

Abstract
Breast cancer progression involves cancer cell heterogeneity, with generation of invasive/metastatic breast cancer cells within populations of nonmetastatic cells of the primary tumor. Sequential genetic mutations, epithelial-to-mesenchymal transition, interaction with local stroma, and formation of hybrids between cancer cells and normal bone marrow–derived cells have been advocated as tumor progression mechanisms. We report herein the spontaneous in vitro formation of heterotypic hybrids between human bone marrow–derived multipotent stromal cells (MSCs) and two different breast carcinoma cell lines, MDA-MB-231 (MDA) and MA11. Hybrids showed predominantly mesenchymal morphological characteristics, mixed gene expression profiles, and increased DNA ploidy. Both MA11 and MDA hybrids were tumorigenic in immunodeficient mice, and some MDA hybrids had an increased metastatic capacity. Both in culture and as xenografts, hybrids underwent DNA ploidy reduction and morphological reversal to breast carcinoma–like morphological characteristics, while maintaining a mixed breast cancer–mesenchymal expression profile. Analysis of coding single-nucleotide polymorphisms by RNA sequencing revealed genetic contributions from both parental partners to hybrid tumors and metastasis. Because MSCs migrate and localize to breast carcinoma, our findings indicate that formation of MSC–breast cancer cell hybrids is a potential mechanism of the generation of invasive/metastatic breast cancer cells. Our findings reconcile the fusion theory of cancer progression with the common observation that breast cancer metastases are generally aneuploid, but not tetraploid, and are histopathologically similar to the primary neoplasm.
Although clonal in origin, breast carcinomas display considerable cell heterogeneity, with intratumoral diversity already apparent at the stage of ductal carcinoma in situ.1,2 The generation of cell subpopulations exhibiting cytogenetic abnormalities, along with invasive/prometastatic features, is inherent to breast cancer progression. Unknown triggers enable some of these cells to break free from the primary tumor, invade the microvasculature, travel, and establish foci at distant sites. Their actual genesis from within populations of nonmetastatic cells of the primary tumor has not yet been clarified. A stochastic mutation of key genes, epithelial-mesenchymal transition (EMT),3,4 and influence of stromal microenvironment at the tumor boundary5–7 have been proposed as underlying mechanisms of breast cancer progression. However, how successive, stepwise mutations of many key genes, necessary for the generation of a metastatic phenotype, might occur in sufficient numbers to overcome the inefficiency of the metastatic process itself is unclear.8 Also, the histopathological similarity of metastases to the primary tumor argues against the EMT theory. A subsequent step, mesenchymal-epithelial transition,9,10 occurring at the metastatic sites, or the possible cooperation between EMT and non-EMT cancer cells11 may resolve this apparent contradiction. The formation of hybrids between cancer cells and normal bone marrow–derived cells, including mesenchymal stem cells or multipotent stromal cells (MSCs), within tumor-associated stroma has been advocated as a nonmutational mechanism that could contribute to aneuploidy and aberrant gene expression patterns associated with highly malignant subpopulations.12–20 However, this theory has encountered much skepticism, mainly because, in most cases, recurrent and metastatic tumors have similar histopathological features as the primary neoplasm and tetraploid metastatic cells are rare. Herein, we show that high-ploidy hybrids with predominantly mesenchymal morphological characteristics, spontaneously formed between human breast cancer cells and MSCs, reacquire, in culture and as xenografts, a ploidy similar to that of the respective parental breast cancer cell line and form tumors histopathologically similar to the original breast carcinoma, while acquiring and maintaining expression of mesenchymal genes.
Materials and Methods
Cell Lines
Human MDA-MB-231 (MDA) breast cancer cells were obtained from the ATCC (Manassas, VA) in 2007. The human MA11 breast carcinoma cell line, established from bone marrow micrometastases of a patient with breast cancer,21,22 was obtained by Øystein Fodstad (Norwegian Radium Hospital, Oslo, Norway) in 2006. Both breast cancer cell lines were used in this study between passages 20 and 30. Breast cancer cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Inc., Norcross, GA), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine. Human MSCs were obtained from Darwin Prockop in 2008. They were isolated from 1- to 4-mL bone marrow aspirates taken from the iliac crest of normal adult donors after informed consent and under a protocol approved by the Tulane University Institutional Review Board, prepared as described by Larson et al,23 and frozen at passage 1. For expansion, MSCs were plated in a 75-cm2 culture dish and incubated for 1 day, to recover viable adherent cells. Cultures contained approximately 50% of rapidly self-renewing cells and 50% of larger, more slowly dividing and more mature cells. MSCs were then replated at 50 cells/cm2 and incubated for 10 days before lentiviral transduction. With time in culture, the percentage of rapidly self-renewing cells decreased progressively to <10% of the total cells. All cell lines were stored in aliquots in liquid nitrogen and kept in culture for <3 months. Complete culture medium for MSCs consisted of α-minimal essential medium (Gibco), 17% fetal bovine serum (lot selected for rapid growth of MSCs) (Atlanta Biologicals), 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L l-glutamine. All cells were tested for Mycoplasma contamination every 6 months and authenticated by a morphological check every 2 weeks.
Fluorescence Microscopy
Confocal images were taken using a homebuilt multiphoton microscope. The excitation wavelength was at 924 nm. The epireflected emission signals arising from enhanced green fluorescence protein (EGFP) and DsRed were collected simultaneously with 500/40-nm and 600/40-nm bandpass filters, respectively. Immunostaining for EGFP on tissue sections, after deparaffinization and boiling in 0.01 mol/L citric acid (pH 6) for antigen retrieval, was performed by overnight 4°C incubation with fluorescein isothiocyanate–EGFP antibody (1:500; GeneTex, San Antonio, TX) in 1% bovine serum albumin–PBS.
Retroviral and Lentiviral Vectors
The EGFP- and DsRed-expressing retroviral vectors used are based on pSF91 (GenBank accession number AJ224005) with the 3′ long terminal repeat of spleen focus-forming virus and the leader of the murine embryonic stem cell virus.24,25 To generate retroviral producers, the Phoenix-gp packaging cell line was transfected with pSF91-DsRed, or pSF91-EGFP, and a plasmid expressing the vesicular stomatitis virus glycoprotein. Culture supernatants containing viral particles were collected at 24 to 48 hours after transfection, passed through 0.22-μm Millex GP filters (Millipore Co, Bedford, MA), and stored at -80°C. For transduction, retroviral supernatants were preloaded onto recombinant fibronectin (Retronectin; Takara Shuzo, Kyoto, Japan) coated plates and centrifuged at 950 × g for 30 minutes at 4°C. The operation was repeated a second time with fresh supernatant. The supernatant was then removed, and the plates were washed with PBS before the addition of cells. After transduction, stable cell lines were isolated via selection with 2 μg/mL puromycin. A few days later, cells were cloned by limiting dilution. MSCs were transduced with vesicular stomatitis virus–pseudotyped lentiviral particles constitutively expressing firefly luciferase (luc) under the control of the cytomegalovirus promoter and the puromycin N-acetyltransferase (pac) gene under the control of the human phosphoglycerate kinase promoter (pCignal-luc) (SA Biosciences, Frederick, MD), using the fibronectin (Retronectin) method, as previously described. Transduced cells were isolated via selection with 2 μg/mL puromycin.
Wound-Healing Assay
Cell motility was measured by a conventional wound-healing assay.26 Briefly, 20,000 cells per well were plated in RPMI 1640 medium supplemented with 10% fetal bovine serum in polystyrene tissue culture-treated six-well plates. After 24 hours, medium was gently aspirated, wounds were generated by yellow tips with constant pressure, and prewarmed (37°C) RPMI 1640 medium supplemented with 1% fetal bovine serum was added. At indicated times, the same areas of each wound were sequentially imaged.
Invasion Assays
In vitro invasion assays were performed in BioCoat invasion chambers with Matrigel-coated positron emission tomographic membrane cell culture inserts (8-μm pore), using noncoated inserts as control (both from BD Biosciences, San Jose, CA), according to the manufacturer's directions. The Matrigel layers of the invasion chambers were rehydrated with serum-free α-minimal essential medium. The lower chambers were filled with α-minimal essential medium containing 5% fetal bovine serum, and equal numbers of cells in serum-free α-minimal essential medium were added to every insert. After a 24-hour incubation at 37°C, the cells on the upper side of the membrane were gently removed with wet cotton swabs. The cells on the lower surface of the membranes were fixed with 4% paraformaldehyde for 10 minutes and then stained with DAPI. The number of cells was counted in 8 to 12 randomly selected microscopic fields per insert (original magnification, ×100) using an Olympus CKX41 fluorescence microscope (Olympus America Corp, Center Valley, PA). To measure invasiveness, the cells that migrated through the Matrigel membranes were counted and expressed as percentage of cells migrated through control insert membranes.
Drug Sensitivity Assay
Puromycin resistance was determined using the methylated tetrazolium salt/phenazine methosulfate microtiter plate assay, as previously described.27
Flow Cytometric Analysis
DNA and marker analyses were performed using an iCyt Reflection flow cytometer (iCyt, Champaign, IL). Throughout all of the experiments, number of cells, concentration of DNA stain, and incubation time and temperature were kept constant, to ensure reproducibility of DNA index (DI) determination. Normal human lymphocytes were stained by the same procedure, with each batch of fresh tumors analyzed and run before the samples to calibrate the instrument for the position of G0/G1 normal DNA-diploid peak. The DI was determined according to Hiddemann et al.28 Briefly, the DI was defined as the mode of the DNA content of the G0/G1 cells of the sample divided by the mode of the DNA measurement of the diploid G0/G1 reference cells. By definition, diploid cells have a DI of 1.0. Analogous results were obtained using fix and perm, followed by RNase treatment or propidium + Nonidet P-40 and RNase.
Tumor Cell Implantation
Animal studies were performed under a protocol approved by the Nevada Cancer Institute Animal Care and Use Committee. Animals included nonobese diabetic (NOD) mice and Cg-Prkdc scid Il2rg tm1Wjl/SzJ (severe combined immunodeficiency γ) mice (The Jackson Laboratory, Bar Harbor, ME). For s.c. injection, cells were resuspended in 100 μL of PBS and injected s.c. into the right flank of the animals. When s.c. tumors reached a volume of 1 cm3, mice were euthanized and tumor, lungs, and brain were collected for histological analysis, fixed, and stained, according to standard techniques.
Deep RNA-Seq
RNA sequencing (RNA-seq)–based mRNA profiling was performed by Ocean Ridge Biosciences (Palm Beach Gardens, FL), essentially as described by Bottomly et al.29 Briefly, the Truseq RNA Sample preparation kit (Illumina, San Diego, CA) was used to perform the following sequential steps: i) isolation of polyadenylated mRNA species, ii) synthesis of double-stranded cDNA, iii) end repair of cDNA, iv) nucleotide addition to cDNA ends, v) ligation of double-stranded adapters to the cDNA, and vi) PCR amplification to select molecules containing adapters on both cDNA ends (Illumina). The amplified libraries were pooled to load two index-tagged samples per lane, diluted to a concentration of 12 pmol/L, and applied to a flow cell using the Cluster Station (Illumina). The generation of clusters on the flow cell was performed using the Cluster Station and Cluster Generation Kit version 5. Sequencing was performed on the GAIIX Sequencer with Paired-End Module (Illumina), using version 5 sequencing kits and protocol 51 + 7 cycle Multiplexed Read 1, which provided for 51 nucleotide single-end reads with decoding of the six-nucleotide multiplex adapter sequences. Image analysis and base calling were performed on the instrument using RTA software version 2.9 (Illumina). Demultiplexing, trimming, and alignment to the Human Genome reference sequence (Human National Center for Biotechnology Information build 37.2) were conducted using CASAVA version 1.8 software (Illumina). The raw results consisted of 21 to 33 million 49-nucleotide reads per sample, with an average quality score of 35. CASAVA version 1.8 software was also used for alignment to the exon model and generation of gene-level counts and single-nucleotide polymorphism (SNP) calls.
Gene Expression Analysis Using RNA-Seq Data
A total of 11,970 of a possible 23,890 Entrez ID genes were selected as expressed based on the criteria that ≥2450 nucleotides of sequence were mapped to that gene in two or more samples. To normalize the data, the raw nucleotides mapped for each gene X sample were converted to reads per 100,000 total reads. Missing data points, in which no reads were mapped for a specific sample X gene, were replaced with a value of 0.08 reads/100,000 total reads. Fold changes were calculated for each specific sample comparison for every gene, in which at least one of the two samples met the threshold of ≥2450 nucleotides of mapped sequences. Otherwise, fold changes were not reported.
SNP Analysis Using RNA-Seq Data
Illumina CASAVA 1.8 software reports the diploid genotype and quality score for each nucleotide position within an mRNA, where the sample sequence differs from the reference sequence. The list of SNP loci was filtered to select for only SNPs with a Q score of >75 in one or more samples. Redundancy was removed to generate a unique list of 37,622 SNPs, and the data were reformatted to produce a table in which each row contained a unique SNP with the Q score and diploid sequence for each sample given in columnar format. For comparison of the SNP genotypes of putative fusion hybrids to the parental cells MA11 and MSC genotypes, we selected 434 loci that had a high-quality score (Q > 75) for both parents, differed between the parents, and also had a high SNP quality score for putative fusion hybrids and two reference cell lines. For comparison of SNP genotypes of putative fusion hybrids to the parent cell MDA and MSC genotypes, we selected a total of 441 SNP loci using similar criteria. Among the selected high-quality SNP calls, the number of diploid matches (ie, both alleles) sequence matches between a potential fusion hybrid or reference cell line and each of the parent cell lines was counted, and these counts were binned by chromosome.
Luciferase Assay
The preparation of cell extracts and measurement of luciferase activity were performed using the Steady-Glo Luciferase Reporter Assay System, according to recommendations by the manufacturer (Promega, Madison, WI). The assays for luciferase activity were performed using a 20/20 luminometer (Turner Biosystems, Sunnyvale, CA).
Immunocytochemistry
Cells were plated onto poly-l-lysine–coated chamber slides, fixed in 4% paraformaldehyde, washed with PBS, permeabilized in 0.2% Nonidet P-40, and blocked with goat serum. After washing with PBS, cells were incubated overnight at 4°C with a 1:100 dilution of anti-human CD29 antibody (Chemicon, Temecula, CA) in 1% bovine serum albumin–PBS, followed by a 45-minute incubation at room temperature with tetrarhodamine isothiocyanate–labeled secondary antibody (1:300; Jackson Immunoresearch, West Grove, PA) in 1% bovine serum albumin in PBS. Cell nuclei were labeled with DAPI (1:1000; Sigma-Aldrich, St. Louis, MO), and cells were viewed under a CKX41 fluorescence-inverted microscope (Olympus, Center Valley, PA).
IHC Data
Sections (4 μm thick) were incubated with mouse anti-pan keratin monoclonal PM011 AE1-AE3 antibody, followed by the MACH 4 universal two-step detection method, and labeled with horseradish peroxidase, as recommended by the manufacturer (Biocare Medical, Concord, CA).
Results
Spontaneous Formation of MSC–Breast Cancer Hybrids
We established co-cultures of human MSCs, derived from bone marrows of healthy adult donors,30 with human MDA or MA11 breast carcinoma cells at a 1:1 ratio (total initial cell density, 1300 cells/cm2). The MSCs and breast carcinoma cells were fluorescently tagged by retroviral transduction with EGFP and DsRed-expressing vectors,25 respectively. In co-culture, after 24 hours, we observed invasion of MSCs by breast cancer cells, resulting in cells with dual-fluorescence and mesenchymal morphological characteristics (Figure 1, A and B). In several cases, the dual-fluorescence cells were able to divide (Figure 1A). To select heterotypic hybrids, we established co-cultures of MSCs with MDA-DsRed or MA11-EGFP cells under conditions favoring the growth and identification of hybrids. For these experiments, MSCs were chemiluminescently tagged and rendered puromycin resistant (MSC-luc) by lentiviral transduction. The tumor cells, seeded at 15,000 cells/cm2, were co-cultured for 3 days with MSC-luc, seeded at 3000 cells/cm2; the MSC–tumor cell hybrids were selected by exposure to 1 μg/mL puromycin, which caused complete disappearance of breast carcinoma cells. After 5 days of selection, 80% ± 14% (SD) of cells in MDA-MSC co-cultures were fluorescent hybrids, versus 30% ± 15% (SD) of cells in MA11-MSC co-cultures, as assessed by flow cytometry (Figure 1C). Interestingly, the different capacity of MDA and MA11 to form hybrids with MSC corresponded roughly to the different capacity of the two breast cancer cell lines to invade Matrigel, a substance mimicking the basement membrane (62% ± 19% versus 15% ± 12% for MDA and MA11 cells, respectively) (Figure 1D).
Figure 1.

Spontaneous formation of proliferating heterotypic hybrids (with MSCs) by breast carcinoma cells. A: Fluorescent micrographs, at successive time points, of a co-culture of human MDA-DsRed (red) and MSC-EGFP (green) in the absence of puromycin, showing formation of double-labeled proliferating hybrid cells with a fibroblastic shape (yellow; arrows). B: Fluorescent micrographs, at time points spaced by 3-hour intervals, of co-cultures of MSC-EGFP (green) with MDA-DsRed or MA11-DsRed (red), showing apparent involvement of cell lysis (arrows) in the interaction of breast cancer cells with MSCs. C: Quantification of hybridization between breast carcinoma cells (MA11-DsRed and MDA-DsRed) and MSC-luc in high-density co-culture. The tumor cells, seeded at 15,000 cells/cm2, were co-cultured for 3 days with MSC-luc, seeded at 3000 cells/cm2, and the MSC–tumor cell hybrids selected by 5-day exposure to puromycin (1 μg/mL). The number of red hybrid cells was then assessed by flow cytometric analysis and expressed as a percentage over the total of puromycin-resistant cells. Data represent the mean ± SD from two experiments. D: Plot of the percentage of cells migrating through polyethylene terephthalate membrane cell culture inserts (8-μm pore) coated with a cell matrix–resembling preparation (Matrigel) for each of the two breast cancer cell lines used in the study. Data are expressed as percentage of Matrigel-migrating cells on total cells migrating through noncoated control insert membranes in 24 hours. The cells on the lower surface of the membranes were fixed with 4% paraformaldehyde for 20 minutes and then stained with DAPI. The number of cells was counted in 8 to 12 randomly selected microscopic fields per insert. Original magnification, ×100. Data represent the mean ± SD of three to four experiments. E: Sensitivity of parental breast carcinoma and hybrid cells to 3-day exposure to different concentrations of puromycin, measured by a tetrazolium-based cell proliferation assay. The units of luciferase (luc) per cell (indicated in parenthesis as mean ± SD) revealed acquisition by the hybrid lines of luciferase expression and puromycin resistance from MSC-luc; by Student's t-test, the difference between the hybrid cells and the parental breast cancer cells was statistically significant (P < 0.01) in the range from 0.1 to 20 μg/mL of puromycin. F: β1-Integrin immunofluorescences on puromycin-selected cells from an MSC-luc-MA11-EGFP co-culture and on parental breast cancer cells, revealing the hybrid nature of the puromycin-selected cells that, in addition to expressing EGFP (green), display high expression of the mesenchymal marker (red) on the cell surface. Nuclei stained with DAPI (blue). Scale bars: 50 μm (A); 20 μm (B); 25 μm (F). *P < 0.01 using the Student's t-test.
Characteristics of MSC–Breast Cancer Hybrids
At 2 to 3 weeks after the end of puromycin treatment, we isolated puromycin-resistant fluorescent hybrids by fluorescence-activated cell sorting. Independently derived hybrid cell lines and their clones, isolated by limiting dilution, displayed mesenchymal morphological characteristics (see Supplemental Figure S1 at http://ajp.amjpathol.org), an increase in DNA content (Table 1), expression of luciferase (Figure 1E), resistance to puromycin (Figure 1E), and high-level expression of β1-integrin (CD29), a stromal marker not expressed or expressed at lower levels in breast cancer cells (Figure 1F). The growth kinetics of the MSC-luc-MA11-EGFP hybrid cell lines and clones were not different from parental MA11-EGFP cells, whereas it was heterogeneous for MSC-luc-MDA-DsRed cell lines and clones, with doubling times ranging from 22 to 55 hours (see Supplemental Figure S2, A and B, at http://ajp.amjpathol.org). At 4 weeks after fusion, the DI was on average 3.2 and 2.0 for MSC-MA11 and MSC-MDA hybrids, respectively, corresponding roughly to the sum of the DI of MA11 (2.18) or MDA (1.25) and of MSC (1.0) (Table 1). The difference in DI between the parental breast carcinoma cell lines and their respective hybrids was statistically significant (unpaired Student's t-test, P < 0.01). MSC-MA11 hybrid clones had a heterogeneous DI, ranging from 2.72 to 4.7, whereas for MSC-MDA, the DI ranged from 1.88 to 2.02 (Table 1). No correlation was observed between the DI and the proliferation rate of the individual hybrid clones.
Table 1.
Ploidy Reduction in Heterotypic Breast Cancer Hybrid Cells in Culture
| Cells | DNA index |
|
|---|---|---|
| D0 | D60 | |
| MSC | 1.0 ± 0.1 | 0.9 ± 0.1 |
| MA11 | 2.2 ± 0.2 | 2.2 ± 0.1 |
| MDA | 1.3 ± 0.1 | 1.3 ± 0.1 |
| Hybrid | ||
| MSC-MA11 | 3.2 ± 0.2⁎ | 2.5 ± 0.2 |
| MSC-MDA | 2.4 ± 0.1⁎ | 1.5 ± 0.1 |
Cells were fixed with 70% ethanol, stained with propidium iodide, and analyzed by flow cytometry, as described in Materials and Methods. The cell content of DNA was measured at D0 and D60. Values are the mean ± SD from three experiments; the values indicated for the hybrids were similar in five clones.
D0, immediately after the end of puromycin selection of the hybrids; D60, 60 days after the end of puromycin selection of the hybrids.
Statistically significant difference by Student's t-test with respect to the corresponding values by the parental tumor cells (P < 0.01).
Intrinsic Fusogenicity of Breast Cancer Cells
In 1:1 co-cultures of MA11-EGFP with MDA-DsRed and of MA11-EGFP with MA11-DsRed, we observed, after 24 to 48 hours, the formation of dual-fluorescence hybrid cells. Figure 2 shows an actively proliferating clone of dual-fluorescence MA11-MSC hybrids.
Figure 2.

Breast carcinoma cells undergo spontaneous homotypic hybridization, producing cells that continue proliferating. Micrographs of double-labeled (EGFP + DsRed) MA11 cells derived from a co-culture of MA11-EGFP (green) and MA11-DsRed (red). Images, taken at 3-day co-culture (D0) and 7 days later (D7), reveal rapid proliferation of yellow hybrid cells. Scale bar = 50 μm.
Tumorigenic and Metastatic Properties of Hybrids
Both MA11-MSC and MDA-MSC hybrids were tumorigenic on s.c. injection in NOD. Cg-Prkdc scid Il2rg tm1Wjl/SzJ (scid γ) mice (Table 2). By using tumor cell inoculum levels resulting in approximately 80% tumorigenicity for both parental breast carcinoma cell lines, we observed that the tumorigenicity of the hybrids was not significantly different from parental breast cancer cells. To examine the capacity of MDA hybrids to form metastasis to the lung in NOD/scid γ mice, 5 × 105 cells of six MDA-DsRed-MSC-luc clones, derived by limiting dilution from two independently derived hybrid cell lines, and six MDA-DsRed clones, derived from parental cells, were implanted s.c. in the right flank (five animals per group). The clones for the in vivo study were selected on the basis of their doubling times in culture (between 22 and 26 hours). Mice were sacrificed when tumors reached a volume of 1.0 cm3. All lungs were examined for metastasis by analyzing 10 stepwise H&E-stained sections (4-μm thick). Statistically significantly more mice implanted with MDA-DsRed-MSC-luc clones than with MDA-DsRed clones developed lung metastases (73% versus 10%; P < 0.01) (Table 2). Also, 7 of the 10 mice implanted with MDA hybrid clones and examined histopathologically for brain metastases were found positive, whereas no brain metastatic foci were found in 10 mice implanted with MDA-DsRed clones. Consistent with previous reports,31 human MSCs, untransduced or transduced with pCignal-luc, were not able to form tumors and/or metastases on s.c. injection in immunodeficient mice.
Table 2.
Tumorigenic and Metastatic Potential of MSC-Breast Carcinoma Hybrids
| Type of tumor | Inoculum of cells |
|||
|---|---|---|---|---|
| 105 |
5 × 105 |
|||
| MA11 | Hybrids | MDA | Hybrids | |
| s.c. Tumor | 4/5 (80) | 6/11 (54) | 25/30 (83) | 27/30 (90) |
| Lung metastasis | 0/5 | 0/11 | 3/30 (10) | 22/30 (73)⁎ |
Data are given as number/total (percentage). Control MSCs implanted alone into 10 mice (inoculum, 500,000 cells/mouse) were nontumorigenic.
Statistically significant difference by two-sided Fisher's exact test with respect to the rate of metastasis by MDA (P < 0.001).
Hybrids Acquire Mesenchymal Characteristics
In view of the increased metastatic capacity of MSC-luc-MDA-DsRed hybrids, we next investigated whether they had acquired higher motility from parental MSCs, which are equipped with motor proteins that enable them to migrate during the development throughout embryonic regions.32 In wound-healing assays, we observed that MSC-luc-MDA-DsRed hybrids closed the gap much faster than parental breast cancer cells. Sequential micrographs from a representative experiment are presented in Figure 3.
Figure 3.

Enhanced motility of MSC-MDA hybrids with respect to parental MDA. Phase-contrast images, spaced by 10-hour intervals, of pipet tip–wounded monolayers of MDA and MSC-MDA heterotypic hybrids in a tissue culture–treated polystyrene plate, showing that the hybrids, similar to parental MSC (data not shown), polarize toward the wound, initiate protrusion, and migrate faster than parental breast carcinoma cells, closing the wound during the time course. Scale bar = 100 μm.
To evaluate whether fusion with MSCs resulted in increased expression of mesenchymal genes, we analyzed the global gene expression profiles of an MSC-luc-MDA-DsRed tumor xenograft and its brain metastasis and compared them with parental MDA-DsRed xenografts and with MSC-luc. To this aim, xenografts and brain metastasis were dissociated, separated from contaminating host cells by fluorescence sorting, and cultured for 3 to 4 days. Their global gene expression profile was analyzed by RNA-seq. The gene expression profile of the hybrids was consistent with their derivation from both parental partners (Figure 4). On average, 1239 and 5345 genes were differentially expressed between MDA hybrid and MDA and between MDA hybrid and MSC, respectively; 1320 and 5297 genes were differentially expressed between MDA hybrid brain metastasis and MDA and between MDA hybrid brain metastasis and MSC, respectively. Interestingly, significant changes in biochemical pathways between MDA hybrid and MDA tumor xenografts were detected by the Kyoto Encyclopedia of Genes and Genomes pathway analysis for cytokine-cytokine receptor interaction: 21 genes overexpressed in the hybrids/91 total; P (P value indicating the significance of enrichment calculated from a hypergeometric test) = 5.37e-08, with extracellular matrix-receptor interaction (12/59; P= 3.17e-05), focal adhesion pathways (22/31; P= 7.09e-06), and cell adhesion molecules (13/14; P= 5.91e-06). Also, several mesenchymal genes33 were overexpressed in hybrids compared with the respective parental MDA cells, including secreted protein, acidic, cysteine-rich (SPARC), fibronectin, and CD109 (see Supplemental Table S1 at http://ajp.amjpathol.org). Interestingly, expression of the aldehyde dehydrogeanse (ALDH) 1A3 isoform, the main ALDH isoform in breast cancer stem cells,34 was increased >10-fold in an MDA hybrid tumor xenograft, and its brain metastasis compared with parental MDA xenografts (17.4- and 13.4-fold, respectively).
Figure 4.
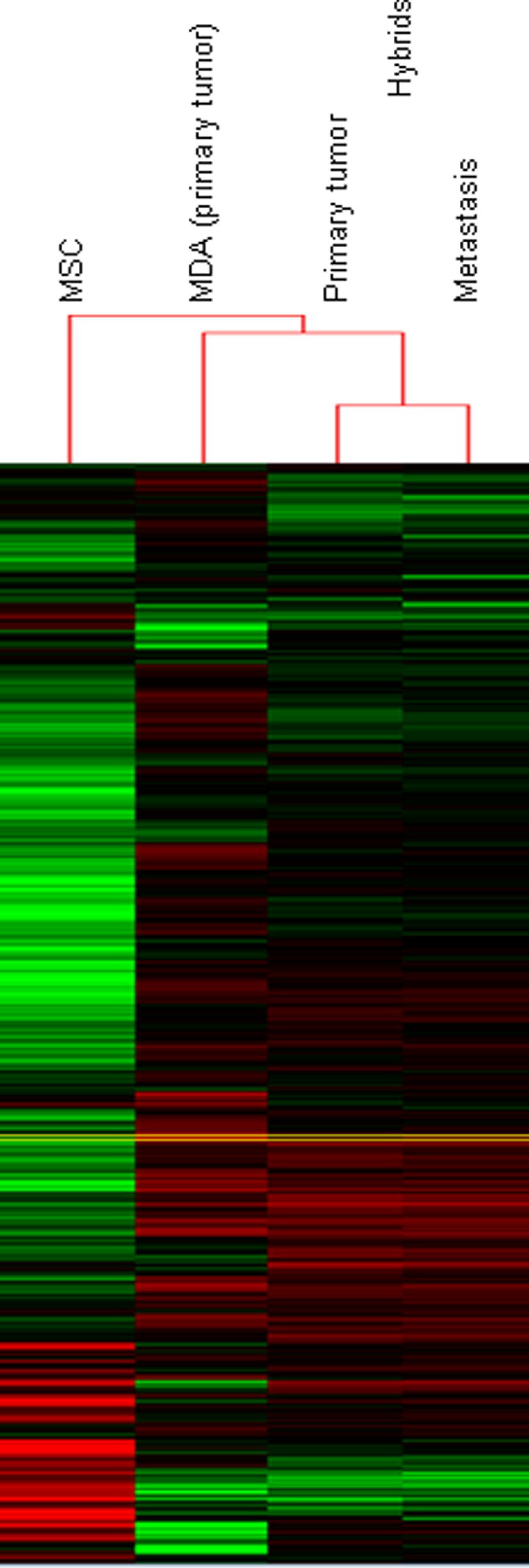
Mixed pattern of gene expression of heterotypic hybrids. Heat maps for MSC-MDA hybrids (from s.c. tumor and metastasis), MSC, and MDA, showing clustered gene-level counts with lower than median (green), higher than median (red), and median (black) levels of expression. Each row represents a single gene. Clustering was performed using centered correlation as distance measure and single linkage as method.
Hybrids Are Genetically Unstable
After 3 months in culture, a generalized decrease in DNA ploidy was observed for both MSC-MA11 and MSC-MDA hybrids (Table 1). The decrease in ploidy occurred even if puromycin-selective pressure was constantly maintained. A similar decrease in DNA ploidy was also observed during growth of tumor xenografts. Multipolar metaphases and unequal divisions with lagging DNA, suggestive of ploidy reduction, were observed in the hybrid cultures (Figure 5A). Interestingly, multipolar mitoses were also observed in homotypic hybrids and, at a lower frequency (P < 0.05), in the parental breast carcinoma cell lines, but not in parental MSCs (Figure 5, B and C).
Figure 5.

Increased frequency of multipolar (tripolar/tetrapolar) mitosis after hybridization. A: Tripolar (left panel) and tetrapolar (right panel) metaphases in MSC-MA11 hybrid cells revealed by DAPI staining of DNA (blue). B: Immunofluorescence for α-tubulin (red), revealing the multipolar configuration of the mitotic spindle in an MSC-MA11-EGFP breast carcinoma hybrid cell. Condensed chromosomes stained with DAPI (blue). C: Graph depicting the percentage of multipolar mitosis in the hybrids of breast carcinoma cells with MSC and the parental tumor cells, showing twofold to threefold greater multipolar mitosis in the heterotypic hybrids. Data represent the mean ± SD from three experiments, each one consisting of 100 metaphases plus anaphases analyzed per cell type. In parental MSCs, no multipolar mitosis was detected. Scale bar = 20 μm.
Hybrids Reacquire Breast Cancer–Like Morphological Characteristics in Culture and in Vivo but Maintain a Mixed Gene Expression Profile
In most cases, a transition from mesenchymal to epithelial-like morphological characteristics was observed in culture within 3 months (Figure 6). However, all hybrids maintained, to a variable extent, puromycin resistance and luciferase expression (data not shown). The phenotypic reversion of hybrids to a breast carcinoma–like appearance after in vivo transplantation and ex vivo culture is shown in Figure 7. Tumor hybrid xenografts were all homogeneously EGFP+ or DsRed+ and formed poorly differentiated adenocarcinomas with high-grade features and a variable extent of necrosis; different from parental xenografts, some of the tumor hybrids showed differentiated features, such as nesting and more cohesive formations (Figure 7). None of the hybrid xenografts had the typical fusiform cell shape and fascicular cellular arrangement characteristic of malignant tumors of mesenchymal origin (fibrosarcomas). Strong diffuse membranous expression of AE1-AE3 cytokeratin in xenografts (Figure 7A), comparable with the expression in parental breast carcinoma xenografts, and cell morphology (Figure 7, B and D) were suggestive of the epithelial nature of the tumors, whereas expression of β1-integrin (CD29) (Figure 7C) and luciferase expression suggested an MSC contribution to the hybrid phenotype. To evaluate whether tumors were transplantable, and to exclude that luciferase activity was the result of contaminating MSC-luc, tumors were disaggregated and cultured for 1 week; 106 cells were re-injected s.c. into NOD/scid γ mice. After 3 to 4 weeks, 1-cm3 tumors were formed that, on dissociation, expressed EGFP and luciferase; the tumors maintained high-level resistance to puromycin (data not shown). Their DI (range, 2.1 to 2.6) was similar to that of parental MA11 cells.
Figure 6.

Reversion of the fibroblastic morphological characteristics of the MSC-MA11 hybrid cells to an epithelial, breast carcinoma–like appearance. A: Micrographs separated by an interval of 90 days of an MSC-MA11 hybrid line in culture showing that the fibroblastic morphological characteristics of the cells reverted, within that period, to an epithelial shape. B: Spontaneous formation of islets of cells with breast cancer–like epithelial morphological characteristics (single asterisk) in four different MSC-MA11 hybrid lines in culture, which, since being formed, were composed only by fibroblast-like cells (indicated by double asterisks). The morphological changes occurred in both the presence and the absence of puromycin. Scale bars: 50 μm (A); 100 μm (B).
Figure 7.

In vivo reversion of MSC–breast carcinoma hybrid cells from fibroblast-like to epithelial morphological characteristics. A: H&E staining (left panel), IHC for keratin (middle panel), and immunofluorescence for EGFP (right panel) on tissue sections of a tumor produced in mice by s.c. implantation of MSC-MA11-EGFP hybrid cells, showing expression by the tumor cells of both EGFP (green) and the epithelial marker, keratin. Nuclei stained with DAPI (blue) in the fluorescence micrograph. B: Tumor xenograft explant of MSC-MA11 hybrid cells reverting from fibroblastic cells with large nuclei to epithelial cells with smaller nuclei. Nuclei stained with DAPI (blue). C: Immunofluorescences for β1-integrin on MSC-MA11-EGFP hybrid cells (right panel), after growth as xenograft tumor and 3 days of ex vivo culture, and an explant of parental MA11-EGFP cells (left panel), showing positivity of heterotypic hybrid cells with both fibroblastic and epithelial morphological characteristics, in contrast to parental cells (red). D: Explants of tumor xenografts of MSC-MA11 hybrid cells (right panel) and parental MA11 cells (left panel) displaying similar breast cancer–like morphological characteristics in ex vivo culture. Scale bars: 40 μm (A); 25 μm (B); 50 μm (C and D).
Genetic Contribution of Both Parental Partners to Hybrid Xenografts and Metastasis
We used analysis of coding SNPs to investigate the genetic contribution of MSCs and breast cancer cells to both MA11 and MDA tumor xenografts and to MDA metastasis. As shown in Figure 8, we analyzed coding SNPs in an MSC-luc-MA11-EGFP xenograft and found SNPs from both parental partners in almost all chromosomes. In the hybrid tumor xenograft, 55% of the 434 SNP loci, which differed between the parental partners, derived from MA-EGFP and 45% derived from MSC-luc. In both MSC-luc-MDA-DsRed tumor xenograft and its brain and lung metastasis, 77% of SNPs derived from MDA-DsRed and 23% derived from MSC-luc (Figure 8). The SNPs of MDA brain and lung metastases were identical.
Figure 8.
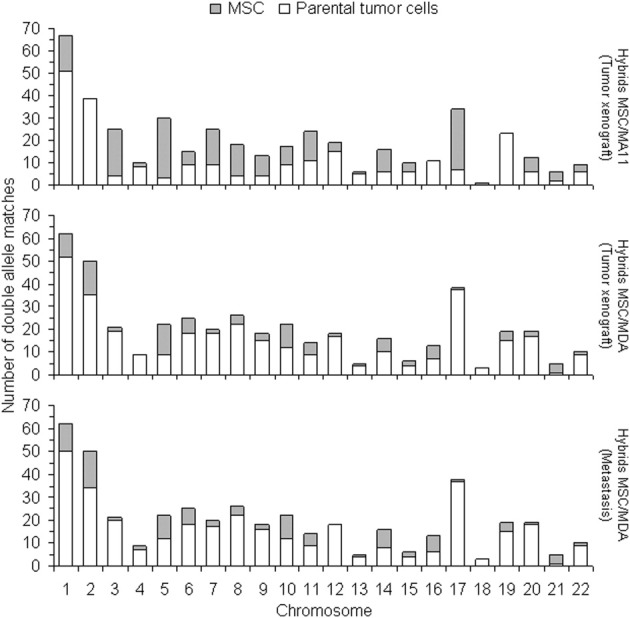
Genetic contribution from both breast carcinoma and MSC to heterotypic hybrids. Comparative SNP analysis between either MSC-MDA or MSC-MA11 hybrids (after growth in mice as xenograft tumors) and their specific parental partners. The 441 and 434 loci differing between the two parental cell types were analyzed for MSC-MDA and MSC-MA11 hybrids, respectively.
Discussion
Herein, we demonstrate that generation of breast cancer–MSC hybrid variants represents a large-scale mechanism of genotypic and phenotypic diversification. High-frequency spontaneous formation of hybrids was surprising, particularly in view of the apparently chaotic nature of aneuploidy.14 One argument against the relevance of heterotypic fusion in cancer progression is the lack of evidence that, during progression, cancer cells become tetraploid.35 Our observation that breast cancer–MSC hybrids undergo multipolar divisions and quickly become aneuploid suggests a logical explanation for this apparent discrepancy. Interestingly, multipolar mitoses were observed in cultures of MA11 and MDA cells, although at a lower rate, but not in MSCs; accordingly, we observed spontaneous formation of tumor-tumor hybrids, suggesting that homotypic, in addition to heterotypic, fusion may contribute to breast cancer heterogeneity. These data are in agreement with recent reports of acquisition of a dual-metastasis organotropism through spontaneous fusion between MDA variants.36 In the present study, we observed invasion of MSCs by breast cancer cells, with a correlation between the invasive capacity of breast cancer cells (measured as the capacity to migrate through Matrigel) and the frequency of formation of hybrids. The active role played by breast cancer cells in the heterotypic hybridization is supported by the observation of formation of homotypic hybrids for both MA11 and MDA cells.
Clearly, the genome of the breast cancer cells contributed tumorigenicity to the hybrids, whereas MSCs contributed expression of mesenchymal genes and, in case of MDA hybrids, increased metastatic potential. As xenografts, and with time in culture, the mixed expression of mesenchymal and breast carcinoma genes was maintained, whereas the morphological characteristics of hybrids became similar to those of the parental breast cancer cells. Interestingly, in a previous study,37 although tumors formed in mice by human fibroblasts cotransduced with E1A and V-Ha-Ras had a histopathological appearance of fibrosarcomas, tumors formed by fibroblast hybrids had an epithelial-like appearance. Instead, malignant transformation of normal mouse stroma–derived cells by fusion with breast cancer epithelium resulted in spindle-shaped fibroblastic morphological features, characteristic of sarcomas.38 Our data are consistent with previous reports39,40 that spontaneously fused bone marrow cells can subsequently adopt the phenotype of the recipient cells. The expression of genes from both parental partners in the MDA-MSC hybrid cells is in agreement with previous findings41,42 in human melanoma–mouse macrophage hybrids. Analysis of coding SNPs in xenografts confirmed their mixed MSC–breast cancer genetic background. Overexpression of mesenchymal genes, such as SPARC,43,44 indicates that breast cancer cells may acquire mesenchymal markers by EMT and by fusion with MSCs; in particular, SPARC has recently been associated with the most aggressive and highly metastatic tumors.45 Up-regulation of SPARC was previously observed by Chakraborty et al46 in macrophage-melanoma hybrids. Interestingly, a subgroup of breast cancer cell lines with enhanced invasive properties and a predominantly mesenchymal gene expression signature has recently been identified.47 Mesenchymal markers were reportedly overexpressed in circulating tumor cells of patients with metastatic breast cancer,48 and residual breast cancer cells surviving after conventional therapy displayed mesenchymal features.49 It is intriguing that ALDH1A3, the main ALDH isoform in breast cancer stem cells, which is reportedly predictive of breast cancer metastasis,34 was increased by 10-fold in MDA hybrid tumor xenografts and its brain metastasis compared with parental MDA xenografts.
Spontaneous fusogenic events between normal stroma–derived cells of mouse mammary glands and mammary cancer cells embedded in the stroma have been previously described.38 The capacity of MSCs to migrate to the site of breast cancer growth,6 and the observation that conditioned medium of breast cancer cells, including MDA, contains signaling molecules that induce MSC chemotaxis,50 reinforces the clinical relevance of the formation of MSC–breast cancer hybrids in the breast. We speculate that these fusogenic events may also occur at metastatic sites, involving a direct interaction between MSCs of the metastatic site and the migrated breast cancer cells. In this case, the hybrid cells may acquire properties from local MSCs that allow them to successfully implant and proliferate at the metastatic site.
Both in vitro and in vivo, DNA ploidy decreased. The decrease was not due to the withdrawal of the selecting agent, because it occurred in hybrid cultures constantly maintained in the presence of puromycin, and did not result in a reversal of the mixed phenotype, as indicated by RNA-seq of xenografts, by immunohistochemistry (IHC) data, and presumably by the acquisition of a metastatic phenotype for MSC-MDA hybrids. In agreement with our data, previous analysis of the ploidy pattern of primary breast carcinoma and corresponding metastatic nodes revealed a much higher incidence of moderate aneuploidy in the metastatic nodes.51 The highly metastatic phenotype that we observed in many MSC-MDA hybrid xenografts suggests that MSC–breast cancer fusion is a potential mechanism of tumor progression. Clearly, not all hybrid cells are tumorigenic and/or have enhanced promalignant properties. In particular, we did not observe formation of metastasis on s.c. implantation of MA11 hybrids. It is conceivable that cell heterogeneity translates in vivo into selection of clones with survival advantages toward adverse conditions in the body and/or to chemotherapeutic/radiotherapeutic treatments. Studies of breast cancer–MSC fusion in vivo are warranted to fully evaluate the clinical relevance and frequency of this phenomenon. Our findings suggest that targeting one or more of the following may prevent acquisition of a more malignant phenotype and/or breast cancer spreading to distant organs: i) fusion process, ii) early and rate-limiting post-fusion events, and iii) mesenchymal gene products. The possibility that MSCs are also implicated in the early phase of tumorigenesis cannot be excluded: initial mutagenic events may act synergistically with the acquisition by fusion with MSCs of mesenchymal traits, such as surface antigen expression, immune tolerance, or stem cell phenotypic features, to allow local tumor growth.
In conclusion, our data show that breast cancer cell fusion with MSCs generates a new hybrid offspring that contributes to breast cancer cell heterogeneity with unforeseen and possibly dangerous consequences. Where such fusions form compatible genetic re-assortments that allow the fusion to be productive, rather than destructive, and acquire prometastatic traits, breast cancer subpopulations with increased malignancy are born.
Acknowledgments
We thank Michael Reiss for critical reading of the manuscript, John Pawelek and Giuseppe Pizzorno for their advice and support, Fabio Anzanello for invaluable technical assistance, Erin Greenwood for help with fluorescence micrographs and growth of hybrids, Christopher Baum for retroviral vectors, Joseph Benito for RNA-seq analysis, David Willoughby for interpretation of RNA-seq results, Darwin Prockop for human MSCs, Jason Gonzales and James Tung for flow cytometric analysis and sorting, and Joseph Khoury for pathological interpretation of slides.
Footnotes
Supported by a grant from the NIH (R01CA133797 to G.R.). Darwin Prockop, who provided human MSCs, was supported by a grant from the National Center for Research Resources, NIH (P40RR017447).
The content herein is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the NIH.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at http://dx.doi.org/10.1016/j.ajpath.2012.02.020.
Current address of all authors: Cancer Research Program, Roseman University of Health Sciences, Henderson, Nevada.
Supplementary data
Mesenchymal morphological characteristics of breast carcinoma–MSC hybrids. Co-cultures of MSC-luc with MDA-DsRed or MA11-DsRed cells resulted in puromycin-resistant fluorescent hybrids that displayed mesenchymal morphological characteristics (arrows).
Growth rates of hybrids. A: Cell growth curves of MSC-luc-MA11-EGFP and MSC-luc-MDA-DsRed2 hybrid lines and clones. B: Doubling times of MA11-EGFP (left panel), MDA-DsRed2 (right panel), MSC-luc, and their respective hybrid cell lines and clones during in vitro and ex vivo culture from xenografts. Doubling times were calculated during growth of duplicate-triplicate cultures by exponential regression analysis.
References
- 1.Park S.Y., Gonen M., Kim H.J., Michor F., Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120:636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allred D.C., Wu Y., Mao S., Nagtegaal I.D., Lee S., Perou C.M., Mohsin S.K., O'Connell P., Tsimelzon A., Medina D. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14:370–378. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- 3.Thiery J.P. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 4.Polyak K., Weinberg R.A. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 5.Schor S.L., Schor A.M. Phenotypic and genetic alterations in mammary stroma: implications for tumour progression. Breast Cancer Res. 2001;3:373–379. doi: 10.1186/bcr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karnoub A.E., Dash A.B., Vo A.P., Sullivan A., Brooks M.W., Bell G.W., Richardson A.L., Polyak K., Tubo R., Weinberg R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 7.Liu S., Ginestier C., Ou S.J., Clouthier S.G., Patel S.H., Monville F., Korkaya H., Heath A., Dutcher J., Kleer C.G., Jung Y., Dontu G., Taichman R., Wicha M.S. Breast cancer stem cells are regulated by mesenchymal stem cells through cytokine networks. Cancer Res. 2011;71:614–624. doi: 10.1158/0008-5472.CAN-10-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambers A.F., Groom A.C., MacDonald I.C. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 9.Chaffer C.L., Brennan J.P., Slavin J.L., Blick T., Thompson E.W., Williams E.D. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271–11278. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 10.Hugo H., Ackland M.L., Blick T., Lawrence M.G., Clements J.A., Williams E.D., Thompson E.W. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 11.Tsuji T., Ibaragi S., Hu G.F. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135–7139. doi: 10.1158/0008-5472.CAN-09-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawelek J.M., Chakraborty A.K. The cancer cell–leukocyte fusion theory of metastasis. Adv Cancer Res. 2008;101:397–444. doi: 10.1016/S0065-230X(08)00410-7. [DOI] [PubMed] [Google Scholar]
- 13.Aichel O. Uber zellverschmelzung mit qualitativ abnormer chromosomenverteilung als ursache der geschwulstbildung. In: Roux W., editor. Vortrage und aufsatze uber entvickelungsmechanik der organismen, Chapter XIII. Wilhelm Engelmann; Leipzig: 1911. pp. 92–111. [Google Scholar]
- 14.Pawelek J.M., Chakraborty A.K. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8:377–386. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 15.Goldenberg D.M., Gotz H. On the “human” nature of highly malignant heterotransplantable tumors of human origin. Eur J Cancer. 1968;4:547–548. doi: 10.1016/0014-2964(68)90011-x. [DOI] [PubMed] [Google Scholar]
- 16.Warner T.F. Cell hybridization: an explanation for the phenotypic diversity of certain tumours. Med Hypotheses. 1975;1:51–57. doi: 10.1016/0306-9877(75)90042-0. [DOI] [PubMed] [Google Scholar]
- 17.Duelli D., Lazebnik Y. Cell fusion: a hidden enemy? Cancer Cell. 2003;3:445–448. doi: 10.1016/s1535-6108(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 18.Lu X., Kang Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009;69:8536–8539. doi: 10.1158/0008-5472.CAN-09-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powell A.E., Anderson E.C., Davies P.S., Silk A.D., Pelz C., Impey S., Wong M.H. Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011;71:1497–1505. doi: 10.1158/0008-5472.CAN-10-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogle B.M., Cascalho M., Platt J.L. Biological implications of cell fusion. Nat Rev Mol Cell Biol. 2005;6:567–575. doi: 10.1038/nrm1678. [DOI] [PubMed] [Google Scholar]
- 21.Rye P.D., Norum L., Olsen D.R., Garman-Vik S., Kaul S., Fodstad O. Brain metastasis model in athymic nude mice using a novel MUC1-secreting human breast-cancer cell line, MA11. Int J Cancer. 1996;68:682–687. doi: 10.1002/(SICI)1097-0215(19961127)68:5<682::AID-IJC20>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 22.Rappa G., Lorico A. Phenotypic characterization of mammosphere-forming cells from the human MA11 breast carcinoma cell line. Exp Cell Res. 2010;316:1576–1586. doi: 10.1016/j.yexcr.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larson B.L., Ylostalo J., Prockop D.J. Human multipotent stromal cells undergo sharp transition from division to development in culture. Stem Cells. 2008;26:193–201. doi: 10.1634/stemcells.2007-0524. [DOI] [PubMed] [Google Scholar]
- 24.Hildinger M., Abel K.L., Ostertag W., Baum C. Design of 5′ untranslated sequences in retroviral vectors developed for medical use. J Virol. 1999;73:4083–4089. doi: 10.1128/jvi.73.5.4083-4089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rappa G., Kunke D., Holter J., Diep D.B., Meyer J., Baum C., Fodstad O., Krauss S., Lorico A. Efficient expansion and gene transduction of mouse neural stem/progenitor cells on recombinant fibronectin. Neuroscience. 2004;124:823–830. doi: 10.1016/j.neuroscience.2003.11.030. [DOI] [PubMed] [Google Scholar]
- 26.Lampugnani M.G. Cell migration into a wounded area in vitro. Methods Mol Biol. 1999;96:177–182. doi: 10.1385/1-59259-258-9:177. [DOI] [PubMed] [Google Scholar]
- 27.Lorico A., Rappa G., Finch R.A., Yang D., Flavell R.A., Sartorelli A.C. Disruption of the murine MRP (multidrug resistance protein) gene leads to increased sensitivity to etoposide (VP-16) and increased levels of glutathione. Cancer Res. 1997;57:5238–5242. [PubMed] [Google Scholar]
- 28.Hiddemann W., Schumann J., Andreef M., Barlogie B., Herman C.J., Leif R.C., Mayall B.H., Murphy R.F., Sandberg A.A., Committee on Nomenclature, Society for Analytical Cytology Convention on nomenclature for DNA cytometry. Cancer Genet Cytogenet. 1984;13:181–183. doi: 10.1016/0165-4608(84)90059-1. [DOI] [PubMed] [Google Scholar]
- 29.Bottomly D., Walter N.A., Hunter J.E., Darakjian P., Kawane S., Buck K.J., Searles R.P., Mooney M., McWeeney S.K., Hitzemann R. Evaluating gene expression in C57BL/6J and DBA/2J mouse striatum using RNA-Seq and microarrays. PLoS One. 2011;6:e17820. doi: 10.1371/journal.pone.0017820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sekiya I., Larson B.L., Smith J.R., Pochampally R., Cui J.G., Prockop D.J. Expansion of human adult stem cells from bone marrow stroma: conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells. 2002;20:530–541. doi: 10.1634/stemcells.20-6-530. [DOI] [PubMed] [Google Scholar]
- 31.Bernardo M.E., Zaffaroni N., Novara F., Cometa A.M., Avanzini M.A., Moretta A., Montagna D., Maccario R., Villa R., Daidone M.G., Zuffardi O., Locatelli F. Human bone marrow derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res. 2007;67:9142–9149. doi: 10.1158/0008-5472.CAN-06-4690. [DOI] [PubMed] [Google Scholar]
- 32.Phinney D.G., Prockop D.J. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair–current views. Stem Cells. 2007;25:2896–2902. doi: 10.1634/stemcells.2007-0637. [DOI] [PubMed] [Google Scholar]
- 33.Mani S.A., Guo W., Liao M.J., Eaton E.N., Ayyanan A., Zhou A.Y., Brooks M., Reinhard F., Zhang C.C., Shipitsin M., Campbell L.L., Polyak K., Brisken C., Yang J., Weinberg R.A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcato P., Dean C.A., Pan D., Araslanova R., Gillis M., Joshi M., Helyer L., Pan L., Leidal A., Gujar S., Giacomantonio C.A., Lee P.W. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 35.Lu X., Kang Y. Cell fusion hypothesis of the cancer stem cell. Adv Exp Med Biol. 2011;714:129–140. doi: 10.1007/978-94-007-0782-5_6. [DOI] [PubMed] [Google Scholar]
- 36.Lu X., Kang Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc Natl Acad Sci U S A. 2009;106:9385–9390. doi: 10.1073/pnas.0900108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duelli D.M., Padilla-Nash H.M., Berman D., Murphy K.M., Ried T., Lazebnik Y. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol. 2007;17:431–437. doi: 10.1016/j.cub.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 38.Jacobsen B.M., Harrell J.C., Jedlicka P., Borges V.F., Varella-Garcia M., Horwitz K.B. Spontaneous fusion with, and transformation of mouse stroma by, malignant human breast cancer epithelium. Cancer Res. 2006;66:8274–8279. doi: 10.1158/0008-5472.CAN-06-1456. [DOI] [PubMed] [Google Scholar]
- 39.Terada N., Hamazaki T., Oka M., Hoki M., Mastalerz D.M., Nakano Y., Meyer E.M., Morel L., Petersen B.E., Scott E.W. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–545. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- 40.Spees J.L., Olson S.D., Ylostalo J., Lynch P.J., Smith J., Perry A., Peister A., Wang M.Y., Prockop D.J. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc Natl Acad Sci U S A. 2003;100:2397–2402. doi: 10.1073/pnas.0437997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pawelek J.M. Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 2005;6:988–993. doi: 10.1016/S1470-2045(05)70466-6. [DOI] [PubMed] [Google Scholar]
- 42.Friedl P. Cell fusion: new mechanisms of plasticity in cancer? Lancet Oncol. 2005;6:916–918. doi: 10.1016/S1470-2045(05)70439-3. [DOI] [PubMed] [Google Scholar]
- 43.Ma L., Teruya-Feldstein J., Weinberg R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- 44.Park S.Y., Lee H.E., Li H., Shipitsin M., Gelman R., Polyak K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin Cancer Res. 2010;16:876–887. doi: 10.1158/1078-0432.CCR-09-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Podhajcer O.L., Benedetti L.G., Girotti M.R., Prada F., Salvatierra E., Llera A.S. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008;27:691–705. doi: 10.1007/s10555-008-9146-7. [DOI] [PubMed] [Google Scholar]
- 46.Chakraborty A.K., de Freitas Sousa J., Espreafico E.M., Pawelek J.M. Human monocyte x mouse melanoma fusion hybrids express human gene. Gene. 2001;275:103–106. doi: 10.1016/s0378-1119(01)00647-3. [DOI] [PubMed] [Google Scholar]
- 47.Neve R.M., Chin K., Fridlyand J., Yeh J., Baehner F.L., Fevr T., Clark L., Bayani N., Coppe J.P., Tong F., Speed T., Spellman P.T., DeVries S., Lapuk A., Wang N.J., Kuo W.L., Stilwell J.L., Pinkel D., Albertson D.G., Waldman F.M., McCormick F., Dickson R.B., Johnson M.D., Lippman M., Ethier S., Gazdar A., Gray J.W. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aktas B., Tewes M., Fehm T., Hauch S., Kimmig R., Kasimir-Bauer S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009;11:R46. doi: 10.1186/bcr2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Creighton C.J., Li X., Landis M., Dixon J.M., Neumeister V.M., Sjolund A., Rimm D.L., Wong H., Rodriguez A., Herschkowitz J.I., Fan C., Zhang X., He X., Pavlick A., Gutierrez M.C., Renshaw L., Larionov A.A., Faratian D., Hilsenbeck S.G., Perou C.M., Lewis M.T., Rosen J.M., Chang J.C. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin S.Y., Yang J., Everett A.D., Clevenger C.V., Koneru M., Mishra P.J., Kamen B., Banerjee D., Glod J. The isolation of novel mesenchymal stromal cell chemotactic factors from the conditioned medium of tumor cells. Exp Cell Res. 2008;314:3107–3117. doi: 10.1016/j.yexcr.2008.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang H.S., Youn Y.K., Oh S.K., Choe K.J., Noh D.Y. Flow cytometric analysis of primary tumors and their corresponding metastatic nodes in breast cancer. Breast Cancer Res Treat. 2000;63:81–87. doi: 10.1023/a:1006470614782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mesenchymal morphological characteristics of breast carcinoma–MSC hybrids. Co-cultures of MSC-luc with MDA-DsRed or MA11-DsRed cells resulted in puromycin-resistant fluorescent hybrids that displayed mesenchymal morphological characteristics (arrows).
Growth rates of hybrids. A: Cell growth curves of MSC-luc-MA11-EGFP and MSC-luc-MDA-DsRed2 hybrid lines and clones. B: Doubling times of MA11-EGFP (left panel), MDA-DsRed2 (right panel), MSC-luc, and their respective hybrid cell lines and clones during in vitro and ex vivo culture from xenografts. Doubling times were calculated during growth of duplicate-triplicate cultures by exponential regression analysis.