Patterson S, Khaw P, Brocchini S. Nat Biotechnol. 2004;22:977–984. doi: 10.1038/nbt995. [DOI] [PubMed] [Google Scholar]
- 13.Fan RH, Achkar M, Hernandez-Torres JM, Wei A. Org Lett. 2005;7:5095–5098. doi: 10.1021/ol052130o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu RH, Chanthamontri C, Han HL, Hernandez-Torres JM, Wood KV, McLuckey SA, Wei A. J Org Chem. 2008;73:6059–6072. doi: 10.1021/jo800713m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu RH, Wei A. J Carbohydr Chem. 2012 doi: 10.1080/07328303.2012.658274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruehl RE, Bertozzi CR, Rosen SD. J Biol Chem. 2000;275:32642–32648. doi: 10.1074/jbc.M001703200. [DOI] [PubMed] [Google Scholar]
- 17.(a) Gordon EJ, Sanders WJ, Kiessling LL. Nature. 1998;392:30–31. doi: 10.1038/32073. [DOI] [PubMed] [Google Scholar]; (b) Gordon EJ, Strong LE, Kiessling LL. Bioorg Med Chem. 1998;6:1293–1299. doi: 10.1016/s0968-0896(98)00122-9. [DOI] [PubMed] [Google Scholar]
- 18.Ziegler T, Hermann C. Tetrahedron Lett. 2008;49:2166–2169. [Google Scholar]
- 19.Hernández-Torres JM, Achkar J, Wei A. J Org Chem. 2004;69:7206–7211. doi: 10.1021/jo048999m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crich D, Smith M. J Am Chem Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Xu Y, Yang B, Tiruchinapally G, Sun B, Liu R, Dulaney S, Liu J, Huang X. Chem Eur J. 2010;16:8365–8375. doi: 10.1002/chem.201000987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Jacquinet J-C. Carbohydr Res. 1990;199:153–181. doi: 10.1016/0008-6215(90)84259-w. [DOI] [PubMed] [Google Scholar]; (b) Haller MF, Boons G-J. Eur J Org Chem. 2002:2033–2038. [Google Scholar]; (c) Shangguan N, Katukojvala S, Greenberg R, Williams LJ. J Am Chem Soc. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 23.Bayley H, Standring DN, Knowles JR. Tetrahedron Lett. 1978:3633–3634. [Google Scholar]
- 24.Vakalopoulos A, Hoffmann HMR. Org Lett. 2000;2:1447–1450. doi: 10.1021/ol0057784. [DOI] [PubMed] [Google Scholar]
- 25.Raghuraman A, Riaz M, Hindle M, Desai UR. Tetrahedron Lett. 2007;48:6754–6758. doi: 10.1016/j.tetlet.2007.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]