


Abstract
Deciphering the structure of gene regulatory networks across the tree of life remains one of the major challenges in postgenomic biology. We present a novel ChIP-seq workflow for the archaea using the model organism Halobacterium salinarum sp. NRC-1 and demonstrate its application for mapping the genome-wide binding sites of natively expressed transcription factors. This end-to-end pipeline is the first protocol for ChIP-seq in archaea, with methods and tools for each stage from gene tagging to data analysis and biological discovery. Genome-wide binding sites for transcription factors with many binding sites (TfbD) are identified with sensitivity, while retaining specificity in the identification the smaller regulons (bacteriorhodopsin-activator protein). Chromosomal tagging of target proteins with a compact epitope facilitates a standardized and cost-effective workflow that is compatible with high-throughput immunoprecipitation of natively expressed transcription factors. The Pique package, an open-source bioinformatics method, is presented for identification of binding events. Relative to ChIP-Chip and qPCR, this workflow offers a robust catalog of protein–DNA binding events with improved spatial resolution and significantly decreased cost. While this study focuses on the application of ChIP-seq in H. salinarum sp. NRC-1, our workflow can also be adapted for use in other archaea and bacteria with basic genetic tools.
INTRODUCTION
The dynamic modulation of gene expression is an important mechanism that allows organisms to sense and respond to changes in their environment. These changes in expression profiles are mediated by dynamic associations of transcription factors and their cognate regulatory regions, collectively known as gene-regulatory networks (GRNs) (1). Regulatory networks integrate complex cellular and environmental cues, orchestrating intricate phenotypes essential for physiology and development. The evolutionary rewiring of these regulatory circuits is thought to be an important driver of speciation (2). Elucidating the structure and function of GRNs is therefore a major research initiative in functional genomics and systems biology (3–8).
The characterization of GRN architecture has been driven by advances in experimental and computational methods for identifying genome-wide protein–DNA interactions (9–13). One such approach is chromatin immunoprecipitation (IP) coupled with high-throughput sequencing (ChIP-seq), a method that provides quantitative genome-wide mapping of target protein-binding events. ChIP-seq identifies protein-binding sites with improved spatial resolution and decreased cost relative to previous microarray-based ChIP-chip technologies (10). While ChIP-seq has become a widely used tool in eukaryotic systems, this method has been applied only once in a bacterial system (14) and there exist no instances of such work in archaea. The small size of bacterial and archaeal genomes makes this high-throughput sequence technology particularly attractive, as sample multiplexing can be used to dramatically reduce costs relative to microarray-based platforms.
Developing a ChIP-seq protocol for archaea would stimulate high-throughput characterization of GRNs, which are a nascent area of study relative to work in the other two domains of life. Archaea are essential drivers of global biogeochemical cycling, integral players in industrial applications and biomedically important organisms. Furthermore, the transcriptional apparatus of archaea exhibits properties of both eukaryotic and bacterial systems, making it an intriguing target for investigating basic principles of regulatory mechanisms across the tree of life (15). Improved understanding of archaeal information processing and transcriptional regulation has widespread applicability.
We present a novel ChIP-seq workflow for the archaea using the model organism Halobacterium salinarum sp. NRC-1 (Hb. NRC-1) and demonstrate its application for mapping the genome-wide binding sites of natively expressed transcription factors. Previous bacterial and archaeal ChIP methods have taken different approaches involving either costly protein-specific antibodies against native proteins (14) or a standard antibody against epitope-tagged target proteins that are constitutively overexpressed from a heterologous plasmid (16,17). This protocol combines these methods by employing a single, commercially available antihemagglutinin (HA) antibody against natively expressed recombinant target proteins. This ChIP-seq method maintains sensitivity and specificity with as little as ∼1 ml of the typical bacterial or archaeal culture, making it suitable for high-throughput analyses. Multiplexing of samples during sequencing significantly decreases experimental costs relative to previous ChIP-chip methods, without diminishing sensitivity and specificity.
A complimentary bioinformatics method is presented for user-friendly identification of binding events using the Pique python package. Integration with the Gaggle toolkit streamlines the exploration and analysis of putative protein-binding sites (18,19). This end-to-end workflow for ChIP-seq of natively expressed proteins provides a suitable platform for large-scale studies of the structure and dynamic remodeling of GRNs. The first protocol of its kind for archaea, this method can be adapted for work in all bacteria and archaea with suitable genetic tools.
MATERIALS AND METHODS
Construction of the pRSK01 ligation-independent cloning vector for Halobacterium salinarum NRC-1
The plasmid pNBK07 (obtained from N. Baliga, Institute for Systems Biology, Seattle, WA) has been previously used to create targeted gene knockouts (17,20–22) in the Hb. NRC-1 ΔpyrF uracil auxotroph strain (Hb. NRC-1 ΔpyrF). For this study, pNBK07 was modified to facilitate Gateway ligation-independent cloning of segments of DNA suitable for chromosomal modification by homologous recombination. Plasmid pNBK07 (sequence and maps in Supplementary Information) was digested with StuI (New England Biolabs cat. #R0187S, Ipswitch, MA), a blunt-end cutting restriction endonuclease. The digested vector was subsequently dephosphorylated with calf intestinal alkaline phosphates (New England Biolabs cat. #M0290S) and purified via agarose gel extraction. PCR primers m13F and m13R (Supplementary Table S1) were used to amplify a fragment of the pDONR221 vector containing an attP1 recombination site, the ccdB gene, a chloramphenicol resistance marker and an attP2 recombination site. This PCR product was ligated into the StuI-digested pNBK07 backbone to create the Gateway (Invitrogen, Carlsbad, CA) compatible pRSK01 vector. The pRSK01 vector was sequenced using the pNBK07F, pNBK07R, ccdB500F and ccdB500B primers. Sequences (plasmid and oligonucleotide) and plasmid maps are provided in Supplementary Information.
Construction of chromosomally tagged transcription factors
Chromosomally epitope-tagged transcription factors were made by way of site-specific homologous recombination in Hb. NRC-1 ΔpyrF. This method is analogous to that used for making in-frame gene knockouts using the pNBK07 vector as previously described (22). Two different approaches were utilized to make the epitope-tagging constructs for this study. In the first approach, PCR-mediated splicing by overlap extension (SOEing) (23) was used to join two PCR products used to add a sequence encoding a region ∼500 bp upstream of the bacteriorhodopsin-activator protein (bat) stop codon, an HA epitope coding sequence, a new stop codon and ∼500 bp downstream of the bat genomic stop codon. PCR primers are listed in Supplementary Table S1. This PCR product was cloned into the StuI site of plasmid pNBK07, which was subsequently transformed into strain Hb. NRC-1 ΔpyrF. A two-step, double-crossover process was followed to chromosomally insert the HA epitope. First crossover recombinants were selected by plating on 2% (w/v) complete media (CM) agar plates containing 20 µg/ml mevinolin. Second crossover recombinants were enriched by selecting on 2% CM agar plates containing 300 µg/ml 5-fluoroorotic acid (5-FOA). The absence of a functional pyrF gene is required for survival on 5-FOA, indicating loss of plasmid.
The second method for chromosomal epitope tagging was used to tag the general transcription factor tfbD. This method takes advantage of commercial DNA synthesis technologies and the new Gateway cloning compatible pRSK01 vector. In this case, a construct consisting of an attB1 recombination site, ∼500 bp upstream of the tfbD stop codon, the sequence encoding an HA epitope tag, a stop codon, ∼500 bp downstream of the tfbD chromosomal stop codon, and an attB2 recombination site were directly synthesized by Geneart (Invitrogen, Carlsbad, CA) and delivered, cloned, in a pANY backbone vector also encoding an ampicillin-resistance marker. This vector was used directly in an in vitro recombination reaction (Gateway cloning, Invitrogen, Carlsbad, CA) with the pRSK01 vector according to manufacturer’s protocols to move the synthetic construct into pRSK01. Once the synthetic construct is inserted into pRSK01, the rest of the tagging procedure is identical to that used for pNBK07-based tagging. We have also used a combination of SOEing and Gateway recombination to directly clone PCR products, flanked by appropriate attB recombination sites, directly into pRSK01 (data not shown).
Verification of chromosomal tagging
The insertion of HA epitopes at the C-terminal ends of the chromosomally encoded bat and tfbD genes was verified both by PCR and DNA sequencing. The following PCR reactions summarized in Supplementary Figure S2 were conducted to verify insertion of the HA epitope.
The initial PCR screen (Reaction 1) verified the presence of the C-terminally tagged gene of interest in the cell using a forward primer (ct_gene_of_interest_a_F) located in the gene and a reverse primer complimentary to the HA epitope tag’s sequence (HA_epiope_R). PCR products of the expected size indicate either successful chromosomal tagging or the presence of residual tagging vector in the cell.
Recombinant strains were also screened for the presence of chromosomally encoded pyrF using primers flanking the chromosomally encoded gene (k_vng167g3_e_F and k_vng1673g_d_R, Reaction 2). The presence of chromosomally encoded pyrF yields a 2050 bp PCR product, while the disrupted pyrF in the Hb. NRC-1 ΔpyrF strain yields a PCR product of 712 bp. Reaction 2 was performed to verify that the pyrF gene from the plasmid has not reintegrated into the chromosome of the Hb. NRC-1 ΔpyrF strain.
A second pyrF PCR screening (Reaction 3) was carried out to confirm that the plasmid carrying the pyrF had been cured and that pyrF had not recombined into the chromosome of the Hb. NRC-1 ΔpyrF strain. Reaction 3 amplifies a region from 465 bp upstream of the pyrF stop codon to 70 bp downstream of the pyrF stop codon (primers k_vng1673g_g_F and k_vng1673g_h_R). This final reaction yields no product in the Hb. NRC-1 ΔpyrF strain and its derivatives. In strains that do carry the pyrF gene, such as wild type Hb. NRC-1, or strains transformed with either the pNBK07 or pRSK01 plasmids, a 535 bp product is formed.
Finally, we screen specifically for plasmid-encoded copies of the pyrF gene using primers that amplify a segment of the pyrF encoded by the pNBK07 or pRSK01 vectors (k_vng1673g_g_F and o_pNBK07_a_R, Reaction 4). The absence of product confirms that the plasmid has been cured, when the reaction is run in conjunction with plasmid-containing positive control.
PCR products derived from PCR reaction using primers ct_gene_of_interest_a_F and ct_gene_of_interest_d_R on strains meeting all the criteria established by the verification Reactions 1–4 above were sequenced via standard Sanger sequencing to verify the integration of the HA epitope. Sequences are provided in the Supplementary Information. Tag integration was further verified by analyzing the genome re-sequencing data for each strain that was generated in the process of the ChIP-seq experiment.
Culture preparation
All cultures were grown in the standard CM for H. salinarum (250 g/l NaCl, 20 g/l MgSO4, 2 g/l KCl, 3 g/L Na–Citrate, 10 g/l Oxoid peptone (Oxoid cat# LP0034) (Oxoid, Basingstoke, UK) supplemented with 50 mg/l uracil and filled to volume with distilled water. Cultures were revived from −80°C freezer stocks and were streaked on agar plates. Starter cultures were inoculated from individual colonies and allowed to reach an optical density at 600 nm of 0.7 before inoculating a culture at a starting optical density at 600 nm of 0.03. Cells were grown under ambient light conditions in unbaffled flasks in a volume equal to 25% of the flask’s maximum volume. All cultures were grown at 37°C and shaken at 150 rpm on a New Brunswick G-53 orbital shaker (New Brunswick, Edison, NJ).
Immunoprecipitation
Biological replicates were conducted as inoculations in separate but identical volumes on the same orbital shaker. Cells were harvested at an OD 600 between 0.9 and 1.0, which corresponds to early stationary phase for Hb. NRC-1. Cells were immediately fixed with 1% (v/v) formaldehyde for 10 min. Fixing was stopped through the addition of glycine to a final concentration of 125 mM. Batches of 1.75 × 1010 cells were removed and pelleted at 5000 ×g. Cell pellets were washed twice with citrate-free basal salts, after which the pellets were frozen at −80°C. Though this was our standard input for the IP reaction, we also examined the effect of decreasing the number of input cells for the IP reaction using the tfbD::HA strain. In addition to 1.75 × 1010 cells, these scaling experiments also used 8.75 × 109, 3.50 × 109, 1.75 × 109 and 3.50 × 108 cells as input material for IP. The rest of the method is described in detail for 1.75 × 1010 cells. Appropriate volumes and quantities of reagents for the scaled-down experiments are reported in Supplementary Table S3.
Cell pellets were resuspended in 1.6 ml of lysis buffer (50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 1% (v/v) Triton X-100, 0.1% (w/v) sodium deoxycholate, pH 7.5) containing protease inhibitors (Roche cat# 04693159001). Resuspended pellets were sonicated using a Bioruptor (Diagenode, Denville, NJ) until DNA fragment size reached an average of ∼500 bp (2–7.5-min cycles, 30 s on/30 s off, high power setting).
Cell lysate was combined with 1 µg of anti-HA antibody (Abcam cat# ab9110) (Abcam, Cambridge, MA) and protein A-conjugated Dynabeads (Invitrogen cat. # 100.2D) preblocked with 5 mg/ml BSA in phosphate-buffered saline and incubated overnight at 4°C. Dynabeads were washed two times with the lysis buffer, two times with 1 ml of the lysis buffer supplemented with 500 mM NaCl, two times with 1 ml wash buffer (10 mM Tris, 250 mM LiCl, 0.5% NP-40 (v/v), 0.5% Na-deoxycholate (w/v), 1 mM EDTA, pH 8.0) and one time with 1 ml TE buffer. Enriched ChIP DNA/transcription factor complexes were eluted by the addition of 50 µl elution buffer (50 mM Tris, 10 mM EDTA, 1% SDS (w/v), pH 8.0) and incubation at 65°C for 10 min. Cross-links were reversed by incubating in TE/SDS (10 mM Tris, 1 mM EDTA, 1% SDS) overnight at 65°C. RNA was digested and DNA sample was subsequently prepared for Illumina single read sequencing.
ChIP-seq library preparation
Individual ChIP samples were blunt ended with T4 DNA polymerase (NEB cat. # M0203L), Klenow large fragment (NEB cat. # M0210L) and T4 polynucleotide kinase (NEB cat. # M0201L) at 20°C for 30 min. Blunt-ended DNA was 3′ A tailed with 3′->5′ exo- Klenow fragment (NEB cat. # M0212L) for 30 min at 37°C. Adapters containing 6 bp barcodes were ligated to the prepared ChIP DNA samples for 15 min at room temperature with T4 DNA ligase (Enzymatics cat. # L603-HC-L). Barcode sequences are provided Supplementary Table S4. A background control of whole cell extract genomic DNA from each sample was prepared as mentioned above. Samples were then used as template for an 18-cycle PCR amplification. PCR products were quantified and visualized with a high-sensitivity DNA chip (Agilent cat. # 5067-4626) on a bioanalyzer (Agilent, Santa Clara, CA). ChIP and background libraries were pooled in equimolar concentrations and loaded onto a single Illumina lane.
Western blotting
Transcription factors were immunoprecipitated under the same conditions as ChIP methods mentioned above. IP samples were run in one dimension on 4–12%, 1.5 mm polyacrylamide gel (Invitrogen cat. # NP0335) in MOPS buffer (Invitrogen cat. # NP0001). Protein was then blotted onto a 0.2 -µm pore size PVDF membrane (Invitrogen cat. # LC2002) at 30 V for one hour in transfer buffer (25 mM Tris, 192 mM glycine, 10% (v/v) methanol, pH 8.4). PVDF was blocked in 0.5% (w/v) casein overnight and subsequently probed with HRP-conjugated anti-HA antibody (Abcam cat. # ab1265). The blot was incubated with GE ECL plus reagents (Amersham cat. # RPN2132) according to the manufacturer’s suggestions and exposed to light-sensitive film.
qPCR verification
qPCR was performed on a Bio-Rad Chromo 4 Real-Time Detector (Bio-Rad, Hercules, CA) using KAPA SYBR® FAST Universal 2× qPCR master mix (Kapa Biosystems cat.# KK4601) (Kapa Biosystems, Woburn, MA) according to the supplied protocol. Primer sets for enriched regions and negative regions were designed using known enriched sites and unenriched sites, respectively, from previous ChIP-seq and ChIP-chip data (see Supplementary Table S1 for primer sequences). Fold enrichment above background was calculated as 2 to the power of cycle threshold difference between a non-enriched region and an expected enriched site. WCE extract, ChIP samples and amplified libraries were all used as template for a qPCR reaction. These were all confirmed by comparing to a set of ChIP-control reactions on the Hb NRC-1 ΔpyrF strain.
Sequencing, processing and ChIP-seq peak calling
Multiplexed samples were sequenced to 40 bp on the Illumina GA-II. Sequences were barcode sorted and quality trimmed (minimum Phred quality 20, minimum length 25 bp) using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) (Gordon,A and Hannon,G.J., unpublished results). Sequencing primer and adapter contamination were filtered using the TagDust package (24). Quality-filtered reads were mapped using Bowtie (25) to the Hb. NRC-1 reference genome with repeat sequences masked, and SAM format sequence files were converted to sorted BAM files using the samtools package (26).
Putative protein–DNA binding events were detected using Pique, a novel microbially focused and freely available peak calling application (available at https://github.com/ryneches/pique, version tag: halo_egw) (Neches,R.Y., Wilbanks,E.G. and Facciotti,M.T., in preparation). Pique is written in Python and makes use of the SciPy signal-processing subroutines (27). Pique is able to operate on systems that have genomic complexities such as IS elements, gene dosage polymorphisms and accessory genomes that cause coverage variations unrelated to ChIP, or in cases where the organism under study is not identical to the reference genome. The resulting enrichment ‘pedestals’ and ‘holes’ can be problematic for accurately detecting binding events and calculating enrichment levels. Hb. NRC-1 has several IS elements and two plasmids that exhibit dosage variations, and so a segmented analysis was performed by providing a genomic map of these features in the reference genome.
ChIP-seq coverage data and candidate peaks were visualized using the Gaggle Genome Browser (18). Shared peaks were assessed using a combination of BEDTools (28) and custom R scripts. To assess the required sequencing depth for accurate and sensitive binding site identification, random subsampling of a 6 million read TfbD ChIP and WCE control runs were performed.
RESULTS
Epitope-tagged strain construction
We developed a protocol for rapidly engineering the strains of the Hb. NRC-1 with epitope-tagged target proteins under the control of their native promoters. Using different classes of transcription factors, we demonstrate this approach’s application and utility. In bacteria and archaea, different transcription factors may bind a wide span of target sites, ranging from one to hundreds. We chose to collect localization data from two extreme cases. The general transcription factor TfbD is known to bind hundreds of promoters (16). As an example of a specific regulator of a smaller regulon, we examined the Bat transcription factor. This transcription factor predicted to bind up to four potential sites and is one of the few haloarchaeal transcription factors with a well-described binding motif (29).
Our epitope-tagging protocol for Hb. NRC-1 employed a homologous recombination method analogous to the gene deletion strategy developed by Peck et al. (30). This epitope knock-in strategy was described by Zhang et al. for ChIP-chip applications in human somatic cell lines (31). For tfbD::HA strain construction, the novel vector pRSK01, which is compatible with Gateway ligation-independent cloning (Invitrogen, Carlsbad, CA), was created to facilitate and further standardize strain construction. This new vector allowed the use of either commercial DNA synthesis technology or PCR-mediated SOEing (23) with Gateway compatible primers to rapidly construct the vectors used for strain construction. While in most instances PCR SOEing is still less expensive, the decreasing cost of DNA synthesis should make this approach a more attractive alternative for future large-scale strain construction projects.
Hb. NRC-1 ΔpyrF was transformed with a recombinant plasmid that contained the terminal 500 bp of the target gene, a hemagglutinin (HA) tag, stop codon and 500 bp downstream sequence (Figure 1). Homologous recombination between the chromosomal target gene and the recombinant plasmid sequence introduced the HA tag to the chromosomal sequence. Successful first recombinants were determined by PCR screening of MevR colonies (Supplementary Figure S1). The plasmid was subsequently resolved using 5-FOA counter-selection as previously described (22,30). Strains with C-terminally HA-tagged target proteins were further verified by PCR and Sanger sequencing (Supplementary Figures S1–S2 and Supplementary Information). For the rapid construction of epitope-tagged transcription factor strains, this general strategy of utilizing DNA synthesis and homologous recombination-based chromosomal modification can be readily extended to any organisms with a system for targeted genetic knockouts.
Figure 1.

Epitope tag-in approach for Hb. NRC-1. The Hb. NRC-1 ΔpyrF strain is transformed with a plasmid containing the mevinolin-resistance determinant (MevR; dark gray box) and the pyrF gene (black box) that confers 5-FOA sensitivity. The plasmid carries an engineered sequence containing the HA epitope sequence (white box) flanked by the last 500 bp of the target gene and the 500 bp downstream of the target gene (light gray boxes). Plasmid sequence is shown as solid line, chromosomal sequence is shown as solid, wavy lines. Cross-over can occur between target gene (light gray box) and flanking sequence (gray wavy line) in the chromosome and the homologous regions in the plasmid sequence, at either position 1 or 2 (position 1 example shown). PCR screening of mevinolin-resistant colonies is used to determine successful first recombinants. Subsequent plating on 5-FOA selects for second recombinants (via counter-selection with the pyrF gene). In this example, a second cross-over at site 2 produces the desired chromosomally integrated recombinant target_gene::HA fusion. PCR screening of these colonies is required to distinguish this desired second recombinant from a second recombinant occurring at position 1. Drawing is not to scale.
Western blotting with anti-HA antibody confirmed the specificity of the ChIP assay in these chromosomally tagged strains (Supplementary Figure S3). The chromosomally integrated, epitope-tagged proteins remain under the control of their native promoters, as observed in the differential expression of the TfbD-HA protein over the course of growth in the recombinant strain ΔpyrF tfbD::HA (Supplementary Figure S4). Increase in the abundance of TfbD during stationary phase is consistent with previous reports of tfbD transcriptional abundance (20). This approach can be used to monitor dynamic changes in the GRN that occur under different physiological conditions.
Identifying target protein DNA-binding sites
We used ChIP-seq to analyze with two different classes of target proteins: the general transcription factor TfbD and the Bat using recombinant strains that natively express the target protein (ΔpyrF bat::HA and ΔpyrF tfbD::HA). From the ChIP-seq datasets of 1.2 million reads for each factor, we identified 380 binding sites for TfbD and two binding sites for Bat (the brp and crtB1 promoters).
These punctate target protein DNA-binding events produce a distinctive bimodal, strand-specific enrichment pattern in sequence coverage (Figure 2). This enrichment pattern was leveraged to identify binding sites using our open source software package Pique, which reports the candidate binding site’s enrichment as the ratio of sequence coverage in the IP data relative to a background control (Neches,R.Y., Wilbanks,E.G. and Facciotti,M.T., in preparation). Pique is implemented from a user-friendly graphical user interface and exports predicted binding sites to the Gaggle Genome Browser (18). From the Gaggle genome browser, users can explore and curate the data before proceeding with analysis via other downstream Gaggle tools (Figure 3).
Figure 2.
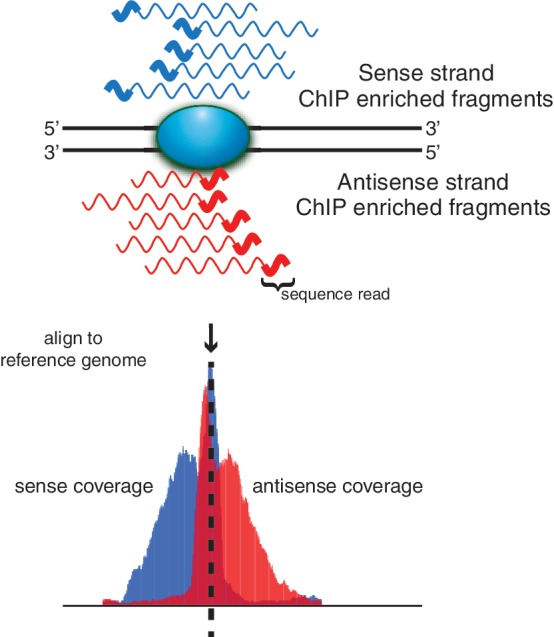
The 5′ to 3′ sequencing requirement and short read length produce stranded bias in sequence coverage. The shaded blue oval represents the protein of interest bound to DNA (solid black lines). Wavy lines represent either sense (blue) or antisense (red) DNA fragments from ChIP enrichment. The thicker portion of the line indicates regions sequenced by short read sequencing technologies. Sequenced tags are aligned to a reference genome and shown below is the strand-specific sequence coverage at each position in the genome. Punctate-binding events (e.g. transcription factors) are characterized by well-defined strand-specific bimodality in sequence coverage.
Figure 3.

The Pique software package processes ChIP-seq coverage data to predict protein-binding sites. Strand-specific coverage data are output as tracks for the Gaggle Genome Browser, and putative-binding sites (peaks) are output as ‘bookmark files’. (A) Screenshot of data browsing in the Gaggle Genome Browser. Green box outlines the navigation window for clicking through bookmarks of predicted binding sites. Details of each site can be displayed (inset). The Gaggle toolbar (shown with black arrow) can be used to broadcast selected data to other ‘geese’ in the gaggle package, programs such as R, cytoscape, BLAST or KEGG. (B) Schematic overview of bioinformatics workflow.
To confirm the specificity of the HA antibody for the ChIP assay on the recombinant HA-tagged strains, we conducted ChIP-seq on the ΔpyrF parent strain where no HA tags were expressed. The data contained a single peak, at chromosomal position 166589, likely because of the presence of a similar epitope in a native protein. This peak was present in all datasets examined and was subsequently filtered from all downstream analysis.
Independent biological replicates of the TfbD ChIP-seq experiment showed good reproducibility (82% overlapping binding sites). Differences between the two replicates are due to the smallest peaks in each dataset, as indicated by examining increasingly stringent enrichment thresholds (Supplementary Figure S5). Both binding sites in the Bat dataset and two example sites in the TfbD datasets (VNG906H and atp_p promoters) were confirmed with a ChIP-qPCR assay. The relationship between the quantification of ChIP enrichment at the binding sites determined by qPCR and sequencing was found to be well correlated across several experiments (Figure 4). The TfbD ChIP-seq binding sites agreed well with previously reported sites from TfbD ChIP-chip experiments (16). For both the ChIP-seq and ChIP-chip methods, 80% of consensus binding sites from biological replicates were identified by at least one of the replicates from the other method.
Figure 4.

ChIP enrichment of binding sites determined by qPCR and sequencing show a linear relationship. Data shown are drawn from multiple ChIP experiments: the Bat ChIP (Pbrp and PcrtB1 closed and open circles) and the TfbD ChIP and the reduced cell number TfbD ChIPs (PVNG906H and Patp_p; closed and open triangles). Differences in enrichment at the TfbD-bound promoters corresponded to changes produced by decreasing the number of cells in the ChIP reaction (see Figure 5 for further details).
Spatial resolution
The spatial resolution of the ChIP-seq binding site prediction was assessed for the Bat and TfbD datasets. As the Bat transcription factor has a well-characterized predicted binding motif (29), we used the distance from our predicted binding site to the motif center as an estimate of the spatial resolution. The binding sites at the brp and crtB1 promoters were found, respectively, at 20 and 27 bp upstream of the predicted Bat-binding motif center (3′ displaced).
As there exists no well-defined binding motif for the general transcription factor TfbD, we used proximity to the nearest predicted transcript start site (TSS) as a measure of spatial resolution for this factor. Archaeal TFB proteins, homologs of the eukaryotic factor TFIIB, canonically bind at B-recognition elements ∼30–50 bp upstream of the TSS, in association with a TATA-binding protein (32). Eighty-six percent of the 312 consensus binding sites from the TfbD ChIP-seq biological replicates were found within 250 bp of a predicted TSS (in either 3′ or 5′ direction). We measured the distance from each of these predicted binding sites to the nearest predicted TSS. These values were compared to the distance to TSS from previously reported TfbD ChIP-chip sites, determined with both 500-bp contiguous and 12-bp overlapped tiling microarray (Figure 5) (16). The distance to TSS for the ChIP-seq binding sites (median = 32, mean = 51) is in agreement with the expected binding pattern for this general transcription factor, and is significantly smaller than that predicted by both resolutions of ChIP-chip microarray (Mann Whitney U-test, p value < 0.005). The variance in these distance measurements provides an estimate of the precision with which each method maps binding sites. The ChIP-seq dataset has significantly decreased variance relative to both ChIP-chip datasets (significance assessed by Bartlett test, p value < 0.005; samples were tested for normality by two-sided Kolmogorov-Smirnov p value < 0.0005). Taken together, these data indicate that the ChIP-seq assay offers improved spatial resolution in mapping the target protein-binding site relative to prior ChIP-chip assays.
Figure 5.

Distance from predicted TfbD-binding site for ChIP-seq (consensus between biological replicates), 500-bp tiling microarray ChIP-Chip (consensus between biological replicates) and 12-bp tiling microarray ChIP-Chip experiments. The observed difference in means was statistically significant (Mann Whitney U-test, p value < 0.005), as is the observed difference in variance (Bartlett test, p value < 0.005).
ChIP assay cell number and sequencing depth
We investigated the number of cells required for ChIP to produce sufficient enrichment at target protein-binding sites. Decreasing the number of cells in the ChIP reaction lowered the observed enrichment at target protein-binding sites; however, enrichment (>5 x) was detectable for highly enriched TfbD-binding sites when as few as 3.50 × 108 cells were used for ChIP, equivalent to 1 ml of a typical culture (Figure 6A). Because of the overall decrease in enrichment, smaller cell number ChIPs were less sensitive in binding site detection but maintained specificity (Figure 6B). The relatively small volume required for this ChIP assay should enable the high-throughput application of this method in the context of dynamic binding studies by allowing for the repetitive sampling of numerous strains with minimal perturbation.
Figure 6.

TfbD ChIP with decreasing numbers of cells. (A) ChIP-qPCR determined fold enrichment at two test promoters (VNG906H, dark bars and atp_p light bars) decreases with lower numbers of cells; however, strong (>5 x) enrichment is still observed with 3.5 × 108 cells. (B) For decreasing cell volume ChIP-seq experiments, fewer peaks could be identified (number of peaks identified, squares and lines), resulting in a significant loss in sensitivity. However, the percentage of identified peaks that were true positives stayed high (% true positives, triangles). True positives were defined as binding sites that were shared with at least one of the optimized 1.75 × 1010 ChIP TfbD biological replicates.
One of the main advantages to the ChIP-seq platform for small microbial genomes is the ability to decrease the experimental cost by multiplexing many samples in a single sequencing lane. We carried out an in silico analysis to determine the depth of sequencing necessary to achieve sensitive and accurate detection of binding sites. Sequence reads were randomly subsampled to decreasing coverage levels from 1.2 M reads (15.4 x coverage) to 10 000 reads (0.13 x coverage). For the TfbD dataset, the number of peaks identified remained stable down to 500K reads (5 x coverage), after which the sensitivity began to decrease (Figure 7). The specificity of binding site identification remained excellent below 500 K reads, even though the sensitivity decreased (Figure 7).
Figure 7.

Subsampling sequence coverage. Sequences were randomly sampled from a TfbD ChIPseq dataset of 6 M reads to create subsampled datasets of decreasing coverage. The number of peaks that could be identified in each subset (filled circles) is shown as a function of the number of sequence reads in the dataset. The specificity of these smaller lists is assessed as the percentage of the identified peaks which overlapped the larger 1.2 M read dataset (solid line).
For the Bat dataset, the more strongly enriched binding site at the brp promoter could be detected in datasets as small as 50 000 reads (0.64 x coverage), whereas the weaker binding at the crtB1 promoter was undetectable below 150 000 reads (1.9 x coverage). No false positives were detected in these lower coverage datasets, with the exception of a single site in the 200 000 read dataset. Examining the effect of decreased coverage on the spatial resolution, we found that the automated prediction of the binding site remained accurate, until just before the site became undetectable (Figure 8).
Figure 8.

Spatial resolution of binding sites calculated from randomly subsampled Bat ChIP-seq datasets of decreasing coverage. The distance from ChIPseq predicted binding site to nearest Bat-binding motif was calculated for both the crtB1 (dark gray bars) and brp (light gray bars) promoter regions. Binding sites for in the crtB1 promoter could not be detected for datasets with 100 000 reads and below (no bars in plot).
DISCUSSION
We report here a workflow for the genome-wide mapping of archaeal natively expressed transcription factors using a standardized, cost-effective, high-throughput ChIP-seq platform. This is the first example of a ChIP-seq protocol for archaea. The development of this high-throughput method for mapping protein–DNA binding events in archaea should catalyze the investigation of the GRNs in this third domain of life.
While major advances have been made in mapping the GRNs of bacteria and eukaryotes, the GRNs of archaea represent a nascent area of exploration, with only a handful of genome-wide experimental studies (8,16,17,33,34). Understanding regulatory mechanisms in archaea will greatly inform our understanding of the basic biology of this important domain, relevant to diverse fields of study from biogeochemistry to biotechnology. The development of improved methods for surveying archaeal GRNs is timely, coinciding with a new wave of archaeal genome sequencing that has suggested many conserved archaeal regulatory mechanisms (34,35).
Archaea also possess an intriguing mosaic transcriptional apparatus that exhibits properties of both eukaryotic and bacterial systems. While the basal transcriptional machinery of archaea is homologous to that of eukaryotes, archaeal transcriptional regulators are more similar to those of bacteria. Studies of archaeal transcription have provided insight into both the mechanisms and evolution of information processing in the three domains of life (32,36). Likewise, deciphering archaeal GRNs holds a great potential for advancing our understanding of fundamental principles employed by GRNs across the tree of life.
The workflow presented here is cost efficient and amenable to high-throughput scaling. To increase throughput and minimize costs, we relied on recombinant strains with low-profile HA epitope-tagged target proteins and a standard anti-HA antibody for the ChIP assay. The HA epitope tag in conjunction with the well-characterized commercial antibody proved efficient for IP while minimally perturbing the target protein. The choice of the sterically slim HA epitope tag can prove quite important; we have found that the larger repeated myc epitope tag (37) can render some DNA-binding proteins nonfunctional. The HA tag does not disrupt Bat function, as seen in its ability to complement a bat knockout mutation and induce bacteriorhodopsin production (Supplementary Figure S6).
A transcription factor’s occupancy of possible binding sites depends, in part, on its concentration within the cell. As our ultimate goal is to map the dynamics of regulatory network rearrangement, native expression of the target transcription factor, rather than constitutive expression from a plasmid construct, was an important feature of our approach. To accomplish this, we used recombinant target proteins that were chromosomally integrated under the control of the wild-type promoter. The construction of these recombinant archaeal strains required the development of a method for generating chromosomally integrated recombinant proteins in Hb. NRC-1, thereby expanding the genetic toolbox available for this model archaeon.
Previous ChIP-chip studies in Hb. NRC-1 used target proteins that were constitutively expressed at nonnative levels from a heterologous plasmid (16,33,38). The resultant protein–DNA associations are, therefore, perhaps best viewed as lists of all possible interactions rather than a snapshot of protein–DNA association network under physiological conditions. While this approach is appropriate for some applications and can offer technical advantages, such as improving ChIP efficiency for proteins present in low abundance, expression of target proteins at nonnative levels can produce artifacts in the list of protein-–DNA binding sites. In the simplest case, constitutive overexpression can drive transcription factor association to weak or nonspecific sites without significantly perturbing expression. Ambiguities concerning which binding sites are physiologically relevant can sometimes be resolved by incorporating data such as transcriptomes and regulatory motifs in the analysis. However, the perturbation of transcription factor expression can also have more serious consequences that cannot be easily resolved, such as unintended protein–protein interactions and changes in the cellular phenotype. Lastly, constitutive nonnative expression precludes investigating the dynamics of transcription factor association, a fundamental aspect in understanding the relationship between the GRN structure and function.
We demonstrated the application of our ChIP-seq protocol on two different classes of archaeal transcription factor: a general transcription factor with many binding sites (TfbD) and a more specific transcriptional activator (Bat). ChIP-seq data were analyzed with the user-friendly, open-source Pique package, designed for identifying protein–DNA binding events in small bacterial and archaeal genomes. Our bioinformatics pipeline integrates with the Gaggle toolkit to facilitate downstream data visualization, curation and analysis.
The predicted binding sites were consistent between biological replicates and with previously published ChIP-chip results for TfbD. We observed few significant trends in the gene classes bound by TfbD, with the exception of gene functionally associated with GTPase activity (p value = 1 × 10−4). The lack of obvious functional partitioning of TfbD target genes is unsurprising, given this factor’s broad role in global transcription initiation. Dynamic ChIP-seq experiments under different physiological conditions would likely be an appropriate future method for determining the potential regulatory roles carried out by TfbD and other archaeal general transcription factors.
The two Bat-binding sites discovered in the brp and crtB1 promoters (Pbrp, and PcrtB1) were verified by qPCR and contain two of the four previously reported occurrences of the Bat regulatory motif (Pbop and Pblp were not bound) (29). It seems initially surprising that Bat binding was not detected upstream of the bacteriorhodopsin apoprotein (bop), a gene it regulates (29,39–41). However, recent research has shown that Bat regulation of bop expression is complex and may work cooperatively with accessory proteins Brz and Brb (42,43).
Interestingly, the Bat-binding motif at Pbrp, and PcrtB1 share a single nucleotide insertion relative to the two unbound motif sites at Pbop and Pblp. Furthermore, the spacer sequence between the Bat motif and the TATA-box is shorter at Pbrp and PcrtB1 sites (2 and 3 bp spacer, respectively), relative to the unbound Pbop and Pblp motif occurrences (5 bp spacer) (29). We note that these small differences in the Bat motif, in concert with our binding data, may provide some preliminary evidence to suggest a way by which Bat (and potential coregulators) distinguishes between the four predicted binding sites in a condition-specific manner.
ChIP-seq identifies protein-binding sites with fine spatial resolution and provides accurate estimates of binding site enrichment. The quantification of enrichment found at protein binding sites calculated by ChIP-seq was very similar to that determined by ChIP-qPCR (Figure 5). Unlike ChIP-chip enrichment values, which become saturated at high levels of enrichment, ChIP-seq has excellent dynamic range, and thus provides an accurate metric for the level of enrichment at target protein-binding sites. A narrow size selection of chromatin immunoprecipitated DNA fragments enhances the enrichment in sequence coverage at target-binding sites. The use of automated size-selection instruments, such as the Pippin Prep® (Sage Science, Beverly, MA), in the preparation of ChIP-seq libraries may improve data quality.
The number of cells required for the ChIP assay was investigated to determine an optimal protocol that balances sensitivity of binding site detection with throughput and ease of sample handling. Decreasing the number of cells for the ChIP assay was found to decrease the enrichment level at target protein-binding sites, and thus the sensitivity of the assay. However, the more strongly enriched sites could still be accurately detected with 3.50 × 108 cells, equivalent to ∼1 ml of a typical culture. The false-positive rate remains very low in the lower cell number ChIP-seq experiments. The ability to use low cell counts as input makes this approach tractable for high-throughput assessment of the more prominent binding sites in the genome, though precludes development of an exhaustive list of possible protein–DNA interactions.
By randomly subsampling deeply sequenced datasets, we determined that the required sequence coverage for the sensitive detection of binding sites corresponds to ∼6.5 x coverage of the complete genome (approximately 500 K reads in Hb. NRC-1). We estimate that for the average sequencing run on the Illumina HiSeq (80 million reads) and the typical bacterial and archaeal genome (∼3 Mb), 130 samples can be multiplexed per lane. With this level of multiplexing, the ChIP-seq assay would cost roughly $15 per sample. The per-sample cost is expected to drop even further with continuing improvements in the output of sequencing technologies. Relative to ChIP-chip, this ChIP-seq workflow greatly reduces the experimental cost of defining the genome-wide binding sites of target transcription factors while also improving spatial resolution. From gene tagging to data analysis, this workflow provides an excellent model for conducting large-scale, dynamic mapping of bacterial and archaeal gene regulator networks.
ACCESSION NUMBERS
All high-throughput sequencing data generated in this work are available via BioTorrents (http://www.biotorrents.net/details.php?id=259) or from our lab website (www.bme.ucdavis.edu/facciotti/resources_data/data/). The open access Pique package and source code can be obtained via github at https://github.com/ryneches/pique. Primer and plasmid sequences used in this study are available in Supplementary Information.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4, Supplementary Figures 1–6, Supplementary Information and Supplementary Reference (44).
FUNDING
UC Davis Startup Funds to M.T.F., an NSF GRFP to E.G.W. and DARPA award (HR0011-05-1-0057) to R.Y.N. Funding for open access charge: UC Davis Startup Funds to M.T.F.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Davidson EH. Genomic Regulatory Systems: Development and Evolution. San Diego: Academic Press; 2001. [Google Scholar]
- 2.Shou C, Bhardwaj N, Lam HY, Yan KK, Kim PM, Snyder M, Gerstein MB. Measuring the evolutionary rewiring of biological networks. PLoS Comput. Biol. 2011;7:e1001050. doi: 10.1371/journal.pcbi.1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. Transcriptional regulatory code of a eukaryotic genome. Nature. 2004;431:99–104. doi: 10.1038/nature02800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peter IS, Davidson EH. Modularity and design principles in the sea urchin embryo gene regulatory network. FEBS Lett. 2009;583:3948–58. doi: 10.1016/j.febslet.2009.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaleta C, Gohler A, Schuster S, Jahreis K, Guthke R, Nikolajewa S. Integrative inference of gene-regulatory networks in Escherichia coli using information theoretic concepts and sequence analysis. BMC Syst. Biol. 2010;4:116. doi: 10.1186/1752-0509-4-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palaniswamy SK, James S, Sun H, Lamb RS, Davuluri RV, Grotewold E. AGRIS and AtRegNet. a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 2006;140:818–29. doi: 10.1104/pp.105.072280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fadda A, Fierro AC, Lemmens K, Monsieurs P, Engelen K, Marchal K. Inferring the transcriptional network of Bacillus subtilis. Mol. Biosyst. 2009;5:1840–52. doi: 10.1039/b907310h. [DOI] [PubMed] [Google Scholar]
- 8.Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, Thorsson V, Shannon P, Johnson MH, Bare JC, et al. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–65. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 9.Hesselberth JR, Chen X, Zhang Z, Sabo PJ, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, et al. Global mapping of protein–DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 2009;6:283–89. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009;10:669–80. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilbanks EG, Facciotti MT. Evaluation of algorithm performance in ChIP-seq peak detection. PLoS One. 2010;5:e11471. doi: 10.1371/journal.pone.0011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, Thorsson V. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biol. 2006;7:R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Jong H. Modeling and simulation of genetic regulatory systems: a literature review. J. Comput. Biol. 2002;9:67–103. doi: 10.1089/10665270252833208. [DOI] [PubMed] [Google Scholar]
- 14.Lun DS, Sherrid A, Weiner B, Sherman DR, Galagan JE. A blind deconvolution approach to high-resolution mapping of transcription factor binding sites from ChIP-seq data. Genome Biol. 2009;10:R142. doi: 10.1186/gb-2009-10-12-r142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bell SD. Archaeal transcriptional regulation—variation on a bacterial theme? Trends Microbiol. 2005;13:262–265. doi: 10.1016/j.tim.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Facciotti MT, Reiss DJ, Pan M, Kaur A, Vuthoori M, Bonneau R, Shannon P, Srivastava A, Donohoe SM, Hood LE, et al. General transcription factor specified global gene regulation in archaea. Proc. Natl Acad. Sci USA. 2007;104:4630–35. doi: 10.1073/pnas.0611663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaur A, Van PT, Busch CR, Robinson CK, Pan M, Pang WL, Reiss DJ, DiRuggiero J, Baliga NS. Coordination of frontline defense mechanisms under severe oxidative stress. Mol. Syst. Biol. 2010;6:393. doi: 10.1038/msb.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bare JC, Koide T, Reiss DJ, Tenenbaum D, Baliga NS. Integration and visualization of systems biology data in context of the genome. BMC Bioinformatics. 2010;11:382. doi: 10.1186/1471-2105-11-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shannon PT, Reiss DJ, Bonneau R, Baliga NS. The Gaggle: an open-source software system for integrating bioinformatics software and data sources. BMC Bioinformatics. 2006;7:176. doi: 10.1186/1471-2105-7-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Facciotti MT, Pang WL, Lo FY, Whitehead K, Koide T, Masumura K, Pan M, Kaur A, Larsen DJ, Reiss DJ, et al. Large-scale physiological readjustment during growth enables rapid, comprehensive and inexpensive systems analysis. BMC Syst. Biol. 2010;4:64. doi: 10.1186/1752-0509-4-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid AK, Reiss DJ, Kaur A, Pan M, King N, Van PT, Hohmann L, Martin DB, Baliga NS. The anatomy of microbial cell state transitions in response to oxygen. Genome Res. 2007;17:1399–1413. doi: 10.1101/gr.6728007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaur A, Pan M, Meislin M, Facciotti MT, El-Gewely R, Baliga NS. A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 2006;16:841–54. doi: 10.1101/gr.5189606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 24.Lassmann T, Hayashizaki Y, Daub CO. TagDust—a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–40. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–79. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peterson P. F2PY: a tool for connecting Fortran and Python programs. Int. J. Comput. Sci. Eng. 2009;4:296–305. [Google Scholar]
- 28.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–42. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baliga NS, Kennedy SP, Ng WV, Hood L, DasSarma S. Genomic and genetic dissection of an archaeal regulon. Proc. Natl Acad. Sci USA. 2001;98:2521–25. doi: 10.1073/pnas.051632498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peck RF, DasSarma S, Krebs MP. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 2000;35:667–76. doi: 10.1046/j.1365-2958.2000.01739.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Guo C, Chen Y, Shulha HP, Schnetz MP, LaFramboise T, Bartels CF, Markowitz S, Weng Z, Scacheri PC, et al. Epitope tagging of endogenous proteins for genome-wide ChIP-chip studies. Nat. Methods. 2008;5:163–65. doi: 10.1038/nmeth1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bell SD, Jackson SP. Transcription in archaea. Cold Spring Harb. Symp. Quant. Biol. 1998;63:41–51. doi: 10.1101/sqb.1998.63.41. [DOI] [PubMed] [Google Scholar]
- 33.Schmid AK, Pan M, Sharma K, Baliga NS. Two transcription factors are necessary for iron homeostasis in a salt-dwelling archaeon. Nucleic Acids Res. 2011;39:2519–33. doi: 10.1093/nar/gkq1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoon SH, Reiss DJ, Bare JC, Tenenbaum D, Pan M, Slagel J, Moritz RL, Lim S, Hackett M, Menon AL, et al. Parallel evolution of transcriptome architecture during genome reorganization. Genome Res. 2011;21:1892–1904. doi: 10.1101/gr.122218.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gelfand MS, Koonin EV, Mironov AA. Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res. 2000;28:695–705. doi: 10.1093/nar/28.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geiduschek EP, Ouhammouch M. Archaeal transcription and its regulators. Mol. Microbiol. 2005;56:1397–1407. doi: 10.1111/j.1365-2958.2005.04627.x. [DOI] [PubMed] [Google Scholar]
- 37.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA-binding proteins. Science. 2000;290:2306–9. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 38.Schmid AK, Reiss DJ, Pan M, Koide T, Baliga NS. A single transcription factor regulates evolutionarily diverse but functionally linked metabolic pathways in response to nutrient availability. Mol. Syst. Biol. 2009;5:282. doi: 10.1038/msb.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gropp F, Betlach MC. The bat gene of Halobacterium halobium encodes a trans-acting oxygen inducibility factor. Proc Natl Acad Sci USA. 1994;91:5475–5479. doi: 10.1073/pnas.91.12.5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leong D, Pfeifer F, Boyer H, Betlach M. Characterization of a second gene involved in bacterio-opsin gene expression in a halophilic archaebacterium. J. Bacteriol. 1988;170:4903–9. doi: 10.1128/jb.170.10.4903-4909.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baliga NS, Pan M, Goo YA, Yi EC, Goodlett DR, Dimitrov K, Shannon P, Aebersold R, Ng WV, Hood L. Coordinate regulation of energy transduction modules in Halobacterium sp. analyzed by a global systems approach. Proc. Natl Acad. Sci. USA. 2002;99:14913–18. doi: 10.1073/pnas.192558999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarasov VY, Besir H, Schwaiger R, Klee K, Furtwangler K, Pfeiffer F, Oesterhelt D. A small protein from the bop-brp intergenic region of Halobacterium salinarum contains a zinc finger motif and regulates bop and crtB1 transcription. Mol. Microbiol. 2008;67:772–80. doi: 10.1111/j.1365-2958.2007.06081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tarasov V, Schwaiger R, Furtwangler K, Dyall-Smith M, Oesterhelt D. A small basic protein from the brz-brb operon is involved in regulation of bop transcription in Halobacterium salinarum. BMC Mol. Biol. 2011;12:42. doi: 10.1186/1471-2199-12-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang CF, Kim JM, Molinari E, DasSarma S. Genetic and topological analyses of the bop promoter of Halobacterium halobium: stimulation by DNA supercoiling and non-B-DNA structure. J. Bacteriol. 1996;178:840–45. doi: 10.1128/jb.178.3.840-845.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.