

Abstract
Small airway-derived pulmonary adenocarcinoma (PAC) and pancreatic ductal adenocarcinoma (PDAC) are among the most common human cancers and smoking is a risk factor for both. Emerging research has identified cAMP signaling stimulated by the stress neurotransmitters adrenaline and noradrenaline as important stimulators of several adenocarcinomas, including PAC and PDAC. The nicotine-derived nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is a potent mutagen and the most powerful tobacco carcinogen. NNK is also an agonist for nicotinic acetylcholine receptors (nAChRs). Using hamster models of NNK-induced PAC and PDAC, we have tested the hypothesis that in analogy to chronic effects of nicotine in the brain, NNK may modulate the α7- and α4β2nAChRs, causing an increase in stress neurotransmitters and decrease in the inhibitory neurotransmitter γ-aminobutyric acid (GABA). In support of our hypothesis, immunoassays showed a significant increase in serum adrenaline/noradrenaline and increased intracellular cAMP in the cellular fraction of blood of NNK treated hamsters. Western blots were done with cells harvested by laser capture microcopy from control small airway epithelia, alveolar epithelia, pancreatic islet and pancreatic duct epithelia and from NNK-induced PACs and PDACs. The GABA synthesizing enzyme glutamate decarboxylase 65 (GAD65) and GABA were suppressed in NNK-induced PACs and PDACs whereas protein expression of the α7nAChR, α4nAChR as well as p-CREB and p-ERK1/2 were upregulated. These findings suggest, for the first time, that NNK-induced alterations in regulatory nAChRs may contribute to the development of smoking-associated PAC and PDAC by disturbing the balance between cancer stimulating and inhibiting neurotransmitters.
Keywords: NNK-induced PAC, NNK-induced PDAC, nicotinic acetylcholine receptor, stress neurotransmitters, GABA
Introduction
Small airway-derived pulmonary adenocarcinoma (PAC) and pancreatic ductal adenocarcinoma (PDAC) are among the most common and most deadly human cancers and smoking is a risk factor for both [1, 2]. The carcinogenic nitrosamine 4-(methylnitrosamino)-1- (3-pyridyl)-1-butanone (NNK) is formed from nicotine by nitrosation during the processing of tobacco and in the mammalian organism [3]. Metabolites formed from NNK are strong mutagens that induce activating point mutations in k-Ras and inactivating mutations in p53 [4], both of which are common in human PAC and PDAC [5–8]. In addition to the mutational activities of its metabolites, the parent NNK itself is an agonist for nicotinic acetylcholine receptors (nAChRs) [9, 10]. This receptor family has been extensively studied in the nervous system where it regulates the synthesis and release of neurotransmitters [11, 12]. The nAChRs also regulate the synthesis and release of the stress neurotranmitters adrenaline and noradrenaline in the adrenal medulla and in sympathetic nerve endings [13–15]. Emerging research suggests that nAChRs are not restricted to the nervous system but are ubiquitously expressed in non-neuronal mammalian cells where they regulate diverse cellular functions, including differentiation, proliferation, apoptosis and angiogenesis [16]. The discoveries that nAChRs regulate the proliferation [17] and apoptosis [18] of small cell lung cancer cells and that NNK is an agonist for nAChRs [9] marked the beginning of intense studies on the role of nAChRs in the regulation of cancer. It has thus been shown that nAChRs stimulate cancer cells via direct intracellular signaling and indirectly, by triggering the release of growth factors and angiogenic factors [19]. On the other hand, recent studies have shown that the stress neurotransmitters adrenaline and noradrenaline stimulate via beta-adrenoreceptor signaling the growth and metastasis of some of the most common human cancers, including PAC [20–22], PDAC [23, 24], gastric cancer [25], colon cancer [26, 27], prostate cancer [28] and ovarian cancer [29, 30].
In the nervous system (Figure 1), the effects of the excitatory neurotransmitters glutamate, noradrenaline, serotonin and dopamine are counteracted by the inhibitory neurotransmitter γ-aminobutyric acid (GABA), which inhibits the formation of adenylyl cyclase-dependent cAMP [31, 32]. The release of the excitatory neurotransmitters from neurons is regulated by the homomeric α7nAChR [13] whereas the heteromeric α4β2nAChR regulates the release of GABA [33]. Chronic exposure to nicotine upregulates both receptors, resulting in an increased number of nAChRs [11, 34]. However, the α4β2nAChR is additionally desensitized by chronic nicotine, resulting in decreased GABA release associated with nicotine addiction [13, 33]. By contrast, the sensitivity of the upregulated α7nAChR to agonists remains undiminished, resulting in increased synthesis and release of excitatory neurotransmitters triggered by the increased number of this receptor [34]. In vitro studies have shown that GABA inhibits the beta-adrenoreceptor-mediated proliferation and migration of colon cancer [35], PDAC [36] and PAC [37] and is under expressed in the majority of human PDACs [36]. These findings suggest that GABA may have tumor suppressor function in beta-adrenoreceptor-regulated cancers. Using well characterized hamster models of PAC [38, 39] and PDAC [40, 41], we have tested the hypothesis that in analogy to the chronic effects of nicotine in the nervous system, NNK may modulate the α7 -and α4β2nAChR in PAC and PDAC, resulting in enhanced stimulatory cAMP signaling and reduced inhibitory GABA.
Figure 1.
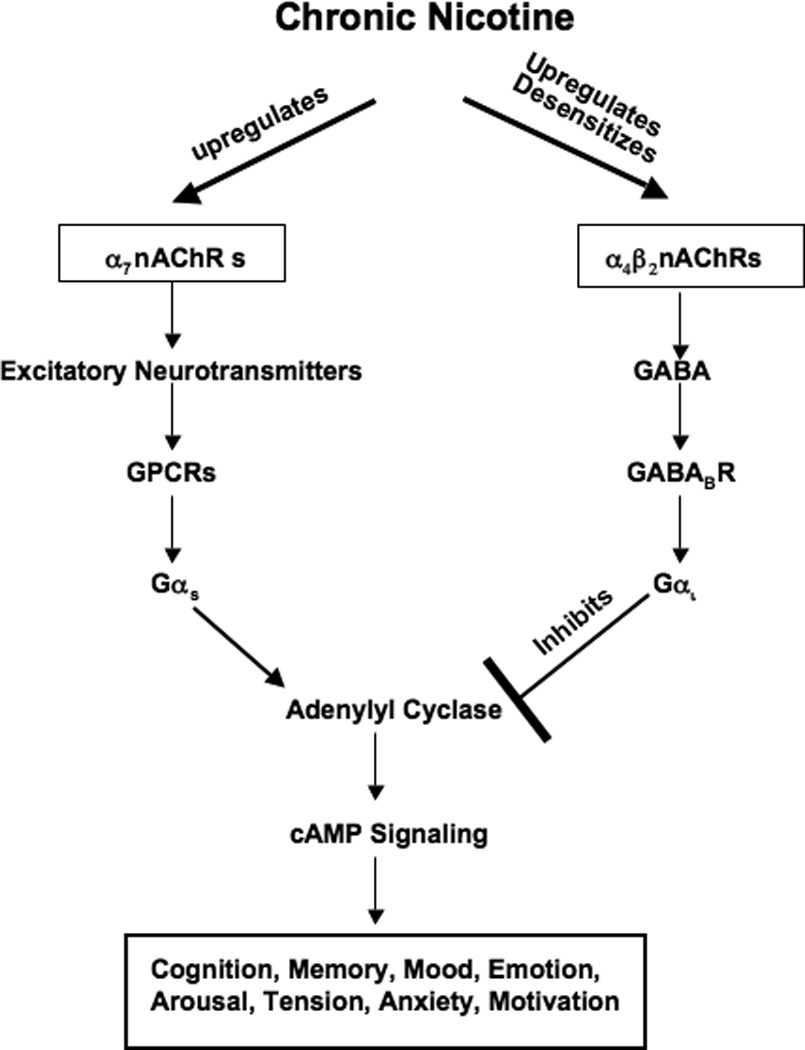
Simplified cartoon, summarizing the effects of chronic exposure to nicotine on excitatory and inhibitory neurotransmitter signaling regulated by nicotinic acetylcholine receptors in the nervous system. Chronic exposure to nicotine upregulates the α7nAChR without desensitizing the receptor[13, 34], resulting in increased release and synthesis of excitatory neurotransmitters glutamate, dopamine, serotonin and noradrenaline stimulated by this receptor. Most of the stimulating neurological and psychological effects of these neurotransmitters are mediated by the activation of adenylyl cyclase downstream of Gαs-coupled receptors. GABA normally balances these effects by inhibiting adenylyl cyclase downstream of the Gαi-coupled GABABR[13, 54]. The α4β2nAChR that stimulates the release and synthesis of GABA is desensitized by chronic exposure to nicotine, thus virtually shutting down all inhibitory GABA signaling[54]. This imbalance in stimulatory and inhibitory brain functions has been implicated in nicotine addiction and withdrawal symptoms [13, 34, 53].
Materials and Methods
Animal experiments
The animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee. Outbred six week old Syrian golden hamsters were purchased from Charles River (Wilmington, DL) and housed one animal per cage in plastic shoebox-type cages under standard laboratory conditions. The animals had free access to food (Purina Rodent Chow, Agri Feed, Knoxville, TN) and tap water. Ten male hamsters were treated by subcutaneous injections with NNK (2.5mg/100g bodyweight three times/week for 20 weeks). This treatment reproducibly induces the development of small airway-derived PAC in 80–100% of the animals [38, 42]. All of these animals developed multiple PACs. For the induction of pancreatic ductal adenocarcinoma (PDAC) in their offspring, pregnant hamsters were given ethanol (10%) in their drinking water from day 5 through the end of their pregnancy and one subcutaneous injection of NNK (5mg/100g) on day 15 of gestation and the offspring were observed until eight moths old. This treatment reproducibly induces pancreatitis-associated PDAC in 50–70% of the animals [40, 41]. Among the 15 surviving offspring of this treatment group, eight developed PDACs. Ten untreated hamsters served as controls. The animals were killed by CO2 asphyxiation. Blood samples were collected from each hamster and pooled for each treatment group for the analysis of three replicate samples per group of adrenaline, noradrenaline, and cAMP. Lungs and pancreas were fixed in 70% ethanol, embedded in paraffin and used for the harvesting of lung and pancreatic cells by laser capture microscopy.
Analysis of adrenaline and noradrenaline in serum
It is well established that binding of nicotine to nAChRs stimulates the release of the stress neurotranmitters adrenaline and noradrenaline, thereby increasing their systemic levels [13, 14]. Since NNK is an agonist for nAChRs [10, 14], we assessed the levels of both neurotransmitters in serum from control hamsters and from hamsters treated for 20 weeks with NNK using an enzyme immunoassay kit according to the manufacture instruction(2-CAT ELISA, Rocky Mountain Diagnostic, Inc., Colorado Spring, CO, USA). Absorbance was read with an ELISA reader at 450 nm. The quantification of the serum samples was achieved by comparing their absorbance with a reference curve prepared with known standard concentrations of adrenaline and noradrenaline. Mean values and standard errors from triplicate samples per treatment group were expressed as fold increase over controls. Statistical significance of data was assessed by one-way ANOVA, Tukey-Kramer multiple comparisons test and two-tailed unpaired t-test.
Analysis of cAMP in blood cells
It has been shown that adrenaline and noradrenaline stimulate the proliferation and migration of numerous human cancers, including PAC and PDAC via cAMP signaling downstream of beta-adrenoreceptors [20–30]. We therefore determined the systemic levels of cAMP in control hamsters and in animals treated for 20 weeks with NNK. The cellular fraction of blood samples comprised of erythrocytes, lymphocytes, granulocytes and thrombocytes was used for this analysis. Each of these blood cells expresses beta-adrenoreceptors, and lymphocytes have previously been used in human patients for the assessment of systemic cAMP-dependent signaling activity [43]. After 3 washes with water, cells were treated with 0.1 M HCL for 20 min, then, lysed by sonication. After centrifugation, samples were analyzed for cAMP levels using a direct cyclic AMP enzyme immunoassay kit according to the manufacturer’s instructions (Assay Designs Inc, Ann Arbor, MI, USA). Reactions were stopped and color intensity was measured immediately at 405 nm. Data are expressed as fold increase over controls of mean values and standard errors from triplicate samples per treatment group. Statistical analysis of data was by one-way ANOVA, Tukey-Kramer multiple comparisons test and two-tailed unpaired t-test.
Laser Capture Micro-dissection
This technique was used to harvest cells from lung and pancreatic tissue sections, using a PixCell IIe Laser Capture Microdissection system (Arcturus, Mountain View, CA. The simple respiratory epithelium (termed “small airway epithelial cells SAECs in this manuscript) comprised of Clara cells and ciliated cells from subsegmental bronchi and bronchioles, alveolar epithelial cells, and cells from PACs were harvested from the lung slides. Cells form pancreatic islets, pancreatic duct epithelia, and cells from PDACs were harvested from the pancreatic slides. The tissue sections were dewaxed by placing them in two baths of xylene for 5 min each, passed through decreasing concentrations of ethanol to rehydrate (100%, 95%, 70%), and water for 30 s each. The tissue was stained according to the procedure outlined in the Histogene Kit (Arcturus). Cells were harvested from histological sections of all hamsters per treatment group to obtain a total of 30,000 cells of each investigated cell type. These cells were allocated to three equal aliquots per treatment group for analysis. The proteins were extracted from the CapSure caps using protein extract buffer (Pierce) within 30 minutes after cell harvesting.
Analysis of protein expression in lung and pancreatic cells
It has been shown in the nervous system and in colon cancer cells that the α7nAChR regulates the synthesis and release of adrenaline and noradrenaline which activate cAMP signaling downstream of beta-adrenoreceptors [13, 14, 27]. In turn, we have shown that the activation of the beta-adrenergic signaling pathway in human PAC and PDAC cells results in the PKA-dependent activation of the transcription factor CREB and transactivation of the EGFR, leading to the phosphorylation of its downstream effector ERK1/2 [24, 44]. The α7nAChR additionally regulates the release of glutamate [33] from which the formation of GABA is catalyzed by the two isoforms of the enzyme glutamate decarboxylase, GAD65 and GAD67 [45]. On the other hand, the synthesis and release of GABA are regulated by the α4β2nAChR [33]. We therefore analyzed the protein expression of nAChRs expressing the α7 or α4 subunits, GABA and its synthesizing enzymes as well as p-CREB and p-ERK1/2 in NNK-induced PAC and PDAC cells, small airway epithelial cells, alveolar epithelial cells, as well as cells from pancreatic ducts and pancreatic islets of control hamsters. Following lysis of the thawed tissues, protein was determined using the BCA Protein Assay (Pierce, Rockford, IL). Equal amounts of protein were treated with loading buffer at 100°C for five minutes and applied to each lane for electrophoresis in 12% polyacrylamide gel. The electrophoresed proteins were transferred onto nitrocellulose membranes in transfer buffer at 100 mV for one hour. After transfer, the membranes were treated with blocking buffer (5% nonfat dry milk in tris buffered saline with Tween 20, TBST) for one hour, and incubated with primary antibody overnight at 4°C. Primary antibodies against the following signaling proteins were used: total CREB (Upstate Biotechnology, Lake Placid, NY, USA), p-CREB, p-ERK1/2 and ERK1/2 (Cell Signaling, Danvers, MA, USA), α4- and α7nAChR, GAD65 and GAD67 and GABA (Millipore, Billerica, MA, USA and beta-actin (Sigma). After being washed with TBST, the membranes were incubated with horseradish peroxidase-labeled secondary antibody (goat anti-mouse, or goat anti-rabbit, Cell Signaling) for one hour. Immunoreactive bands were detected using a chemiluminescent reaction (ECL, Amersham Biosciences, Piscataway, NJ) via autoradiography on Kodak Bio-Max XAR film. Three separate Western blots were conducted for each antibody per sample and yielded similar data. Relative densities of the bands were determined by image analysis using NIH SCION image analysis software. Mean values and standard errors from five densitometric readings per band were analyzed by one way ANOVA and Tukey-Kramer multiple comparison test.
Results
Analysis of serum samples by immunoassay showed that noradrenaline was increased 3.8-fold (p<0.001) and adrenaline 2.3-fold (p<0.001) in the NNK treated hamsters (Figure 2). These NNK-induced changes in neurotransmitter levels were accompanied by a significant (p<0.001) increase (2.2-fold) of cAMP levels in the cellular fraction of blood samples (Figure 2). These findings indicate, that similar to nicotine, NNK increases the systemic levels of stress neurotransmitters which in turn trigger a generalized increase in intracellular cAMP downstream of adrenoreceptors.
Figure 2.
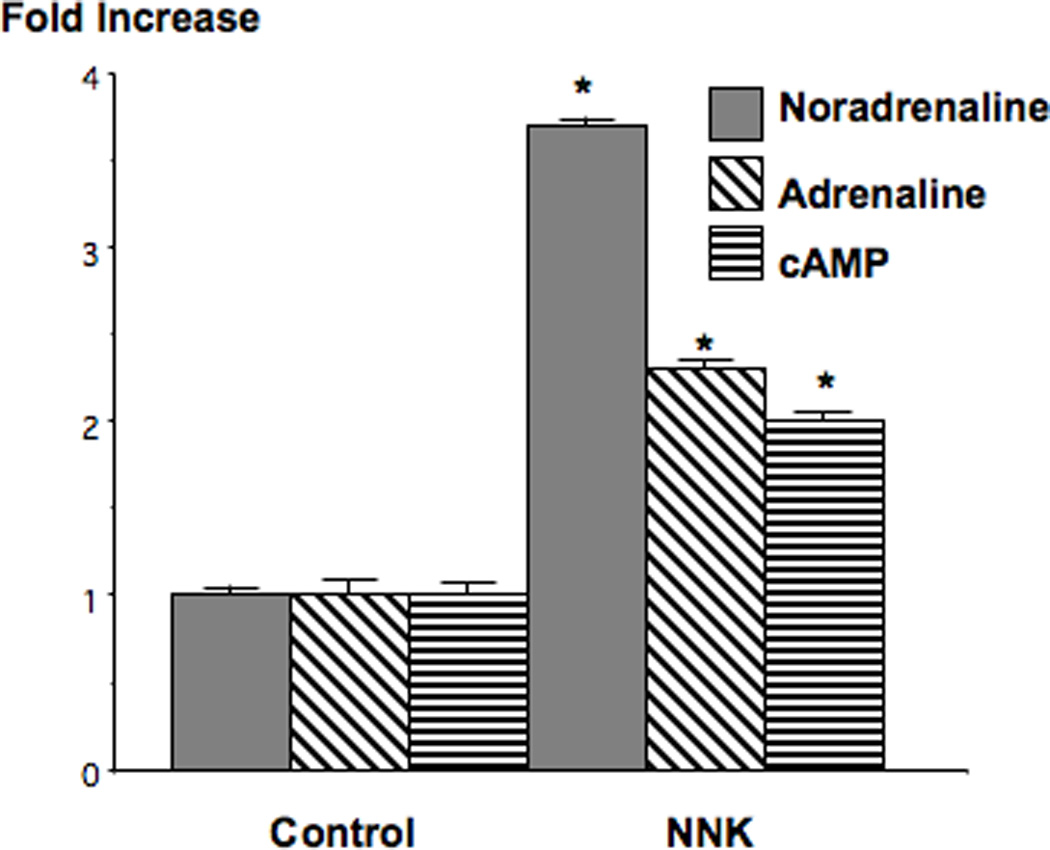
Results of immunoassays demonstrating the modulation of serum adrenaline/noradrenaline levels and cAMP in blood cells of hamsters treated for 20 weeks with NNK. There was a 3.8-fold increase in noradrenaline and a 2.3-fold increase in adrenaline while cAMP was increased 2.2-fold. Data are mean values and standard errors of triplicate samples from the pooled blood of 10 hamsters per treatment group. Asterixes indicate values significantly (p<0.001) different from controls.
Western blots of protein samples from cells harvested by laser capture microscopy showed that nAChRs expressing the α4 subunit predominated over the α7nAChR in normal lung tissues (Figure 3). Small airway epithelial cells, from which NNK-induced PAC arises in this species [38], expressed significantly (p<0.001) higher protein levels of either receptor than alveolar epithelial cells. Both receptors were prominently upregulated (p<0.001) in NNK-induced PACs with a 3.2-fold increase over the levels observed in small airway epithelial cells in α4nAChR protein expression and a 3.4-fold increase in α7nAChR protein (Figure 3). These findings indicate that chronic exposure to NNK has similar upregulating effects on nAChRs in lung cells as the reported upregulation of this receptor family by chronic nicotine exposure in the brain [11].
Figure 3.
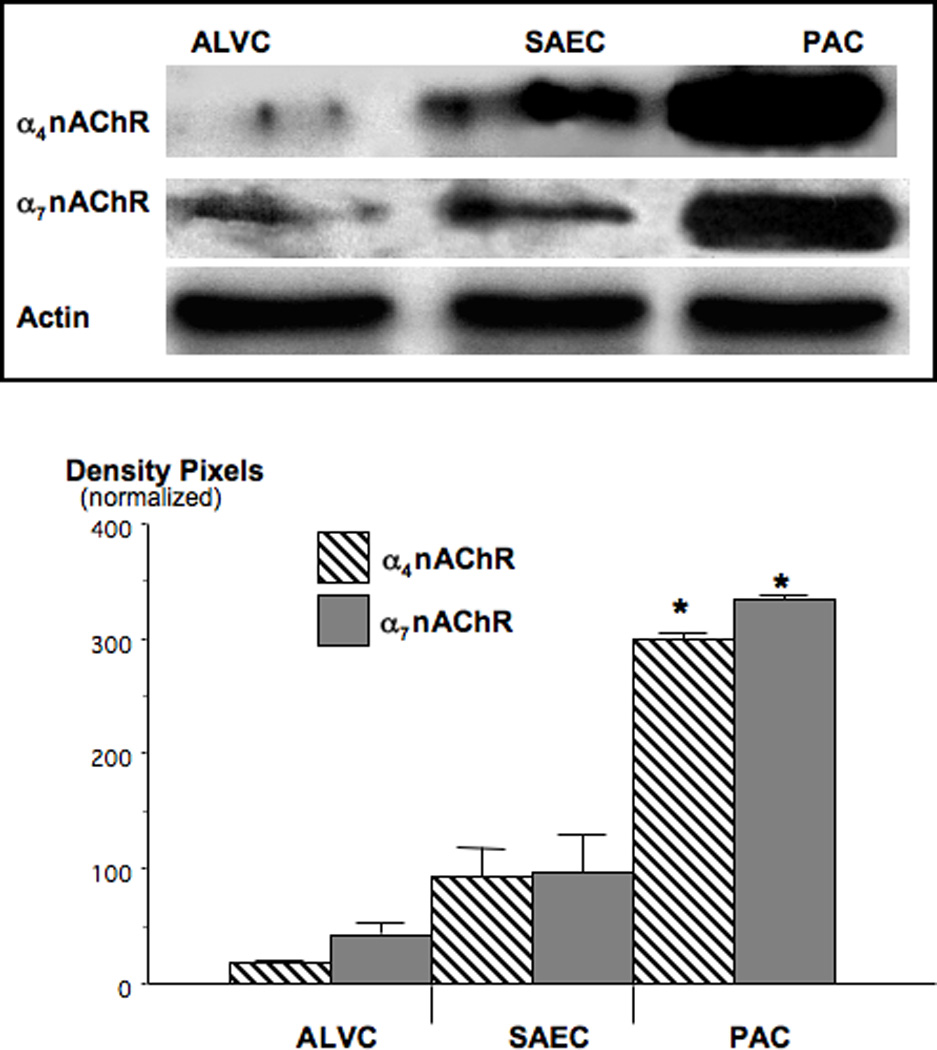
Western blots, illustrating the upregulation of α7- and α4nAChR protein in NNK-induced PAC. In comparison with their normal cells of origin, small airway epithelial cells (SAEC), α4subunit protein was increased 3.2-fold (*: p<0.001) and α7 subunit protein 3.4-fold (*: p<0.001). Data in the graph are mean values and standard errors of ratios of nAChR-protein over actin from five densitometric readings per band and normalized with expression levels in SAECs set at 100%. Each Western blot was conducted three times with similar data.
Both GAD isozymes as well as GABA were expressed in all investigated lung cells, with the highest levels of all three proteins observed in small airway epithelial cells from control hamsters (Figure 4). GAD65 was the predominating isozyme in lung cells. The expression levels of GAD65 and GABA were significantly (p<0.001) suppressed below the levels of small airway epithelial cells in NNK-induced PAC (Figure 4; GAD65: 25% of small airway epithelial cells, GABA: 26% of small airway epithelial cells). Expression levels of GAD67 were less affected by NNK (reduction to 73% of small airway epithelial cell levels). In contrast to the observed suppression of GABA and its synthesizing enzymes, both, p-CREB (1.7-fold) and p-ERK1/2 (1.8-fold) were significantly (p<0.001) induced in the PAC cells from NNK-treated hamsters while expression of the unphosphorylated proteins remained unchanged (Figure 5).
Figure 4.
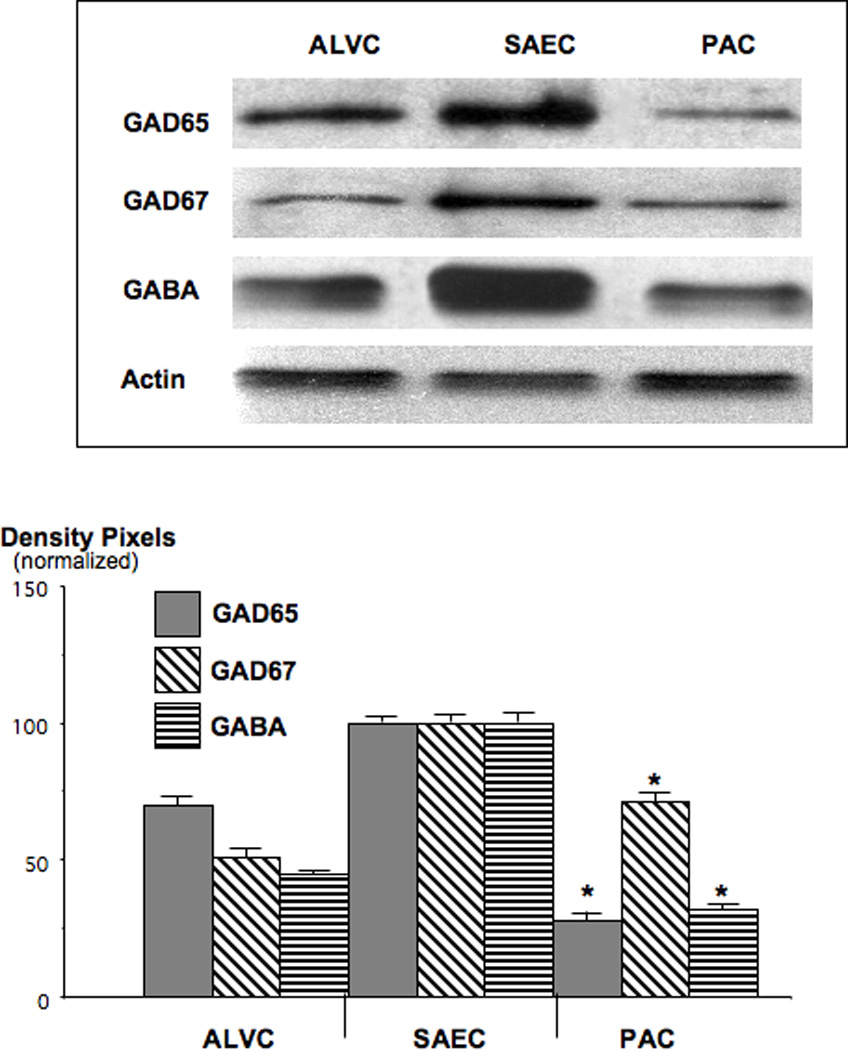
Western blots, exemplifying the suppression of GAD and GABA in NNK-induced PAC. GAD65 was reduced to 25% (*: p<0.001) and GABA to 26% (*: p<0.001) of the levels observed in SAECs. Data in the graph are normalized (expression levels in SAEC=100%) mean values and standard errors of ratios of GAD or GABA over actin from five densitometric readings per band. Each Western blot was conducted three times with similar data.
Figure 5.
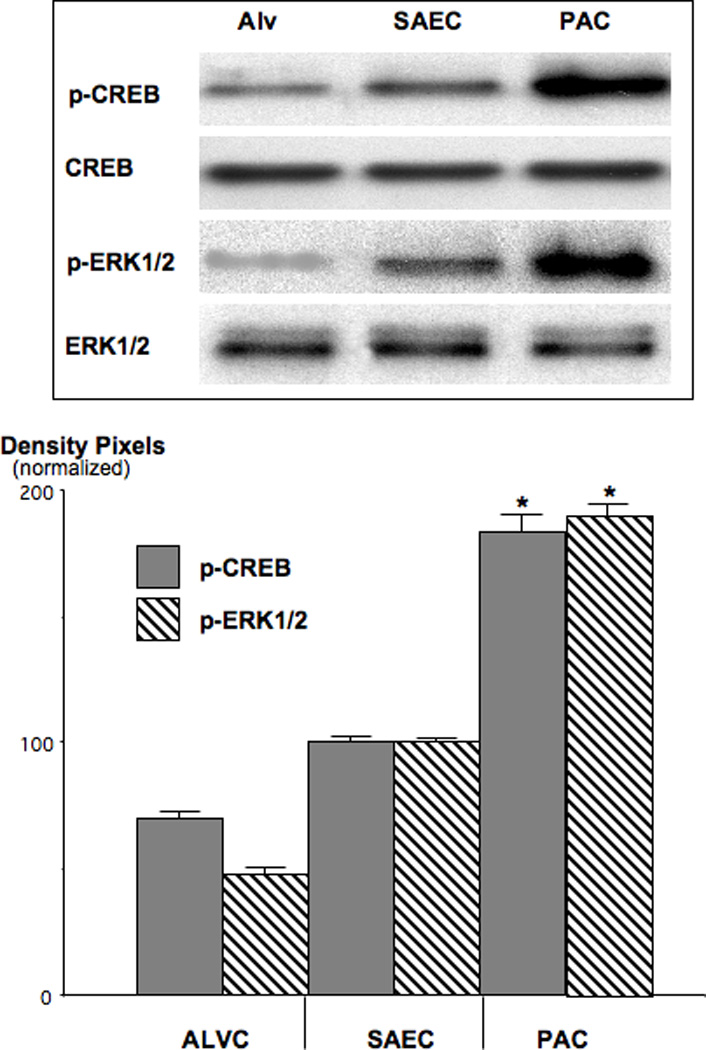
Western blots, documenting the induction of p-CREB and p-ERK1/2 in NNK-induced PAC. The increase in p-CREB expression was 1.7-fold (*: p<0.001) and that of p-ERK1/2 1.8-fold (*: p<0.001) over the levels in SAECs. Data in the graph are normalized (expression levels in SAEC=100%) mean values and standard errors of ratios of p-CREB over CREB and p-ERK1/2 over ERK1/2 from five densitometric readings per band. Each Western blot was conducted three times with similar data.
In cells harvested from pancreatic tissues, Western blots identified the α7nAChR as the predominating nAChR in normal duct cells from control hamsters (Figure 6). Significantly (p<0.001) higher levels of both nAChRs were observed in duct epithelial cells than in islet cells. Similar to the observations in PAC cells, each of the two investigated nAChRs was significantly (p<0.001) upregulated in the NNK-induced PDACs (Figure 6; α4nAChR: 2.3-fold, α7nAChR: 1.8-fold). The upregulation of these receptors was contrasted sharply by prominent (p<0.001) suppression of GABA and GAD65 to barely detectable levels in PDAC cells (Figure 7). GAD65 predominated in control duct epithelial cells whereas GAD67 predominated in control islet cells (Figure 7). Similar to the observations in PAC cells, p-CREB (2.65-fold) and p-ERK1/2 (2.55-fold) were significantly (p<0.001) induced in the PDAC cells from NNK-treated hamsters (Figure 8) whereas the levels of the unphsophorylated proteins remained unchanged.
Figure 6.
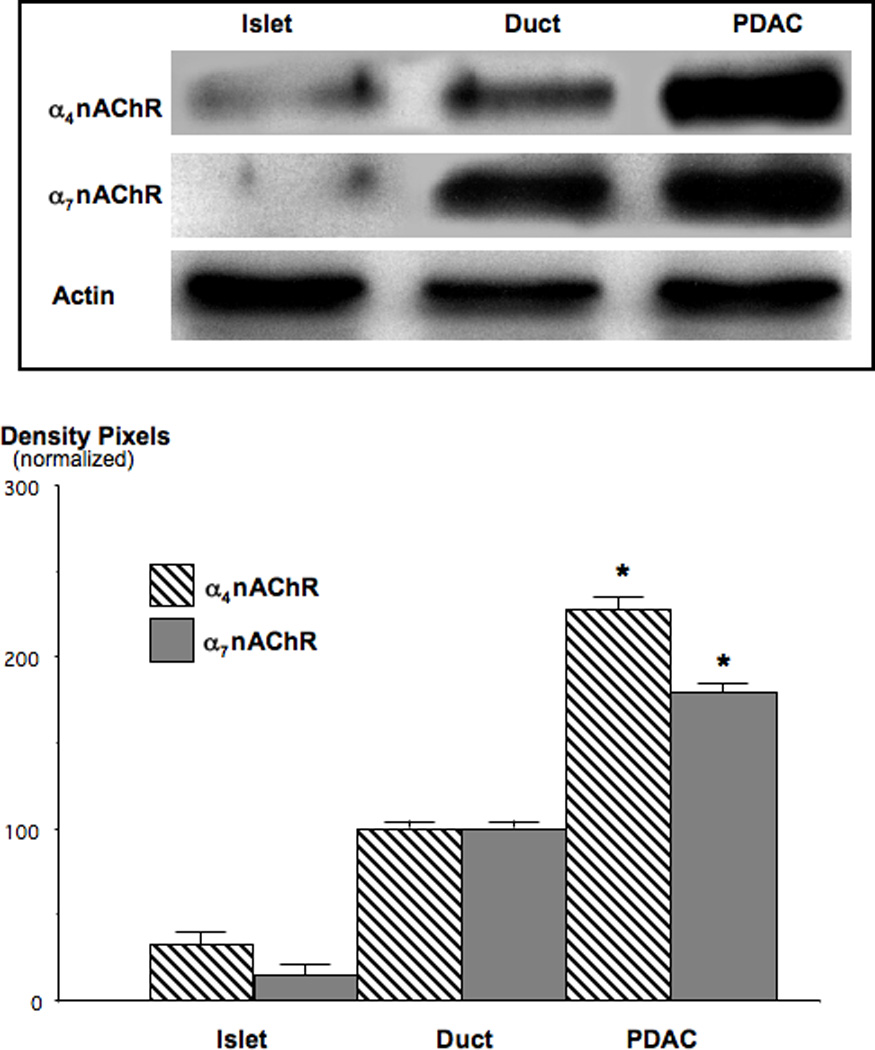
Western blots, showing the upregulation of α4-and α7 nAChR subunit proteins in NNK-induced PDAC. Expression levels of the α4nAChR was increased 2.3-fold (*: p<0.001) and that of the α7nAChR 1.8-fold (*: p<0.001) over levels observed in duct epithelial cells. Data in the graph are mean values and standard errors of five densitometric readings per band of ratios of nAChR protein over actin and normalized with levels in duct epithelial cells set as 100%. Each Western blot was conducted three times with similar data.
Figure 7.
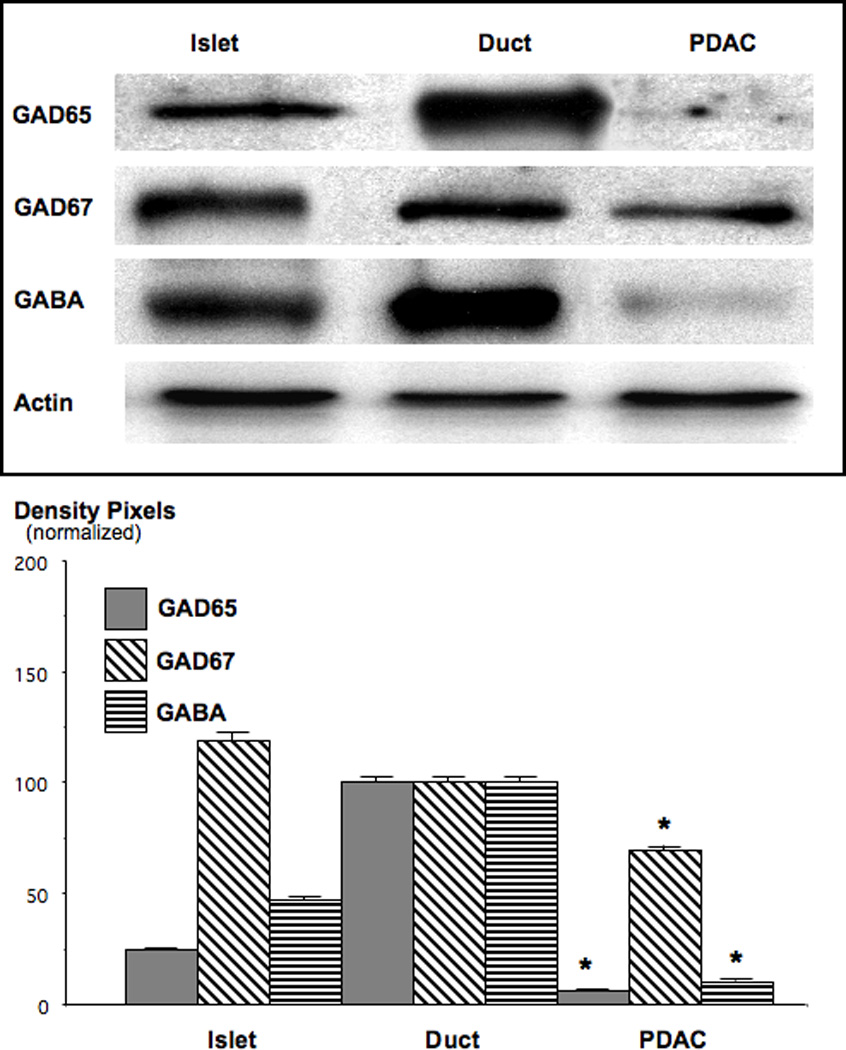
Western blots, exemplifying the suppression of GAD and GABA in NNK-induced PDAC. Both, GAD65 and GABA were reduced to barely detectable levels of below 10% of control duct epithelial cells (*: p<0.001). Quantitative data in the graph are mean values and standard errors of ratios of GAD or GABA over actin and are normalized with levels in duct epithelial cells set at 100%. Each Western blot was conducted three times with similar results.
Figure 8.
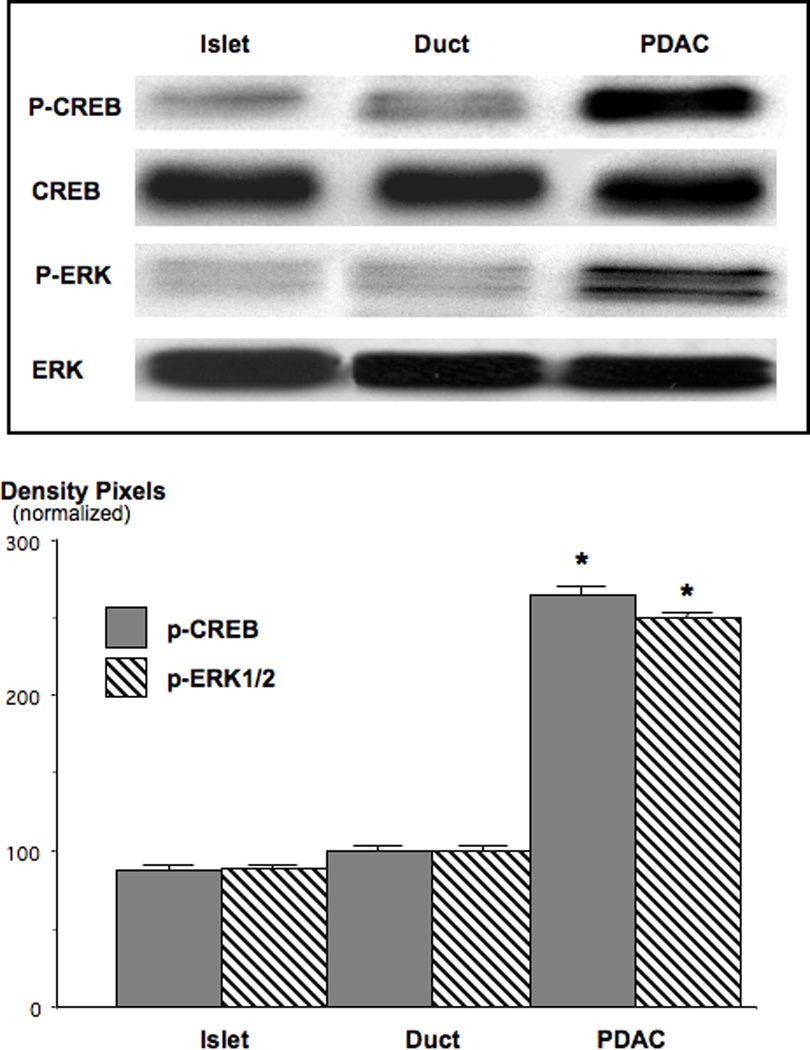
Western blots, showing the induction of p-CREB and p-ERK1/2 in NNK-induced PDAC. Both phosphorylated proteins were barely detectable in control islet and duct cells. P-CREB was increased 2.65-fold (*: p<0.001) and p-ERK1/2 2.55-fold (*: p<0.001) over the levels observed in duct epithelial cells. Data in the graph are normalized (expression levels in duct cells=100%) mean values and standard errors of ratios of p-CREB over CREB and p-ERK1/2 over ERK1/2 from five densitometric readings per band. Each Western blot was conducted three times with similar data.
Collectively, NNK upregulated the investigated nAChRs and representative phosphorylated proteins of the cAMP (p-CREB) and EGFR (p-ERK1/2) pathways while suppressing GAD65 and GABA in both types of NNK-induced adenocarcinomas.
Discussion
Our data show, for the first time, that the nicotine-derived carcinogenic nitrosamine NNK causes a systemic increase in the stress neurotransmitters adrenaline and noradrenaline associated with a generalized increase in intracellular cAMP in cells expressing β-adrenoreceptors. While in vitro studies have shown that NNK can stimulate PAC [21, 22, 44] and PDAC [23, 24] cells directly by binding as an agonist to their beta-adrenoreceptors (Figure 9), the observed systemic responses add a novel aspect to the mechanisms of action of this powerful tobacco carcinogen. These findings are in accord with reports on significant stimulating effects of these stress neurotransmitters on the proliferation and metastasis of adenocarcinoma of the colon [26, 27], stomach [25], prostate [28] and ovary [29, 30]. Accordingly, the NNK-induced systemic increase in these neurotransmitters may contribute to the development of these cited tumors as well as PAC and PDAC in smokers. In addition, smoking-induced increase in systemic adrenaline/noradreanline is an important factor in the etiology of cardiovascular disease that has been solely attributed to the nicotine contained in cigarette smoke [46]. By contrast, our data suggest that NNK also contributes to these adverse cardiovascular effects observed in smokers.
Figure 9.
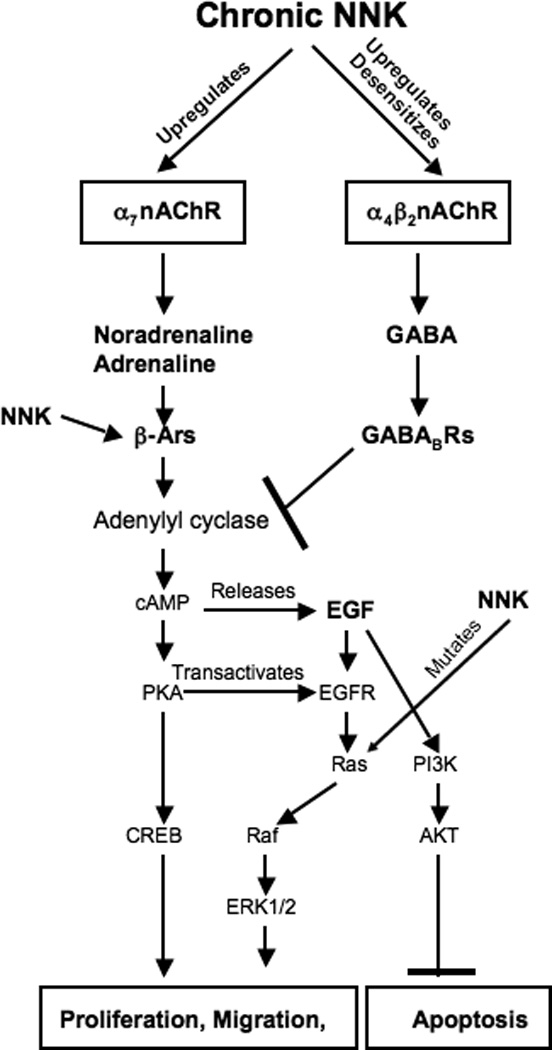
Simplified cartoon illustrating the proposed effects of chronic exposure to NNK on neurotransmitter signaling in PAC and PDAC and their microenvironment. Based on the finding that NNK is an nAChR agonist [9], in conjunction with the data reported in this publication, we propose that chronic exposure to this carcinogenic nitrosamine has effects on nAChRs in PAC and PDAC similar to those reported for nicotine in the nervous system (Figure 1). In peripheral non neurogenic tissues [27] and sympathetic nerve endings [55] the α7nAChR stimulates the release of noradrenaline from which adrenaline is formed. The observed increase in these stress neurotransmitters due to upregulated α7nAChRs enhances cancer stimulating cAMP signaling [21, 23, 24, 44] downstream of β-adrenergic receptors (β-ARs), an effect further intensified by direct stimulation of these receptors by NNK [21, 23] and by NNK-induced activating point mutations in K-ras [39, 56]. At the same time, inhibition of tumor stimulating cAMP signaling by GABA [37, 47] is virtually shut down due to desensitization of α4β2nAChRs.
The observed significant upregulation of the α7-and α4nAChRs in NNK-induced PAC and PDAC cells is in accord with the finding that this nitrosamine is an nAChR agonist that causes similar responses downstream of these receptors as nicotine [9, 10]. Chronic exposure to nicotine typically upregulates all nAChRs (Figure 1) [11] and our data show that NNK has similar effects (Figure 9). In analogy to the effects of chronic nicotine on brain cells [33] the simultaneous strong suppression of GAD65 and GABA in NNK-induced PAC and PDAC suggests that the upregulated α4nAChR is desensitized. This nAChR regulates the synthesis and release of GABA in brain cells and its desensitization has been associated with reduced GABA levels that contribute to nicotine addiction and withdrawal symptoms [33]. It has been shown that GABA acts as an important tumor suppressor in beta-adrenoreceptor-regulated cancers such as PAC, PDAC [37, 47], as well as adenocarcinoma of the colon [35], and breast [48]. Chronic agonist-induced desensitization of its upstream regulator, the α4β2nAChR, and the resulting GABA deficiency may thus contribute to the development of these cancers in smokers.
The observed significant increases in p-CREB and p-ERK1/2 in both investigated NNK-induced cancers is in accord with in vitro studies that have identified beta-adrenoreceptor-mediated signaling via cAMP/PKA/p-CREB and PKA-dependent transactivation of the EGFR as a key regulatory cascade in human PAC [21, 44, 49] and PDAC [23, 24] cells (Figuren9). Additional in vitro studies have shown that agents that increase intracellular cAMP enhance the responsiveness of this signaling cascade [49–52] whereas inhibitors of adenylyl cyclase, cAMP or PKA had blocking effects [24, 36, 44, 53]. These data identify GABA, which inhibits the adenylyl cyclase-dependent formation of camp by binding to the Gαi-coupled GABABR [31], as a key physiological defense against the hyperactive cAMP signaling that appears to drive the development and progression of PAC and PDAC (Figure 9). While the focus of prior investigations has been on beta-adrenoreceptors as the regulators of these signaling events, the current data suggest that the α7nAChR acts upstream of these receptors by regulating the systemic levels of adrenaline and noradrenaline. This interpretation is in accord with the documented regulatory role of this receptor for adrenaline/noradrenaline in the brain and sympathetic nerves [13, 14] and is supported by a recent report of α7nAChR-mediated synthesis of these neurotransmitters in colon cancer cells [27]. It remains to be investigated if PAC and PDAC cells are also able to synthesize adrenaline and noradrenaline.
In conjunction with the cited literature, the current data suggest that due to their centralized regulatory functions, the upregulation of α7nAChRs and the upregulation and desensitizion of the α4β2nAChR associated with nicotine addiction [33, 34] may also play crucial roles in the development and progression of smoking-associated PAC and PDAC and possibly other cancer stimulated by the stress neurotransmitters and camp signaling.
Acknowledgments
Financial support: RO1CA042829 and RO1CA096128 with the National Cancer Institute and grant 003650 with the National Lung Cancer Partnership.
Footnotes
Conflict of interest: None to declare.
References
- 1.Devesa SS, Bray F, Vizcaino AP, Parkin DM. International lung cancer trends by histologic type: male:female differences diminishing and adenocarcinoma rates rising. Int J Cancer. 2005;117(2):294–299. doi: 10.1002/ijc.21183. [DOI] [PubMed] [Google Scholar]
- 2.Talamini G, Bassi C, Falconi M, Sartori N, Salvia R, Rigo L, et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig Dis Sci. 1999;44(7):1303–1311. doi: 10.1023/a:1026670911955. [DOI] [PubMed] [Google Scholar]
- 3.Hecht SS, Hoffmann D. N-nitroso compounds and tobacco-induced cancers in man. IARC Sci Publ. 1991;(105):54–61. [PubMed] [Google Scholar]
- 4.Hecht SS. Recent studies on mechanisms of bioactivation and detoxification of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific lung carcinogen. Crit Rev Toxicol. 1996;26(2):163–181. doi: 10.3109/10408449609017929. [DOI] [PubMed] [Google Scholar]
- 5.Mitsudomi T, Viallet J, Mulshine JL, Linnoila RI, Minna JD, Gazdar AF. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6(8):1353–1362. [PubMed] [Google Scholar]
- 6.Luttges J, Schlehe B, Menke MA, Vogel I, Henne-Bruns D, Kloppel G. The K-ras mutation pattern in pancreatic ductal adenocarcinoma usually is identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer. 1999;85(8):1703–1710. [PubMed] [Google Scholar]
- 7.Iwakawa R, Kohno T, Anami Y, Noguchi M, Suzuki K, Matsuno Y, Mishima K, et al. : Association of p16 homozygous deletions with clinicopathologic characteristics and EGFR/KRAS/p53 mutations in lung adenocarcinoma. Clin Cancer Res. 2008;14(12):3746–3753. doi: 10.1158/1078-0432.CCR-07-4552. [DOI] [PubMed] [Google Scholar]
- 8.Koliopanos A, Avgerinos C, Paraskeva C, Touloumis Z, Kelgiorgi D, Dervenis C. Molecular aspects of carcinogenesis in pancreatic cancer. Hepatobiliary Pancreat Dis Int. 2008;7(4):345–356. [PubMed] [Google Scholar]
- 9.Schuller HM, Orloff M. Tobacco-specific carcinogenic nitrosamines. Ligands for nicotinic acetylcholine receptors in human lung cancer cells. Biochem Pharmacol. 1998;55(9):1377–1384. doi: 10.1016/s0006-2952(97)00651-5. [DOI] [PubMed] [Google Scholar]
- 10.Arredondo J, Chernyavsky AI, Grando SA. Nicotinic receptors mediate tumorigenic action of tobacco-derived nitrosamines on immortalized oral epithelial cells. Cancer Biol Ther. 2006;5(5):511–517. doi: 10.4161/cbt.5.5.2601. [DOI] [PubMed] [Google Scholar]
- 11.Lindstrom J, Anand R, Gerzanich V, Peng X, Wang F, Wells G. Structure and function of neuronal nicotinic acetylcholine receptors. Prog Brain Res. 1996;109:125–137. doi: 10.1016/s0079-6123(08)62094-4. [DOI] [PubMed] [Google Scholar]
- 12.Gotti C, Fornasari D, Clementi F. Human neuronal nicotinic receptors. Prog Neurobiol. 1997;53(2):199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- 13.Barik J, Wonnacott S. Indirect modulation by alpha7 nicotinic acetylcholine receptors of noradrenaline release in rat hippocampal slices: interaction with glutamate and GABA systems and effect of nicotine withdrawal. Mol Pharmacol. 2006;69(2):618–628. doi: 10.1124/mol.105.018184. [DOI] [PubMed] [Google Scholar]
- 14.Haass M, Kubler W. Nicotine and sympathetic neurotransmission. Cardiovasc Drugs Ther. 1997;19(6):657–665. doi: 10.1007/BF00053022. [DOI] [PubMed] [Google Scholar]
- 15.Tachikawa E, Mizuma K, Kudo K, Kashimoto T, Yamato S, Ohta S. Characterization of the functional subunit combination of nicotinic acetylcholine receptors in bovine adrenal chromaffin cells. Neurosci Lett. 2001;312(3):161–164. doi: 10.1016/s0304-3940(01)02211-x. [DOI] [PubMed] [Google Scholar]
- 16.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154(8):1558–1571. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuller HM. Cell type specific, receptor-mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem Pharmacol. 1989;38(20):3439–3442. doi: 10.1016/0006-2952(89)90112-3. [DOI] [PubMed] [Google Scholar]
- 18.Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci U S A. 1990;87(9):3294–3298. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuller HM. Neurotransmission and cancer: implications for prevention and therapy. Anticancer Drugs. 2008;19(7):655–671. doi: 10.1097/CAD.0b013e3283025b58. [DOI] [PubMed] [Google Scholar]
- 20.Park PG, Merryman J, Orloff M, Schuller HM. Beta-adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: implications for individuals with preexisting chronic lung disease. Cancer Res. 1995;55(16):3504–3508. [PubMed] [Google Scholar]
- 21.Schuller HM, Tithof PK, Williams M, Plummer H., 3rd The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999;59(18):4510–4515. [PubMed] [Google Scholar]
- 22.Majidi M, Al-Wadei HA, Takahashi T, Schuller HM. Nongenomic beta estrogen receptors enhance beta1 adrenergic signaling induced by the nicotine-derived carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in human small airway epithelial cells. Cancer Res. 2007;67(14):6863–6871. doi: 10.1158/0008-5472.CAN-07-0483. [DOI] [PubMed] [Google Scholar]
- 23.Weddle DL, Tithoff P, Williams M, Schuller HM. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis. 2001;22(3):473–479. doi: 10.1093/carcin/22.3.473. [DOI] [PubMed] [Google Scholar]
- 24.Askari MD, Tsao MS, Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol. 2005;131(10):639–648. doi: 10.1007/s00432-005-0002-7. [DOI] [PubMed] [Google Scholar]
- 25.Shin VY, Wu WK, Chu KM, Koo MW, Wong HP, Lam EK, et al. Functional role of beta-adrenergic receptors in the mitogenic action of nicotine on gastric cancer cells. Toxicol Sci. 2007;96(1):21–29. doi: 10.1093/toxsci/kfl118. [DOI] [PubMed] [Google Scholar]
- 26.Masur K, Niggemann B, Zanker KS, Entschladen F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers. Cancer Res. 2001;61(7):2866–2869. [PubMed] [Google Scholar]
- 27.Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol Appl Pharmacol. 2007;221(3):261–267. doi: 10.1016/j.taap.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 28.Palm D, Lang K, Niggemann B, Drell TLt, Masur K, Zaenker KS, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 2006;118(11):2744–2749. doi: 10.1002/ijc.21723. [DOI] [PubMed] [Google Scholar]
- 29.Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, et al. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. 2006;12(2):369–375. doi: 10.1158/1078-0432.CCR-05-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- 31.Vanhoose AM, Ritchie MD, Winder DG. Regulation of cAMP levels in area CA1 of hippocampus by Gi/o-coupled receptors is stimulus dependent in mice. Neurosci Lett. 2004;370(1):80–83. doi: 10.1016/j.neulet.2004.07.093. [DOI] [PubMed] [Google Scholar]
- 32.Hirano T, Watanabe D, Kawaguchi SY, Pastan I, Nakanishi S. Roles of inhibitory interneurons in the cerebellar cortex. Ann N Y Acad Sci. 2002;978:405–412. doi: 10.1111/j.1749-6632.2002.tb07583.x. [DOI] [PubMed] [Google Scholar]
- 33.Markou A. Review. Neurobiology of nicotine dependence. Philos Trans R Soc Lond B Biol Sci. 2008;363(1507):3159–3168. doi: 10.1098/rstb.2008.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawai H, Berg DK. Nicotinic acetylcholine receptors containing the alpha 7 subunits on rat cortical neurons do not undergo long lasting inactivation even when upregulated byc chronic nicotine exposure. J Neurochem. 2001;78(6):1367–1378. doi: 10.1046/j.1471-4159.2001.00526.x. [DOI] [PubMed] [Google Scholar]
- 35.Joseph J, Niggemann B, Zaenker KS, Entschladen F. The neurotransmitter gamma-aminobutyric acid is an inhibitory regulator for the migration of SW 480 colon carcinoma cells. Cancer Res. 2002;62(22):6467–6469. [PubMed] [Google Scholar]
- 36.Schuller HM, Al-Wadei HA, Majidi M. GABA(B) receptor is a novel drug target for pancreatic cancer. Cancer. 2008;112(4):767–778. doi: 10.1002/cncr.23231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuller HM, Al-Wadei HA, Majidi M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis. 2008;29(10):1979–1985. doi: 10.1093/carcin/bgn041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuller HM, Witschi HP, Nylen E, Joshi PA, Correa E, Becker KL. Pathobiology of lung tumors induced in hamsters by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the modulating effect of hyperoxia. Cancer Res. 1990;50(6):1960–1965. [PubMed] [Google Scholar]
- 39.Oreffo VI, Lin HW, Padmanabhan R, Witschi H. K-ras and p53 point mutations in 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced hamster lung tumors. Carcinogenesis. 1993;14(3):451–455. doi: 10.1093/carcin/14.3.451. [DOI] [PubMed] [Google Scholar]
- 40.Schuller HM, Jorquera R, Reichert A, Castonguay A. Transplacental induction of pancreas tumors in hamsters by ethanol and the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1993;53(11):2498–2501. [PubMed] [Google Scholar]
- 41.Schuller HM, Zhang L, Weddle DL, Castonguay A, Walker K, Miller MS. The cyclooxygenase inhibitor ibuprofen and the FLAP inhibitor MK886 inhibit pancreatic carcinogenesis induced in hamsters by transplacental exposure to ethanol and the tobacco carcinogen NNK. J Cancer Res Clin Oncol. 2002;128(10):525–532. doi: 10.1007/s00432-002-0365-y. [DOI] [PubMed] [Google Scholar]
- 42.Schuller HM, Porter B, Riechert A, Walker K, Schmoyer R. Neuroendocrine lung carcinogenesis in hamsters is inhibited by green tea or theophylline while the development of adenocarcinomas is promoted: implications for chemoprevention in smokers. Lung Cancer. 2004;45(1):11–18. doi: 10.1016/j.lungcan.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Dizimiri N, Basco C, Moorji A, Afrane B, Al-Halees Z. Characterization of lymphocyte beta- 2-adrenoreceptor signaling in patients with left ventricular volume overload disease. Clin Exp Pharmacol Physiol. 2002;29(3):181–188. doi: 10.1046/j.1440-1681.2002.03625.x. [DOI] [PubMed] [Google Scholar]
- 44.Laag E, Majidi M, Cekanova C, Masi T, Takahashi T, Schuller HM. NNK activates ERK1/2 and CREB/ATF-1 via beta-1-AR and EGFR signaling in human lung adenocarcinoma and small airway epithelial cells. Int J Cancer. 2006;119(7):1547–1552. doi: 10.1002/ijc.21987. [DOI] [PubMed] [Google Scholar]
- 45.Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7(1):91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- 46.Benowitz NL. Cigarette smoking and cardiovascular disease: pathophysiology and implications for treatment. Prog Cardiovasc Dis. 2003;46(1):91–111. doi: 10.1016/s0033-0620(03)00087-2. [DOI] [PubMed] [Google Scholar]
- 47.Schuller HM, Al-Wadei HA, Majidi M. GABA B receptor is a novel drug target for pancreatic cancer. Cancer. 2008;112(4):767–778. doi: 10.1002/cncr.23231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drell TLt, Joseph J, Lang K, Niggemann B, Zaenker KS, Entschladen F. Effects of neurotransmitters on the chemokinesis and chemotaxis of MDA-MB-468 human breast carcinoma cells. Breast Cancer Res Treat. 2003;80(1):63–70. doi: 10.1023/A:1024491219366. [DOI] [PubMed] [Google Scholar]
- 49.Al-Wadei HA, Majidi M, Tsao AS, Schuller HM. Low concenrtations of beta-carotene stimulate the proliferation of human pancreatic duct epitheial cells in a PKA-dependent manner. Cancer Genomics Proteomics. 2007;4(1):35–42. [PubMed] [Google Scholar]
- 50.Al-Wadei HA, Schuller HM. Cyclic adenosine monophosphate-dependent cell type-specific modulation of mitogenic signaling by retinoids in normal and neoplastic lung cells. Cancer Detect Prev. 2006;30(5):403–411. doi: 10.1016/j.cdp.2006.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Wadei HA, Takahashi T, Schuller HM. Growth stimulation of human pulmonary adenocarcinoma cells and small airway epithelial cells by beta-carotene via activation of cAMP, PKA, CREB and ERK1/2. Int J Cancer. 2006;118(6):1370–1380. doi: 10.1002/ijc.21537. [DOI] [PubMed] [Google Scholar]
- 52.Al-Wadei HA, T T, Schuller HM. Caffeine stimulates the proliferation of human lung adenocarcinoma cells and small airway epithelial cells via activation of PKA, CREB and ERK1/2. Oncol Rep. 2006;15(2):431–435. [PubMed] [Google Scholar]
- 53.Schuller HM, Al-Wadei HA, Majidi M. Gamma-aminobutyric acid, a potential tumor suppressor for small airway-derived lung adenocarcinoma. Carcinogenesis. 2008 doi: 10.1093/carcin/bgn041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53(4):606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- 55.Mozayan M, Lee TJ. Statins prevent cholinesterase inhibitor blockade of sympathetic alpha7nAChR-mediated currents in rat superior cervical ganglion neurons. Am J Physiol Heart Circ Physiol. 2007;293(3):H1737–H1744. doi: 10.1152/ajpheart.00269.2007. [DOI] [PubMed] [Google Scholar]
- 56.Belinsky SA, Devereux TR, Anderson MW. Role of DNA methylation in the activation of proto-oncogenes and the induction of pulmonary neoplasia by nitrosamines. Mutat Res. 1990;233(1–2):105–116. doi: 10.1016/0027-5107(90)90155-w. [DOI] [PubMed] [Google Scholar]