

Abstract
Mortalin was over expressed in tumor cells and bind to p53 protein. This interaction was suggested to promote sequestration of p53 in the cytoplasm, thereby inhibiting its nuclear activity. The p53 is a tumor suppressor that is essential for the prevention of cancer development and loss of p53 function is one of the early events in immortalization of human cells. Therefore, abrogation p53-mortalin interaction using small molecule is guaranteed stop cancer cell grow. However study interaction of p53-mortalin, and its inhibition using small molecule is still challenging because specific site of mortalin that bind to p53, vice versa, is still debatable. This study has aims to analyze the p53-binding site of mortalin using molecular docking and to screen drug-like compounds that have potential as inhibitors of p53-mortalin interaction using virtual screening. The result showed that the lowest energy binding of p53-mortalin complex is -31.89 kcal/mol, and p53 protein bind to substrate binding domain of mortalin (THR433; VAL435; LEU436; LEU437; PRO442; ILE558; LYS555). Furthermore, the p53-binding domain of mortalin was used as receptor to screen 9000 drug-like compounds from ZINC database using molecular docking program Auto Dock Vina in PyRx 0.8 (Virtual Screening Tools). Here, we have identified three drug-like compounds that are ZINC01019934, ZINC00624418 and ZINC00664532 adequate to interrupt stability of p53-mortalin complex that warrant for anticancer agent.
Keywords: Mortalin, p53, Virtual screening, Docking, Drug-like compounds
Background
Protein–protein interactions (PPIs) are essential components of all biological processes such as oncogenesis [1]. Several observations suggested that mortalin is involved in the transformation of normal cells to cancer-cells [2], which mortalin interacted with p53 that promotes sequestration of p53 in the cytoplasm, thereby inhibiting its nuclear activity [3] and induce the resistance of tumors in radiotherapy and chemotherapy. Mortalin changes its sub-cellular location from mitochondria of normal cells to cytosol, of cancerous cells [4]. Protein of p53, the “guardian of the genome,” is a major player in cell cycle arrest and apoptosis in response to the diverse endogenous and exogenous stress signals [5]. The p53 is a tumor suppressor that is essential for the prevention of cancer development [6] . Loss of p53 function is one of the early events in immortalization of human cells [7]. Another function of p53, independent to its nuclear localization, is to regulate mitochondrial membrane potential by interactions with the mitochondrial proteins bcl2 and mortalin [8]. Furthermore inhibition of p53-mortalin complex using small molecule is promising strategy to find drug materials for cancer therapy since the small molecules able to inhibit interaction between two proteins [9]. However study interaction p53-mortalin and its inhibition using small molecule is still challenging because specific site of mortalin that bind to p53, vice versa, is still debatable. Experimental analysis by truncated protein recombinant suggested that mortalin-binding site of p53 was localized to the tetramerization domain, namely amino acids 323–337 residues [5]. Conversely, the binding site for p53 on mortalin was mapped to amino acids 253–282 residues, in the ATPase domain [10]. Otherwise Iosefson demonstrated that p53 binds to the peptide-binding domain of mortalin and not to the ATP-binding site of mortalin [11]. P53 harbors two binding sites for mortalin. The first one was previously determined to be in the tetramerization domain [5], and the second is found in the C-terminal domain [11]. Moreover we elucidated the binding site in both of the protein (p53 and mortalin) by molecular docking method. The docking method was powerful to predict specific binding site of protein-protein complex [12]. Here we demonstrated that mortalin-binding site of p53 reside in 326 to 341 residues, and p53-binding site of mortalin hold in range 423 to 450 residues. Based on the binding activity, then we analyzed its binding stability when been interfered with small molecule to obtain drug candidate for cancer therapy. Furthermore we identified small molecules inhibitor of p53-mortalin complex. Here we found 3 drug-like compounds from Zinc database that potentially abrogate p53-mortalin interaction using Virtual Screening.
Methodology
Retrieval of Target Sequence:
The amino acid sequence of mortalin ID: NP_000537.3 was obtained from the sequence database of NCBI (http://www.ncbi.nlm.nih.gov), and model of p53 (1AIE) was from PDB BANK (http://www.rscb.org).
Modeling proteins mortalin:
The tertiary structure of mortalin is not available in the protein structure databases, and then the structure was predicted by comparative modeling using MODELLER (PS2 Server) [13]. The predicted three dimension (3D) structures were validated using the PROCHECK to evaluate backbone conformation based on Psi/Phi Ramachandran plot analysis.
Docking protein – protein:
We used cons-PPISP to predict a site for interaction of p53 and mortalin. The input is the unbound structure of a protein, which is known to bind another protein [14]. Then we used the PatchDock algorithm (rigid docking) to compute possible interaction p53 with mortalin based on its interaction site. Output of rigid docking was refined using FireDock. FireDock refines the candidate models according to an energy function [15]. Thereafter, we used FiberDock software to simulate most possible native complex structure of mortalin-p53 in flexible mode, which are include both backbone and side-chains movements [16].
Ligand Preparation:
About 9000 drug-like compounds were downloaded from zinc database (http://zinc.docking.org), then its energy form were minimized and converted to pdbqt format by Open Babel in PyRx 0.8 [12] as ligand for virtual screening.
Virtual Screening:
Identifications small molecules targeted mortalin was done by docking of the entire 9000 drug-like compounds from ZINC database against p53-binding domain of mortalin using AutoDock Vina in PyRx 0.8 (Virtual Screening Tools) [12]. Virtual screening is now established as an effective paradigm for filtering compounds for drug discovery process [17]. The grid for docking calculations was centered on p53-binding domain of mortalin. The best three of drug-like compounds from virtual screening were taken on the basis of higher scoring function for pharmachopore modeling, which is one of the most powerful techniques to classify and identify key features from a group of molecules. LigandScout was used to develop pharmacophore and molecular interaction of these three druglike.
Molecular Dynamics Simulation:
The binding stability of p53-mortalin complex before and after interference by the small molecules was simulated using GroGUI 0.5.2 version of GROMACS MD simulation package. The first steps are preparation of the atomic and topology of ligand, receptor and the small molecules. Second steps are made box water for simulation and then energy minimization is performed in order to eliminate potential geometric problems in the protein and ligand structures, such as unrealistic bond distances, bond angles and torsion angles. Next step involved the equilibration of the solute molecules with a fixed configuration of the solvent molecules in which the system was heated 300K with Berendsen temperature coupling and Pressure coupling with Parrinello-Rahman (isotropic). The Particle Mesh Ewald (PME) method was used to treat longrange electrostatic interactions. The coordinates of the trajectory was sampled every 2.5 ps for analysis of the energy stabilization Plots for potential energy, RMSD fluctuations, involving p53- mortalin and other complexes [18].
Visualization:
All the visualization of the structure files were done using PyMol molecular graphics system (www.pymol.org).
Discussion
Designing anti-cancer drug targeted p53 protein [19] or p53 binding-domain of mortalin is very promising [10]. However the p53-binding site of mortalin still debatable [5] [10], then we analyzed the binding site based on their interaction using molecular docking. Since the three dimensions (3D) structure of mortalin was still not available yet then we used 3D model of mortalin to determine it's binding to p53 protein. Model 3D of mortalin was determined by comparative modeling using MODELLER (PS2 Server). The quality and reliability of the model was ensured by assessing the backbone conformation, angels and bond lengths based on Psi/Phi Ramachandran plot using PROCHECK, it showed a total of 93.7% residues in the most favored regions (Figure 1).
Figure 1.
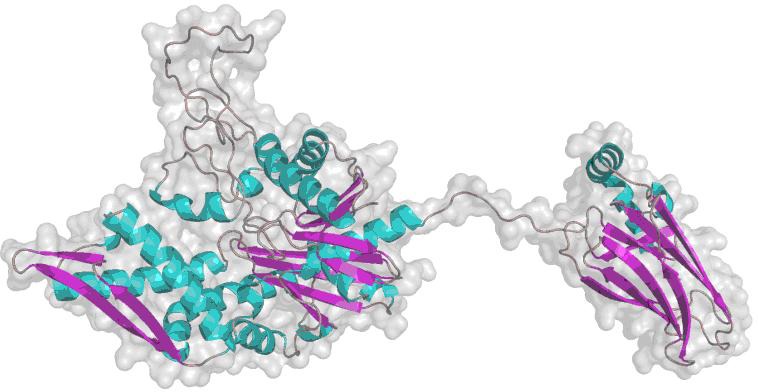
3D model of mortalin.
Furthermore, we used the valid model to analyze p53-binding site of mortalin based on the flexible docking. The result showed that the lowest energy binding of p53-mortalin complex is -31.89 kcal/mol, and p53 protein bind to substrate binding domain of mortalin (THR433; VAL435; LEU436; LEU437; PRO442; ILE558; LYS555) (Figure 2). The result suggested that mortalin-binding site of p53 reside in 326 to 341 residues, in accordance with opinion Kaul et al (1998) [5], and p53-binding site of mortalin in 423 to 450 residues that it strengthens Iosefson data (2010) [11].
Figure 2.
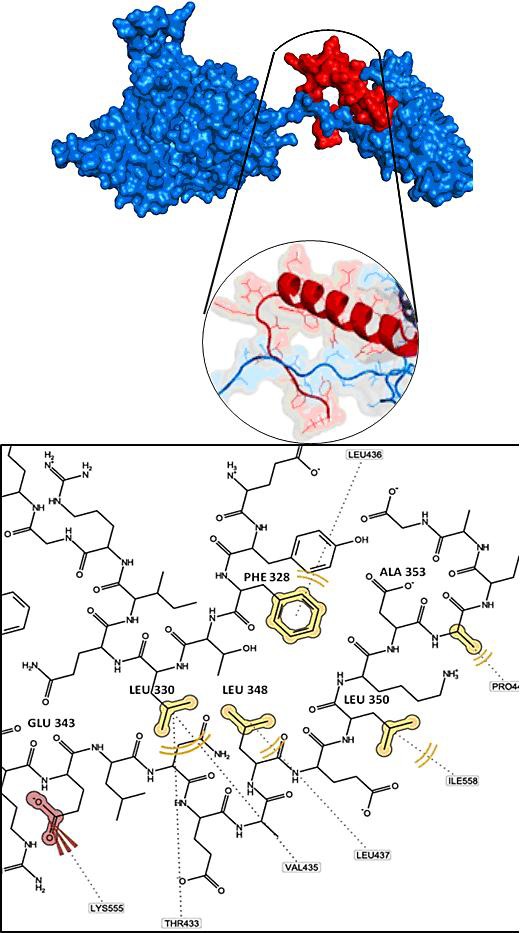
p53-mortalin complex. p53 (red color) binds to mortalin (blue color) on Substrate Binding Domain (THR433; VAL435; LEU436; LEU437; PRO442; ILE558; LYS555).
The p53-binding domain of mortalin was used as receptor to screen 9000 drug-like compounds (small molecules) from ZINC database. The screening was performed using molecular docking program AutoDock Vina in PyRx 0.8 (Virtual Screening Tools) [12]. The virtual screening obtained the best three druglike molecules that are ZINC01019934, ZINC00624418 and ZINC00664532, which are require smaller energy than other molecules to bind with mortalin (Figure 3). These data indicated that the three molecules were able bond strongly to p53-binding site of mortalin and its binding will inhibit p53 protein interact to mortalin. Then we evaluated interaction of the three molecules to the p53-mortalin complex by docking followed with ligandscout. The result demonstrated that the three molecules bound to amino acid in center of the active site of both mortalin and p53, which are essential for establishment p53-mortalin complex (Figure 4). Hereafter, we evaluated the binding stability of the three molecules to p53-mortalin complex using GroGUI 0.5.2 version of GROMACS molecular dynamics (MD) simulation package [18]. MD simulations have provided detailed information on the fluctuations and conformational changes in protein complex [20].
Figure 3.
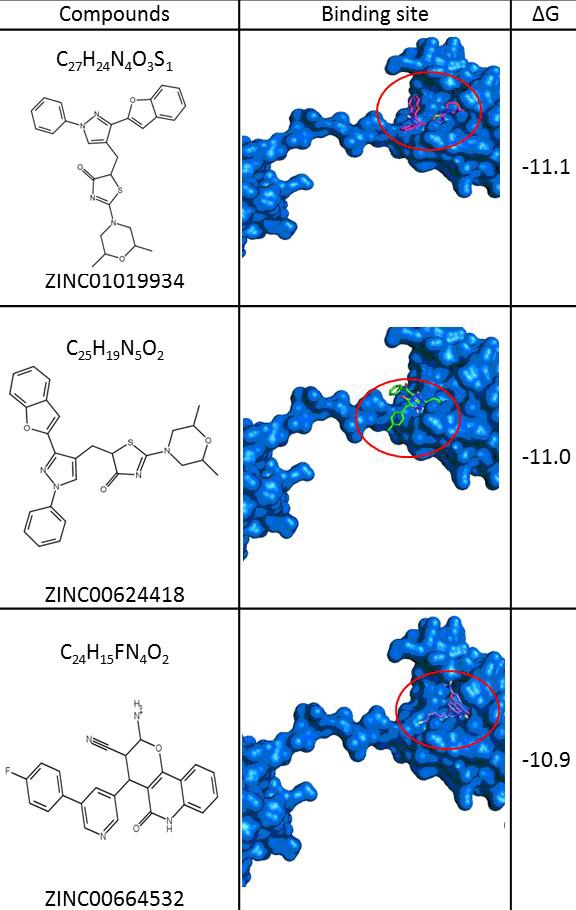
The best three small molecules bind to p53-binding site of mortalin (ZINC01019934, ZINC00624418, and ZINC00664532).
Figure 4.
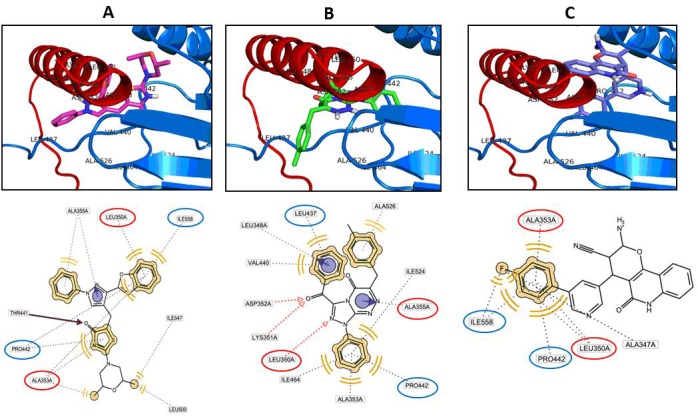
Ligands bind to complex p53-mortalin. A. ZINC01019934 blocks amino acid LEU 350 and ALA 353 of p53 protein and blocks amino acid PRO 442 and ILE 558 of mortalin protein. B. ZINC00624418 blocks amino acid LEU 350 and ALA 353 of p53 protein and blocks amino acid LEU 437 and PRO 442 of mortalin protein. C. ZINC00664532 blocks amino acid LEU 350 and ALA 353 of p53 and blocks amino acid PRO 442 and ILE 558 of mortalin.
The simulation described that the three molecules interrupted stability of p53-mortalin complex. This is indicated by an increase binding energy of mortalin-p53 complex and root mean square deviations (RMSD) of the protein complexes have changed significantly after added with the small molecule (Figure 5). Taken together the results suggested that the three molecules have potency to abrogate p53-mortalin interaction that warrant for anticancer agent. Therefore further experiment to examine its activities in vitro and in vivo to cure cancer is still shall to be done.
Figure 5.
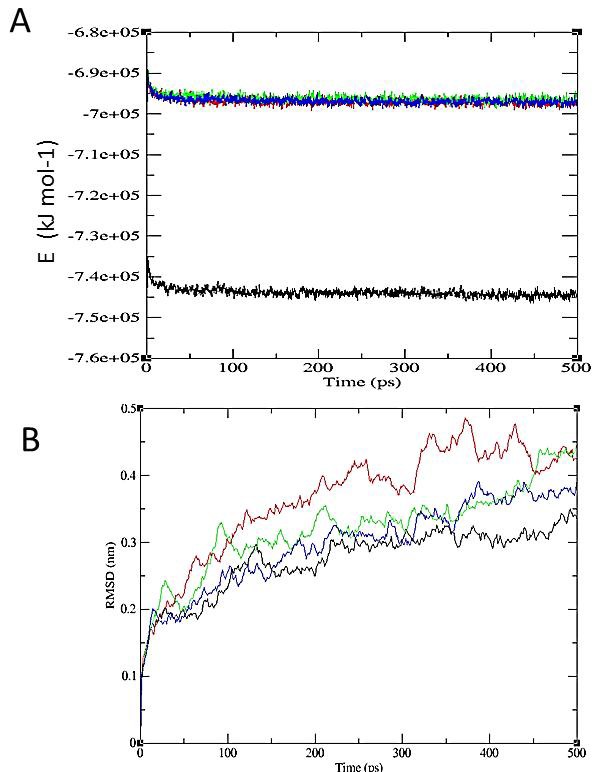
The stability of p53-mortalin complexes. A. Energy stability of p53-mortalin complex (black line) increase after added the small molecules (blue, red and green line). B. RMSD of p53-mortalin complex (black line) has more unstable after interfered with the small molecules (green, blue and red line). The red color indicates ZINC01019934, green is ZINC00624418, and blue is ZINC00664532.
Conclusion
These studies could be concluded that p53-binding site of mortalin reside in substrate binding domain, 423 to 450 residues. We have identified three drug-like molecules that are ZINC01019934, ZINC00624418 and ZINC00664532 adequate to interrupt stability of p53-mortalin complex that warrant for anticancer agent.
Footnotes
Citation:Utomo et al, Bioinformation 8(9): 426-429 (2012)
References
- 1.LL Blazer, RR Neubig. Neuropsychopharmacology. 2009;34:126. [Google Scholar]
- 2.R Wadhwa, et al. Int J Cancer. 2006;118:2973. doi: 10.1002/ijc.21773. [DOI] [PubMed] [Google Scholar]
- 3.SC Kaul, et al. Exp Gerontol. 2007;42:263. doi: 10.1016/j.exger.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 4.R Wadhwa, et al. J Biol Chem. 1998;273:29586. doi: 10.1074/jbc.273.45.29586. [DOI] [PubMed] [Google Scholar]
- 5.SC Kaul, et al. Oncoqene. 1998;17:907. [Google Scholar]
- 6.AJ Levine. Cell. 1997;88:323. [Google Scholar]
- 7.X Yi, et al. Mol Cell Proteomics. 2008;7:315. doi: 10.1074/mcp.M700116-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.SC Kaul, et al. FEBS Lett. 2000;474:159. doi: 10.1016/s0014-5793(00)01594-5. [DOI] [PubMed] [Google Scholar]
- 9.PJ Hajduk, DJ Burns. Comb Chem High Throughput Screen. 2002;5:613. doi: 10.2174/1386207023329996. [DOI] [PubMed] [Google Scholar]
- 10.SC Kaul, et al. Exp Cell Res. 2003;286:96. doi: 10.1016/s0014-4827(03)00101-0. [DOI] [PubMed] [Google Scholar]
- 11.O Iosefson, A Azem. FEBS Lett. 2010;584:1080. doi: 10.1016/j.febslet.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 12.O Trott, AJ Olson. J Comput Chem. 2010;31:455. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.CC Chen, et al. Nucleic Acids Res. 2006;34:W152. [Google Scholar]
- 14.H Chen, HX Zhou. Proteins. 2005;61:21. [Google Scholar]
- 15.E Mashiach, et al. Nucleic Acids Res. 2008;36:W229. [Google Scholar]
- 16.E Mashiach, et al. Nucleic Acids Res. 2008;36:W2296. [Google Scholar]
- 17.M Jalaie, V Shanmugasundaram. Mini Rev Med Chem. 2006;6:1159. doi: 10.2174/138955706778560157. [DOI] [PubMed] [Google Scholar]
- 18.Jr De Azevedo WF. Curr Med Chem. 2011;18:1353. doi: 10.2174/092986711795029519. [DOI] [PubMed] [Google Scholar]
- 19.T Nurseta, et al. Indonesian Journal of Obstetrics and Gynecology. 2011;35:1. [Google Scholar]
- 20.H Benyamini, et al. Proteins. 2009;77:602. doi: 10.1002/prot.22473. [DOI] [PubMed] [Google Scholar]