

Initial crystallographic studies of the X. fastidiosa small heat-shock protein HSP17.9 are reported.
Keywords: heat-shock proteins, Xylella fastidiosa, HSP17.9
Abstract
The ORF XF2234 in the Xylella fastidiosa genome was identified as encoding a small heat-shock protein of 17.9 kDa (HSP17.9). HSP17.9 was found as one of the proteins that are induced during X. fastidiosa proliferation and infection in citrus culture. Recombinant HSP17.9 was crystallized and surface atomic force microscopy experiments were conducted with the aim of better characterizing the HSP17.9 crystals. X-ray diffraction data were collected at 2.7 Å resolution. The crystal belonged to space group P4322, with unit-cell parameters a = 68.90, b = 68.90, c = 72.51 Å, and is the first small heat-shock protein to crystallize in this space group.
1. Introduction
Heat-shock proteins (HSPs) are molecular chaperones that prevent protein misfolding and aggregation of newly synthesized proteins. Expression of HSPs may be induced as a stress response to temperature or chemical agents and in the pathogenesis process of infectious organisms. The HSP superfamily is divided according to the molecular mass and sequence identity of its members (Lindquist, 1992 ▶; Li & Srivastava, 2003 ▶).
The small heat-shock proteins (smHSPs) are included in a family of HSPs containing proteins of molecular mass from 12 to 43 kDa (de Jong et al., 1998 ▶) that are structurally divergent from other HSPs. They are ATP-independent proteins that form large and dynamic oligomers. Members of this family present a characteristic α-crystallin structural domain flanking their carboxy- and amino-terminal ends. The α-crystallin fold consists of two β-sheets composed of 3–4 antiparallel β-strands which form strong hydrogen-bond networks within each sheet, a structural organization that is related to the resistance of the smHSPs to high temperatures and other stress agents (Poulain et al., 2010 ▶; Stamler et al., 2005 ▶; Kim et al., 1998 ▶; van Montfort et al., 2001 ▶). The N-terminal region contains structurally diverse elements with no identified correlation among the members of the smHSP family. This region is mainly polar and highly disordered. Recently, a conserved hydrophobic region was found at the C-terminal end of smHSP members which is related to the high propensity of smHSP to form oligomers (Poulain et al., 2010 ▶).
A small number of smHSP crystal structures have been reported, most of which display hexagonal symmetry, the exceptions being the orthorhombic crystal structures of TSP36 from Taenia saginata (PDB entry 2bol; Stamler et al., 2005 ▶) and HSP14.0 from Sulfolobus tokodaii (PDB entry 3aab; also crystallizes in the tetragonal space group P41, PDB entry 3aac; Takeda et al., 2011 ▶) and the monoclinic α-crystallin domain of rat HSP20 (PDB entry 2wj5; Bagnéris et al., 2009 ▶). Cryo-electron microscopy structures are also available, including an oligomeric arrangement of yeast HSP26 formed by 24 subunits (White et al., 2006 ▶) and a dodecameric HSP16.3 oligomer from Mycobacterium tuberculosis (Kennaway et al., 2005 ▶).
The smHSP from Xylella fastidiosa, the infectious agent of citrus variegated chlorosis (CVC), was identified after genomic sequencing of the phytopathogen by the Brazilian Consortium ONSA (Simpson et al., 2000 ▶). HSP17.9 is the gene product of ORF XF2234 in the X. fastidiosa 9a5c genome, which results in a protein of 17.9 kDa (Azzoni et al., 2004 ▶). Citrus culture plays an important role in the Brazilian economy and infection caused by X. fastidiosa is responsible for a loss of approximately US $280–320 million per year (http://www.fundecitrus.com.br/). Therefore, it is important to understand the molecular roles of the proteins involved in X. fastidiosa pathogenesis, with the aim of developing biotechnological tools to prevent and combat this infection.
In the present work, we report the crystallization, surface atomic force microscopy (AFM) crystal analysis, X-ray data collection and initial structure solution of HSP17.9 in a crystal form which has not been observed for other members of the smHSP family.
2. Material and methods
2.1. Crystallization and X-ray data collection
2.1.1. Crystallization
Protein cloning, expression and purification were conducted as described by Azzoni et al. (2004 ▶). Briefly, the full-length HSP17.9 gene (UniProtKB Q9PBB0) was subcloned into pET32-Xa/LIC (Novagen) and gene integrity was confirmed by DNA sequencing. HSP17.9 was expressed in Escherichia coli BL21 (DE3) cells (Novagen) and purified using Ni–NTA resin (Qiagen). To remove the thioredoxin-His6 tag, the pooled fractions were cleaved by factor Xa (New England BioLabs) overnight followed by inactivation of the protease by PMSF, and loaded onto an Ni–NTA column to remove the fusion tag. In the final step, HSP17.9 (residues 1–160) was dialyzed into 5 mM Tris–HCl pH 7.5. The identity of the target protein as well as its molecular mass was confirmed by mass spectrometry.
Purified HSP17.9 samples were submitted to initial crystallization experiments using the JBScreen Classic Kits 1–10 (Jena Bioscience) and the sitting-drop vapour-diffusion method in 24-well plates; manually prepared drops composed of 2 µl protein solution at 8 mg ml−1 (in 5 mM Tris–HCl pH 7.5) and 2 µl well solution were equilibrated against 300 µl well solution. Small and weakly diffracting crystals (resolution limit lower than ∼3.6 Å) were obtained at 291 K after 1–2 weeks in condition B5 of JBScreen Classic Kit 9 [0.2 M sodium citrate tribasic dehydrate, 0.1 M HEPES sodium salt pH 7.5, 20%(v/v) 2-propanol]. In parallel, a Honeybee 963 Pipettor robot (Genomic Solutions) at the Laboratório Nacional de Biociências (LNBio, Campinas, Brazil) was used to find further crystallization conditions with the sitting-drop vapour-diffusion method in 96-well plates. The commercial crystallization kits Crystal Screen, Crystal Screen 2 and SaltRx (Hampton Research), Wizard I and II and Precipitant Synergy (Emerald BioSystems) and PACT/JCSG+ (NeXtal/Qiagen) were used in the screens (544 conditions in total); drops consisting of 1 µl well solution and 1 µl protein solution were equilibrated against 80 µl well solution at 291 K. These experiments were not successful in finding another crystal form, as crystals only appeared in condition No. 27 of Crystal Screen, which is exactly the same as the JBscreen condition mentioned above.
Attempts to improve crystal size and diffraction were performed by changing the buffer pH, the precipitant and protein concentration in manual mode using the hanging-drop technique. Tissue-culture test plates (TPP, 24 wells) were prepared with 500 µl crystallization solution in the well and drops consisting of 2 µl protein solution and 2 µl well solution. The temperature was maintained at 291 K. Screenings with various additives were also conducted using detergents, organic solvents, carbohydrates, cryoprotective agents and salts. Compared with the initial crystallization conditions, an improvement in the diffraction patterns was observed for crystals grown in a cryosolution containing ethylene glycol, as described below.
2.1.2. X-ray data collection
X-ray data collection was performed on the W01B-MX2 beamline of the Laboratório Nacional de Luz Síncrotron (LNLS; Campinas, Brazil; Guimarães et al., 2009 ▶). Diffraction images were recorded using a MAR Mosaic CCD 225 detector with a crystal-to-detector distance of 160 mm. 1° oscillation frames were collected with 60 s exposure time to give a total of 247 images. To prevent radiation damage, the crystal was flash-cooled in a nitrogen-gas stream at 100 K (Oxford Cryosystems). Several cryoprotectant agents were screened and different strategies were tested. The best diffracting crystals (Fig. 1 ▶) were grown in the presence of ethylene glycol and were obtained from hanging drops consisting of 2 µl HSP17.9 at 10 mg ml−1 and 2 µl well solution [0.1 M HEPES sodium salt pH 7.5, 0.3 M sodium citrate, 20%(v/v) 2-propanol, 15%(v/v) ethylene glycol].
Figure 1.

A typical crystal of HSP17.9, with maximum dimension 100 µm.
2.2. Microscopy of HSP17.9 crystals
Protein crystals were analyzed by AFM in their original mother solutions. Measurements were carried out with an AutoProbe CP (ThermoMicroscopes, Minnesota, USA) using Si cantilevers (k ≃ 3.2 N m−1) in liquid medium (nonreplenishing open cell). Topographic images were acquired using a noncontact mode. To avoid dampening effects arising from the viscous solution in which imaging took place, crystals lying close to the surface of the liquid were scanned with only the tip of the probe immersed in the solution. In some cases, images were acquired repeatedly for long periods of time (a few hours) to check for crystal stability. Height profiles and r.m.s. roughness were used for surface analysis.
3. Results and discussion
3.1. Microscopic analysis
The size, resolution limit and mosaicity of protein crystals are impaired by the presence of impurities in the growth solution; this is a major source of defects and disorder which affect crystal diffraction properties (Malkin & Thorne, 2004 ▶; McPherson et al., 2000 ▶). A better understanding of the crystallization process may lead to alternative approaches in which these problems may be prevented or ameliorated, ultimately resulting in higher quality crystals and more accurate structures (Malkin & Thorne, 2004 ▶; McPherson et al., 2004 ▶). AFM is a relatively simple technique which has been successfully used to follow and evaluate macromolecular crystal growth. Such analysis reveals growth patterns and defects, providing insights into improving growth, diffraction and cryoprotection, particularly in the case of crystals with high solvent content, as these are particularly affected by cryocrystallographic techniques (Malkin & Thorne, 2004 ▶; McPherson et al., 2001 ▶). As part of our efforts to obtain better diffracting crystals, surface AFM experiments were carried out in an attempt to gain information on the crystallization process.
The AFM topography of HSP17.9 crystals shows smooth surfaces with surface meandering terraces (Fig. 2 ▶ a). These topographical characteristics are typical of a two-dimensional growth mode (McPherson et al., 2003 ▶; Durbin & Carlson, 1992 ▶; Land et al., 1995 ▶). Step edges in these images are about ∼3 nm high. One would usually expect the AFM step height to be similar to (or a multiple of) the unit-cell parameter (d) perpendicular to the crystallographic surface in the image (Land et al., 1995 ▶, 1997 ▶; Durbin & Carlson, 1992 ▶). The unit-cell parameters obtained by X-ray crystallographic analysis are about 70 Å (Table 1 ▶), indicating that the step edges found in the AFM measurements correspond to approximately half of the unit-cell axis. There have been a few cases of the observation of step heights arising from the presence of a screw axis in the crystal structure (Malkin & Thorne, 2004 ▶).
Figure 2.

AFM topographies of an HSP17.9 crystal in the solution in which growth took place. (a) Image of the crystal surface, showing terraces, step edges and a depression (indicated by an arrow). (b, c) A closer look at the anisotropic structures found at the depression site. (d) Anisotropic structures located at the top of the crystal.
Table 1. Statistics of data collection and processing.
Values in parentheses are for the highest resolution shell.
| Beamline | W01B-MX2, LNLS |
| Wavelength (Å) | 1.4586 |
| Space group | P4322 |
| Unit-cell parameters (Å) | a = b = 68.90, c = 72.51 |
| Molecules in asymmetric unit | 1 |
| Solvent content (%) | 74.1 |
| Resolution limits (Å) | 40.45–2.90 (3.06–2.90) |
| No. of images | 247 |
| No. of reflections | 76140 (10879) |
| Unique reflections | 4089 (565) |
| Multiplicity | 18.6 (19.3) |
| Completeness (%) | 98.0 (97.5) |
| Rmerge† (%) | 11.5 (67.3) |
| 〈I/σ(I)〉 | 17.3 (3.5) |
| Wilson plot B factor (Å2) | 90.2 |
R
merge = 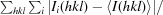
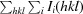 , where Ii(hkl) is the intensity of the ith observation of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl.
Typical macroscopic growth features associated with the presence of defects such as screw dislocations could not be identified in the images from several different crystals. On the other hand, depressions (Fig. 2 ▶ a, arrow) could occasionally be observed in specific surface regions. Taking a closer look at these regions, a different surface structure appears both within the depressions (Figs. 2 ▶ b and 2 ▶ c) and at the top surface of the crystal (Fig. 2 ▶ d). These structures are anisotropic in shape (hundreds of nanometres long and up to 5–6 nm high) and do not show any preferred direction on the surface; in fact, in some cases they seem to be fully confined to the top incomplete terraces forming the surface, as shown in Fig. 2 ▶(c). Also, in the region where it is present, surface roughness is usually greater than in the regions of the larger crystal terraces. Step-bunching mechanisms during crystal growth could give rise to such anisotropic structures (Schwoebel & Shipsey, 1966 ▶; Land et al., 1995 ▶; Malkin et al., 1997 ▶), but their apparent random direction and positioning on the surface, as well as the greater roughness values, are not consistent with this picture. These structures could also be an artifact resulting from surface wear owing to scanning as well as dehydration because of the diminishing amount of liquid during image acquisition. However, imaging the same region for longer periods of time did not show any significant topography.
Another interpretation of this anisotropic crystal shape is a different route for crystallization of the molecules in which strings of proteins are randomly distributed on the surface. The nucleation could follow a different path, for instance driven by changes in the environment where growth takes place. If this is the case these structures should be formed at lower supersaturations, when the solution is closer to protein depletion and contaminants can have a stronger effect on crystal growth (Caylor et al., 1999 ▶; De Yoreo & Dove, 2004 ▶). They could either be deposited from the solution or self-assemble on the surface from individual molecules. This latter scenario is more likely to explain the greater roughness observed around the anisotropic structures, as well as the depressions where they are usually found. Moreover, this would be compatible with the known propensity of smHSPs to form oligomers which, in principle, could act as macromolecular contaminants, a process that is difficult to avoid during crystal growth. In fact, it has been shown that one of the most harmful impurities which limits crystal quality consists of molecules exhibiting structural variability, including clusters or aggregates (McPherson et al., 2004 ▶). In this sense, it is noteworthy that an interesting case studied by AFM has been reported in which competition between different oligomeric forms might be responsible for the disorder and low resolution limit of the diffraction patterns (Larson et al., 2005 ▶). In the present study, the information obtained from the AFM analysis, although limited, helps us to understand the most probable reason for the relatively poor diffraction properties of the HSP17.9 crystals.
3.2. Diffraction data collection and processing
A HSP17.9 data set was collected as described in §2. A representative diffraction pattern with well defined spots to 2.7 Å resolution is presented in Fig. 3 ▶. Data were integrated with MOSFLM and scaled using SCALA (Winn et al., 2011 ▶). The results of data processing and the data statistics are summarized in Table 1 ▶. Initial indexing conducted with MOSFLM unequivocally indicated a tetragonal Bravais lattice. The possible symmetry of the diffraction patterns was assessed using POINTLESS (Evans, 2006 ▶). The systematic absences and probability analysis indicated P422 as the most probable point group, with a Laue-group confidence of higher than 0.99.
Figure 3.

A diffraction pattern from the crystal used in data collection. A higher resolution region of the image is shown with increased contrast. Rings are drawn at 2.4, 3.2, 4.8 and 9.7 Å resolution.
The space group was determined during phasing by molecular replacement conducted with the online BALBES pipeline (Long et al., 2008 ▶). At this stage, the HSP17.9 sequence and the scaled structure factors were submitted and the eight possibilities within point group P422 were tested. No structure with a similar unit cell and symmetry was found. The best solution was obtained in space group P4322, with a template model based on chain A of the crystal structure of an smHSP from Xanthomonas axonopodis pv. citri (PDB entry 3gla; Hilario et al., 2006 ▶, 2011 ▶), which has a sequence similarity to HSP17.9 of 88%. An HSP17.9 monomer was found in the asymmetric unit, corresponding to a solvent content of 74.1%. Automated restrained refinement using REFMAC (Murshudov et al., 2011 ▶; Winn et al., 2011 ▶) was performed as part of the BALBES protocol. The final R factor and R free (5% of the total reflections) were 29.2% and 32.1%, respectively. A clear contrast among the tested point groups, with a BALBES Q-factor parameter of 0.834 for the template model and a probability of 99% for the solution obtained, further supports its correctness. It is interesting to note that although HSP17.9 exhibits a tendency to form oligomeric complexes (Azzoni et al., 2004 ▶), in its function as a chaperone, a single monomer was found in the crystal asymmetric unit. Most probably, as in the case of other smHSPs, monodisperse oligomers might be assembled from dimers in solution (Takeda et al., 2011 ▶; Bagnéris et al., 2009 ▶; Stamler et al., 2005 ▶), thus providing a necessary condition for crystals to grow. Small changes in the chemical environment of the protein molecules can induce significant changes in the crystal characteristics (Malkin & Thorne, 2004 ▶). Conceivably, our inability to control the oligomerization process and thus the degree of monodispersity of the growing solution was the major impediment in improving the quality of HSP17.9 crystals.
4. Conclusion
To the best of our knowledge, this is the first example of an smHSP that crystallized in space group P4322. The step edges observed by AFM agree with the unit-cell dimensions obtained from the X-ray diffraction data. A flexible N-terminal domain and the tendency of HSP17.9 to form oligomers may be related to the anisotropic crystal shape and disorder, as reflected by the moderate X-ray data resolution. A correct molecular-replacement solution was found and the space group was unequivocally assigned. The refined structure will contribute to a better understanding of HSP17.9 and may provide further insights into the oligomerization of small heat-shock proteins.
Acknowledgments
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). RA, MAC and APS were also supported by a researcher grant from CNPq. We gratefully acknowledge LNLS for W01B-MX2 beamline time and LNBio for use of the Honeybee 963 Pipettor robot facility.
References
- Azzoni, A. R., Tada, S. F. S., Rosselli, L. K., Paula, D. P., Catani, C. F., Sabino, A. A., Barbosa, J. A. R. G., Guimarães, B. G., Eberlin, M. N., Medrano, F. J. & de Souza, A. P. (2004). Protein Expr. Purif. 33, 297–303. [DOI] [PubMed]
- Bagnéris, C., Bateman, O. A., Naylor, C. E., Cronin, N., Boelens, W. C., Keep, N. H. & Slingsby, C. (2009). J. Mol. Biol. 392, 1242–1252. [DOI] [PubMed]
- Caylor, C. L., Dobrianov, I., Lemay, S. G., Kimmer, C., Kriminski, S., Finkelstein, K. D., Zipfel, W., Webb, W. W., Thomas, B. R., Chernov, A. A. & Thorne, R. E. (1999). Proteins, 36, 270–281. [PubMed]
- De Yoreo, J. J. & Dove, P. M. (2004). Science, 306, 1301–1302. [DOI] [PubMed]
- Durbin, S. D. & Carlson, W. E. (1992). J. Cryst. Growth, 122, 71–79.
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Guimarães, B. G., Sanfelici, L., Neuenschwander, R. T., Rodrigues, F., Grizolli, W. C., Raulik, M. A., Piton, J. R., Meyer, B. C., Nascimento, A. S. & Polikarpov, I. (2009). J. Synchrotron Rad. 16, 69–75. [DOI] [PubMed]
- Hilario, E., Martin, F. J. M., Bertolini, M. C. & Fan, L. (2011). J. Mol. Biol. 408, 74–86. [DOI] [PubMed]
- Hilario, E., Teixeira, E. C., Pedroso, G. A., Bertolini, M. C. & Medrano, F. J. (2006). Acta Cryst. F62, 446–448. [DOI] [PMC free article] [PubMed]
- Jong, W. W. de, Caspers, G. J. & Leunissen, J. A. (1998). Int. J. Biol. Macromol. 22, 151–162. [DOI] [PubMed]
- Kennaway, C. K., Benesch, J. L. P., Gohlke, U., Wang, L., Robinson, C. V., Orlova, E. V., Saibil, H. R., Saibi, H. R. & Keep, N. H. (2005). J. Biol. Chem. 280, 33419–33425. [DOI] [PubMed]
- Kim, K. K., Kim, R. & Kim, S.-H. (1998). Nature (London), 394, 595–599. [DOI] [PubMed]
- Land, T. A., De Yoreo, J. J. & Lee, J. D. (1997). Surface Sci. 384, 136–155.
- Land, T. A., Malkin, A. J., Kuznetsov, Y., McPherson, A. & De Yoreo, J. J. (1995). Phys. Rev. Lett. 75, 2774–2777. [DOI] [PubMed]
- Larson, S. B., Kuznetsov, Y. G., Day, J., Zhou, J., Glaser, S., Braslawsky, G. & McPherson, A. (2005). Acta Cryst. D61, 416–422. [DOI] [PubMed]
- Li, Z. & Srivastava, P. (2003). Current Protocols in Immunology, Appendix 1T. doi:10.1002/0471142735.ima01ts58. [DOI] [PubMed]
- Lindquist, S. (1992). Curr. Opin. Genet. Dev. 2, 748–755. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Malkin, A. J., Kuznetsov, Y. G. & McPherson, A. (1997). Surface Sci. 393, 95–107.
- Malkin, A. J. & Thorne, R. E. (2004). Methods, 34, 273–299. [DOI] [PubMed]
- McPherson, A., Kuznetsov, Y. G., Malkin, A. & Plomp, M. (2003). J. Struct. Biol. 142, 32–46. [DOI] [PubMed]
- McPherson, A., Kuznetsov, Y. G., Malkin, A. J. & Plomp, M. (2004). J. Synchrotron Rad. 11, 21–23. [DOI] [PubMed]
- McPherson, A., Malkin, A. J. & Kuznetsov, Y. G. (2000). Annu. Rev. Biophys. Biomol. Struct. 29, 361–410. [DOI] [PubMed]
- McPherson, A., Malkin, A. J., Kuznetsov, Y. G. & Plomp, M. (2001). Acta Cryst. D57, 1053–1060. [DOI] [PubMed]
- Montfort, R. L. van, Basha, E., Friedrich, K. L., Slingsby, C. & Vierling, E. (2001). Nature Struct. Biol. 8, 1025–1030. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Poulain, P., Gelly, J.-C. & Flatters, D. (2010). PLoS One, 5, e9990. [DOI] [PMC free article] [PubMed]
- Schwoebel, R. L. & Shipsey, E. J. (1966). J. Appl. Phys. 37, 3682–3686.
- Simpson, A. J. et al. (2000). Nature (London), 406, 151–159.
- Stamler, R., Kappé, G., Boelens, W. & Slingsby, C. (2005). J. Mol. Biol. 353, 68–79. [DOI] [PubMed]
- Takeda, K., Hayashi, T., Abe, T., Hirano, Y., Hanazono, Y., Yohda, M. & Miki, K. (2011). J. Struct. Biol. 174, 92–99. [DOI] [PubMed]
- White, H. E., Orlova, E. V., Chen, S., Wang, L., Ignatiou, A., Gowen, B., Stromer, T., Franzmann, T. M., Haslbeck, M., Buchner, J. & Saibil, H. R. (2006). Structure, 14, 1197–1204. [DOI] [PubMed]
- Winn, M. et al. (2011). Acta Cryst. D67, 235–242.