

Abstract
The efficacy of immunotherapeutic TLR7/8 activation by resiquimod (R848) was evaluated in vivo, in the CNS-1 rat glioma model syngeneic to Lewis rats. The immune treatment was compared with cytotoxic cyclophosphamide chemotherapy, and as well, was compared with the combination cytotoxic and immunotherapeutic treatments. We found that parenteral treatment with the TLR7/8 agonist, resiquimod, eventually induced complete tumor regression of CNS-1 glioblastoma tumors in Lewis rats. Cyclophosphamide (CY) treatment also resulted in dramatic CNS-1 remission, while the combined treatment showed similar antitumor effects. The resiquimod efficacy appeared not to be associated with direct injury to CNS-1 growth, while CY proved to exert tumoricidal cytotoxicity to the tumor cells. Rats that were cured by treatment with the innate immune response modifier resiquimod proved to be fully immune to secondary CNS-1 tumor rechallenge. They all remained tumor-free and survived. In contrast, rats that controlled CNS-1 tumor growth as a result of CY treatment did not develop immune memory, as demonstrated by their failure to reject a secondary CNS-1 tumor challenge; they showed a concomittant outgrowth of the primary tumor upon secondary tumor exposure. Rechallenge of rats that initially contained tumor growth by combination chemo-immunotherapy also failed to reject secondary tumor challenge, indicating that the cytotoxic effect of the CY likely extended to the endogenous memory immune cells as well as to the tumor. These data demonstrate strong therapeutic antitumor efficacy for the immune response modifier resiquimod leading to immunological memory, and suggest that CY treatment, although effective as chemotherapeutic agent, may be deleterious to maintenance of long-term antitumor immune memory. These data also highlight the importance of the sequence in which a multi-modal therapy is administered.
Keywords: TLR agonist, brain tumor, glioma, immunotherapy
Introduction
Glioblastoma multiforme (GBM) is the most common malignant brain tumor in adult patients. Due to its highly infiltrative nature GBM is notoriously difficult to treat and complete surgical resection is difficult. GBM tumors are inevitably recurrent either locally, close to the original tumor, or at distant sites. Moreover, chemotherapy and/or radiotherapy have shown only limited success. As a result the overall prognosis for this tumor has changed little over the last two decades. Today the average survival time for a newly diagnosed patient is between 12 and 15 mo and new forms of therapy are desperately needed to change the clinical course of this highly malignant tumor. GBM therefore requires additional forms of therapy to prolong the lifespan and quality of life of patients. Immunotherapy is now emerging as a novel fourth option for clinicians.
Immunotherapy stimulates and teaches the patient’s immune system to recognize and eradicate malignant tumor cells. If successful it has the added advantage of generating a memory response to prevent tumor reoccurrences after cessation of treatment. Our immune system has evolved to protect our body by eliminating pathogens and abnormal cells with minimal damage to healthy tissues. It is complex and has multiple levels of regulation to guarantee the appropriate balance between immune activation and immune suppression. Over the last two decades the fundamentals of this regulation have become more clear.
It has been amply shown that the immune system can prevent the emergence and growth of cancer. For example, an adaptive immune response influences the behavior of human colon cancer tumors as evidenced by in situ analysis of tumor-infiltrating immune cells.1,2 Also, in ovarian cancer the presence of intraepithelial tumor infiltrating lymphocytes is associated with prolonged clinical remission and improved survival.3 Similar observations have been made for other tumors, such as renal carcinoma,4 prostate cancer5 and breast carcinoma.6 These observations in patients are further supported by the observations in experimental systems that an impaired immune system is less able to protect the host against the development of spontaneous and chemically-induced tumors.7-9 In addition, individuals with cancer sometimes develop spontaneous reactivity against the antigens of the tumor.10 Hence, it can be concluded that tumors can be recognized and eliminated as a result of natural tumor-specific immune responses that develop in the host.
The observations that the capacity to mount natural immune responses is linked to improved survival supports the concept to develop immune activating tumor vaccines, stand alone immune activators, or biological response modifiers. Presently, such strategies are tested in clinical trials for the treatment of different types of solid tumors, including glioblastoma, as recently reviewed by Hofman and coworkers.11 To date, these approaches have provided only modest clinical results. Nevertheless, they have shown promise by successfully generating antigen specific effector T cells capable of reacting with the tumor, and significant survival advantage or improved quality of life in subgroups. Remarkably, effector immune cells may fail to produce tumor regression because newly triggered and successfully expanded tumor-specific lymphocytes are actively inhibited within the draining lymph nodes or upon entrance into the tumor. In recent years it has become well-established that many tumors, including GBM, use various mechanisms of immune suppression or evasion, including immunediting, and the generation of T regulatory (Treg) and myeloid derived suppressor cells (MDSC).12
These cells act to inhibit the beneficial effects of immune activation13 by direct cell contact mechanisms or by secretion of inhibitory molecules, such as IL-10, and TGFβ.14,15 As a result, the suppression of immunity in tumors may present a major challenge to clinicians interested in using tumor vaccines or other methods of immune activation to treat tumors at the time of diagnosis. Immunotherapy may be further complicated in situations where the immune system promotes tumor development by selecting for tumor escape variants with reduced immunogenicity.16
Hence, successful immunotherapy for the treatment of solid tumors may require two entirely different steps: (1) the use of potent immune activators such as single immunostimulants or tumor vaccines comprising suitable adjuvants; and (2) reagents that can reverse immune suppression induced by the tumor. In the last two decades there has been a strong interest in using toll-like receptor (TLR) agonists as immunostimulants and adjuvants for therapeutic vaccines because of their stimulatory effects on innate immune responses which precedes the shaping of adaptive immune effector and memory cells.17,18 TLRs are so-called pattern recognition receptors that are found on a variey of innate immune cells17 and able to recognize pathogen-specific molecular patterns (PAMPS). They discriminate these PAMPS from invading pathogens as non-self molecules, which represent a signal for the receptor-expressing immune cells to become activated and produce pro-inflammatory cytokines and costimulatory molecules resulting in recruitment and activation of immune cells.
The imidazoquinoline-based small molecules imiquimod and resiquimod are synthetic ligands that have been shown to activate human TLR 7 and 8, and TLR7 in mice and rats. TLR8 is not functional in mice. These TLRs also recognize viral and synthetic single-stranded RNAs. The TLR7/8 agonist R837 (imiquimod) has been licensed as a key ingredient in Aldara cream for the topical treatment of genital warts, basal cell carcinoma and bladder cancer.19-21 In mouse studies, imidazoquinolines were able to act as adjuvants promoting adaptive immune response to co-administered prophylactic antigens.22,23 This observation is in line with the notion that single-stranded RNA induces an antigen-specific immunity characterized by a potent cytotoxic T-cell response.24
However, very little is known about how systemically administered TLR7/8 agonists affect immune responses in general and anti-tumor immunity to glial brain tumors. Interestingly, Xiong and Ohlfest recently showed that topical imiquimod (Aldara) applied on the skin has therapeutic and immunomodulatory effects against intracranial tumors in a mouse model.25 Weekly application increased survival of mice against implanted syngeneic GL261 glioma tumors. Resiquimod is related to imiquimod, as they are both synthetic small molecules that activate Toll-like receptor (TLR) 7.26
In the present study, we investigated the anti-tumor immune effects of parenterally injected resiquimod (R848) in a CNS-1 glioma model in immunocompetent Lewis rats. This model represents a valuable in vivo system for preclinical studies because of histopathological and pathological features which highly resemble human GBM.27 Our results show that TLR7/8 agonist resiquimod (R848) affects immune responses leading to growth arrest of large established glioma tumors, and that R848 treatment, at the concentrations used, does not inhibit CNS-1 tumor growth directly. Remarkably TLR7/8 activation by R848, as a therapeutic stand-alone therapy, is able to reject smaller established CNS-1 tumors, leading to solid, immunological memory against tumor rechallenge. Hence, this TLR7/8 activation approach provides a new opportunity for rational therapeutic immune interventions based on strengthened anti-tumor immune responses that may translate into successful clinical outcome in patients affected by glioblastoma.
Results
Therapeutic adminstration of the TLR7 immunostimulant resiquimod arrests growth of large (35-day old) CNS-1 tumors
We sought to determine immunotherapeutic strategies for controlling the malignant growth of syngeneic CNS-1 glioma tumor cells in Lewis rats using the newly described small molecule TLR7/8 agonist resiquimod, also referred to as R848.
To examine whether immunocompetent Lewis rats, which had developed large syngeneic CNS-1 tumors, would benefit from R848 treatment (100 µg/kg, 30 µg/dose), we started to treat established, large five week-old, 10- 20,000 mm3, CNS-1 tumors, at 38 days after tumor implantation. Indeed, resiquimod treatment was able to arrest or slow tumor growth in 2/3 animals, as shown in Figure 1, however, complete regression of these large tumors was not noted within the observation period.
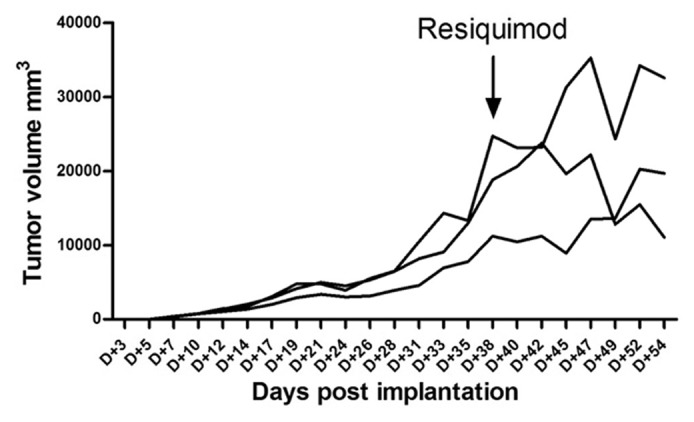
Figure 1. Indication of inhibition of established large CNS-1 tumors in Lewis rats (n = 3) treated with resiquimod starting at day 38. Individual growth as a function of time after implanation is shown for each individual animal. The arrow indicates the start of resiquimod treament.
We next decided to investigate the antitumor activity of resiquimod against lower tumor burden in rats with less advanced CNS-1 tumors. In a subsequent pilot dose-finding experiment we noticed a dose-dependent inhibition of CNS-1 tumor growth, when treatment was started earlier at day 10 after tumor implantation. Injection of a low dose of immunostimulatory resiquimod (3.3 µg/kg = 1 µg/dose) did not inhibit CNS-1 tumor growth, while a higher dose of either 10 or 50 μg/dose, representing 33.3 and 166.6 μg/kg respectively, clearly evoked reduction in tumor growth (data not shown). We therefore decided to test robustness of this protective treatment in a larger experiment. Figure 2A shows that administration of R848, at a dose of 100 μg/kg (this is about 30 μg/ dose), when given three times per week (Monday, Wednesday and Friday) profoundly reduces tumor growth, relative to a control group receiving no treatment (p < 0.001). We stopped the therapeutic weekly treatment regimen after day 42 (week 6) and further continued to monitor tumor growth.
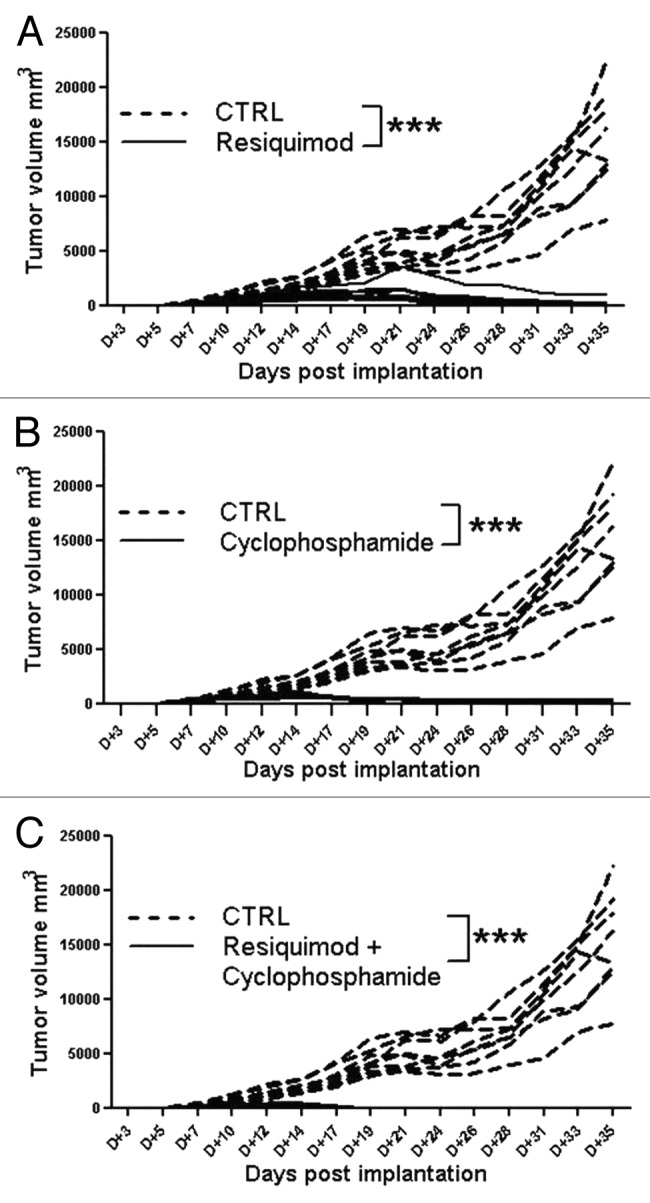
Figure 2. Therapeutic administration of TLR7/8 agonist resiquimod strongly inhibits tumor growth. CNS-1 tumor development after implantation in groups of rats (n = 8) treated with either resiquimod (A), Cyclophosphamide (B) or a combination of resiquimod and cyclophosphamide (C). Individual growth as a function of time is shown. The stippled lines show individual tumor growth of untreated controls.
Therapeutic administration of high dose cyclophosphamide alone or in combination chemo-immunotherapy of CY with resiquimod protects against CNS-1 tumor growth
We next examined whether resiquimod immunotherapy combined with chemotherapy further improves anti-tumor immunity. Hence, in a parallel arm of the same animal experiment we investigated the therapeutic efficacy of the cytotoxic alkylating agent cyclophosphamide (CY), given once every two weeks, which is a well-know direct cytostatic antitumor agent, but has also been shown to mediate immune suppression or even tumor regression by abrogation of immunosuppressive T regulatory cell function.28 In addition, we tested the combined treatment of CY plus R848 to investigate beneficial synergy of CY with active immunotherapy, as a chemo-immunotherapy variant.29,30Figure 2 shows that inhibition of CNS-1 glioma tumor growth was observed for a group which received CY alone (Fig. 2B) or resiquimod in conjunction with CY at a 100 mg/kg dose (Fig. 2C; p < 0.001). The combined administration of CY and R848 seemed to further inhibit tumor development as noted by earlier tumor regression. Poly I:C injected in a similar regimen, at a dose of 30 or 50 µg, did not inhibit CNS-1 growth (data not shown). A lower dose of 30 mg/kg of CY alone also failed to inhibit tumor growth.
TLR7 agonist does not directly inhibit CNS-1 tumor growth
To examine whether parenterally injected resiquimod may have been able to directly affect CNS-1 tumor growth we tested the cytotoxicity of resiquimod in vitro, in parallel to cyclophosphamide and poly I:C, a prototype TLR3 agonist. Figure 3 shows a direct cytotoxicicty of cyclophosphamide when used at high dose, and no apparent direct growth inhibition by resiquimod, at concentrations reflecting the in vivo dose. The effective concentration of resiquimod of 30 μg/dose used in vivo is not directly cytotoxic in vitro and is therefore very unlikely to evoke direct contralateral tumor killing, while the in vivo concentration of 100 mg/kg (30 mg/dose) is clearly cytotoxic for CNS-1 cells cultured in vitro (200,000 cells /well).

Figure 3. CNS-1 cells (200,000 cells per well) were exposed for 24 hours to increasing concentrations per well of either resiquimod (A), or CY (B). Viability was measured (in triplicate) in a standard (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) bromide assay, and was expressed as a percentage of viability measured for cells cultured in medium only (control). Representative data of two experiments are shown.
CNS-1 glioma cells do not express TLR 7/8
We checked expression of TLR7 and TLR8 by CNS-1 cells using RT-PCR, but where unable to detect receptor expression, while expression of both receptors could be detected in rat spleen tissue (Fig. 4).
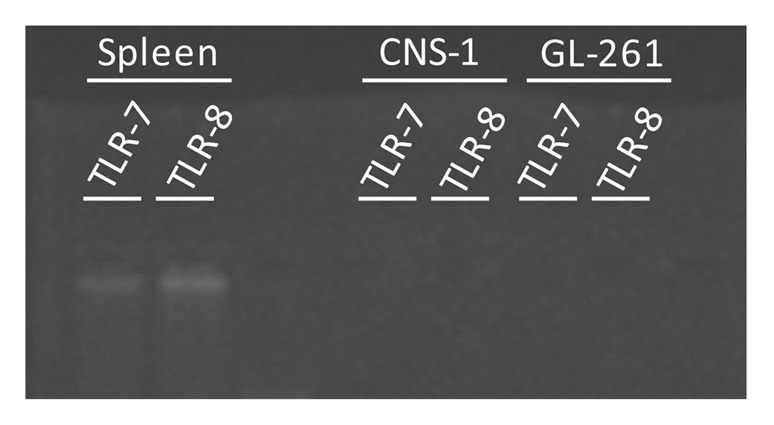
Figure 4. Lack of detection of TLR 7 and TLR 8 expression in both rat glioma CNS-1 cells and murine glioma GL261 cells. However, in rat spleen tissue, used as positive control, expression of both TLR7 and TLR8 can be detected.
Glioma tumors cured by resiquimod therapy alone evoke immunological memory when rechallenged with syngeneic CNS-1 tumor
To examine whether immunocompetent Lewis rats, which had rejected CNS-1 cells as a result of resiquimod treatment, had developed immunological memory against CNS-1 tumor cells, they were re-challenged at day 49 after the first tumor implantation (one week after treatment arrest and wash out of R848) using a tumor dose of CNS-1 which evoked tumor growth in naive age-matched control rats.
As shown in Figure 5 all recipients of R848 therapy which had eliminated the first CNS-1 tumors, completely rejected the secondary CNS-1 challenge, with no evidence of measurable secondary tumor growth (Fig. 5B, p < 0.01), while the same dose of CNS-1 tumor cells induced reproducible tumor development in all untreated age-matched Lewis rats (Fig. 5A). This observation was confirmed in a subsequent experiment as evidenced by a complete rejection of a secondary tumor challenge, after cessation of treatment for a period of three months, in a group of four R848 treated Lewis rats, which had rejected the first CNS-1 tumor. Again this additional tumor challenge evoked progressive tumor growth in naive age-matched controls.

Figure 5. Individual CNS-1 tumor development as a function of time after secondary tumor implantation (A–D) in groups of rats (n = 8) treated either with Resiquimod alone (B), Cyclophosphamide alone (C) or a combination of Resiquimod and cyclophosphamide (D). The CNS-1 implanted, untreated age-matched group is shown in (A) (n = 4). Mean tumor growth of (A) is shown as a stippled line for the untreated control group in (B–D). (E–G) show the growth of the recurrent individual primary CNS-1 tumors at the original inoculation site, for the resiquimod treated (E), Cyclophosphamide treated (F) and combination group (G) at the days post primary tumor challenge.
Glioma tumors controlled by treatment with CY alone or by CY-resiquimod combination therapy fail to evoke immunological memory against rechallenge with syngeneic CNS-1 tumor
Lewis rats that had initially rejected CNS-1 cells as a result of CY treatment alone, or after CY-R848 combination treatment, were tested for immunological memory against CNS-1 tumor cells, by a re-challenge at day 49 after the first tumor implantation. Upon secondary tumor challenge both, the CY only treated, as well as the CY-R848 combination treated hosts, displayed an initial inhibition of secondary tumor development (Fig. 5C and D, p < 0.01 and < 0.05, respectively), but eventually progressive CNS-1 growth was noted in most animals. This indicated a deleterious effect of the CY on the endogenous immune cell component that was initially engendered, sensitized to and keeping the CNS-1 tumor cell growth suppressed. Further substantiating this conclusion, the CY-treated rats, alone as well as in combination therapy, also showed a recurrence of their primary tumor, which apparently was not completely resolved (Fig. 5F and G, respectively). In both groups, seven out of eight animals showed progressive tumor growth.
Safety
In our studies we did not observe any signs of toxicity in rats treated with the systemic resiquimod.
Discussion
In the present study we demonstrate that immunotherapy based on the innate immune cell activator resiquimod, is effective as a treatment modality for eradication of established CNS-1 glioma tumors.
Our CNS-1 glioma cell implants are syngeneic, haplotype RT-1l, for Lewis rats and represent an excellent in vivo glioma model, because of its glial phenotype, reproducible in vivo growth rates and histological features that closely resemble human glioma.31 It has been demonstrated that CNS-1 tumor cells are immunoreactive for glial fibrillary acidic protein (GFAP), S100 and vimentin, as well as neuronal adhesion molecule, retinoic acid receptor α, intracellular adhesion molecule and neuron specific enolase.31 This model therefore provides an excellent in vivo model in which to investigate immunotherapeutic intervention strategies against glioblastoma multiforme in immunocompetent hosts.
Natural immune responses against glioma tumors are often elicited as demonstrated by histological evidence of local inflammation and tumor-specific lymphocytes, likely directed against tumor specific antigens. However, the GBM tumor microenvironment is characterized by the presence of a variety of immunosuppressive cells and their inhibitory products, which may eventually result in the escape of the tumor from immune surveillance.32-35 However, when an effective therapeutic dose of resiquimod was injected three times per week, we observed a dramatic reduction in tumor volume. While most untreated or control tumor-bearing animals had to be sacrificied, either due to massive tumor volumes or due to ulceration of the tumor, the groups receiving a dose of more than 10 µg resiquimod per injection eventually showed complete regression of the tumor volumes. When therapeutic treatment was arrested, at day 49 after implantation, the tumor had shrunk to minute or nonmeasurable sizes. In vitro studies revealed that resiquimod (0.01 or 0.1 mg/ml), in contrast to CY, did not directly inhibit CNS-1 tumor cell growth.
These results may seem contradictory with other data showing that when tumor cells express TLR7/8, activation of this TLR type leads to cell survival and chemo resistance.36 We have therefore checked expression of TLR7/8 by CNS-1 cells by RT-PCR, but where unable to detect receptor expression by PCR. However, even if TLR7/8 activation by resiquimod would have stimulated tumor growth the net effect in vivo would apparently still be tumor regression.
Interestingly, all rats proved immune to re-challenge with CNS-1 glioma cells (Fig. 5) as evidenced by complete inhibition of tumor development. Immune memory against rechallenge was confirmed for rats which received the additional tumor inoculation even after three months of treatment arrest, while naive rats developed tumors. In view of the short half-life of the imiquimod family members of only few hours it is very unlikely that resiquimod had some remnant activity after a three month resting period before administration of a tumor rechallenge. The complete inhibition of secondary tumor growth suggest that immunotherapeutic treatment during the first tumor growth, using resiquimod, a known innate immune response agonist activating TLR7/8, results in tumor regression that results in the development of T cells with immune memory. Hence this innate immune triggering acts as an in situ therapeutic vaccine, alerting the adaptive immune system to recognize and eliminate the syngeneic secondary CNS-1 brain tumor. In future studies we will set out to decipher the exact mechanism underlying this intriguing observation of in situ immune memory priming.
In addition, we evaluated the effects of CY on CNS-1 tumor development. Cyclophosphamide (CY), although primarily used as cytotoxic therapy and expected to suppress the immune system, has been shown to abrogate immunosuppressive T reg function, and beneficially synergize with active immunotherapy when used at an appropriate dose and timed correctly.29,37,38
CNS-1 tumors regressed, as a result of CY treatment, and even faster after combined CY-R848 chemo-immunotherapeutic treatment. Importantly, the administration of CY, after the animals had developed immunity to CNS-1, was deleterious (Fig. 4). The explanation for why, after tumor rechallenge, the animals that were treated with CY only or by the CY-R848 combination were not able to inhibit secondary tumor development relates to the CY also causing damage to the CTL that had developed in situ at the beginning of the treatment. Additionally, it provides an explanation for why the CY-treated rats also exhibited recurrence of the primary tumor. These data highlight the need to carefully arrange the administration of combined therapeutics involving cytotoxic chemotherapeutic agents with immunotherapeutic agents so one agent does not interfere with the effects derived from the other. However, the delay in tumor growth after rechallenge of the cyclophosphamide group, suggests that there is an immune effect, which is most likely dependent on T cells, although a memory response by B cells cannot be excluded formally. In both scenarios T cells are necessary for T help and likely also for T-cell effector function. The effect of T cell depletion will be subject of follow-up studies addressing the biological mechanism of action responsible for rechallenge immunity.
These results provoke two intriguing questions. How does R848 eradicate CNS-1 tumors, and how does immune memory develop during this treatment? In addition, it is of interest to know how CY hampers antitumor immunity. The exact mode of action and associated immune pathway responsible for the observed resiquimod-mediated anti-tumor immunity needs to be defined in detailed follow-up studies. Most likely resiquimod-based immunotherapy is able to activate a spontaneous, natural, innate anti-tumor immune response, that under normal circumstances is unable to control tumor growth, likely as a result of delayed or actively suppressed immune control. Non-specific immune attack of the tumor evoked by TLR7/8 activating resiquimod, but not by poly I:C treatment activating TLR3 (data not shown), may release tumor antigens into the surrounding tumor environment which are sampled by locally attracted antigen presenting cells and which allow presentation to and priming of adaptive immune lymphocytes, in the draining lymph nodes. Alternatively, or in parallel, an in situ “vaccination” occurs as a result of R848 therapy. TLR7 activation by the related imiquimod causes human and rodent dendritic cells to become tumoricidal.39 Eventually, a sufficient number of tumor-specific naive adaptive immune cells, such as cytolytic T cells, are triggered and expanded in draining lymph nodes as a result of parenteral R848 immunotherapy and enabled by activated antigen-presenting cells. These presumed cytolytic T cells selectively recognize and eliminate the tumor and provide immunological memory, as illustrated by the rejection of secondary tumor cell implants. However, dedicated follow-up studies need to address to involvement of anti-tumor killer macrophages or NK cells, or IFNs for the resiquimod-induced glioma growth regression and immune memory.
In conclusion, our data show that injection of the innate immune cell receptor agonist resiquimod as a therapeutic TLR7/8 activating stand-alone therapy, is able to cure established CNS-1 tumor growth in Lewis rats. They suggest that immunotherapeutic parenteral treatment of established glioma tumors by resiquimod, as defined in the protocol, significantly improves anti-brain tumor immunity in a way that leads to immune memory, which is superior to CY treatment alone. Our studies have thereby identified a promising novel antitumor immunotherapy which may lead to clinical benefit.
Materials and Methods
Tumor model
Rat CNS-1 cells (2 × 105 cells/200 ul) were implanted subcutaneously (SC) using a 21 gauge needle into the right flank of 8–12 week-old (300 g body weight) male Lewis rats. For each treatment group and control, 4–8 rats/group were used. The same tumor implantation procedure was performed during re-challenge experiments, on the contralateral side, for rats which had controlled the tumor growth after first exposure. All animal studies were approved by an independent ethical committee.
Monitoring tumor growth
The sizes of the CNS-1 tumor volumes were measured using a caliper three times per week on Mondays, Wednesdays and Fridays to monitor the effects of each treatment group.
Completion of experiment
Tumor implanted rats were sacrificed if they showed unfavorable signs of discomfort, as defined by the ethical committee. For example if they appeared moribund due to weight loss, lethargy, ruffled fur, or when tumors showed ulceration. A mixture of Rompun and ketamine was used for anesthesia, followed by a dose of sodium pentobarbital for euthansia.
Chemicals and reagents
Immunomodulators and potentiators
Rats were subcutaneously (SC) injected in the flank, contralateral to the tumor-implanted side, with resiquimod (R848) (purchased from Invivogen, catalog number tlrl-r848), a Toll-like receptor 7/8 agonist, in a range of 3.3–166.6 µg/kg, corresponding to 1–50 µg/dose, three times per week on Mondays, Wednesdays and Fridays. Resiquimod(R-848, S-28463) was shown to be more soluble and more potent in inducing cytokine expression than its family member imiquimod which has a half-life of 2–3 h in humans40.
In a parallel arm of the experiment we evaluated the effect of cyclophosphamide administration on CNS-1 glioma development. Cyclophosphamide (CalBiochem, 239785) was given at 30–100 mg/kg. CY was injected once every two weeks on Fridays.
Cytotoxicity assay
The direct cytotoxicity of resiquimod, CY and Poly I:C (Invivogen, Tlrl-pic, tlrl-pic-5), which was included as a reference TLR-3 agonist, was determined by exposing CNS-1 cells at a concentration of 200,000 cells per well in a 96-well plate in DMEM culture medium (30-2002, ATCC), supplemented with 10% fetal bovine serum (FBS; Lonza, DE14-801E), for 24 h. The viability of CNS-1 cells, measured in triplicate, was measured in a standard (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) bromide assay, absorbance was read at 590 nm, and was expressed as a percentage of viability measured for cells cultured in medium only.
TLR 7 and TLR 8 detection by RT-PCR
Samples collection
Normal spleen tissue was obtained by surgical resection from a male non-treated Lewis rat and cut in pieces of 1 mm3 with a sterile surgical blade. CNS-1 and GL-261 cell lines were cultured as described above and a pellet of 1 × 106 cells was used. Cells or tissue sample were put in lysis buffer using the SV Total RNA Isolation System (Promega Corp., Leiden, The Netherlands).
RNA extraction and reverse transcription
After extraction of total RNA it was reverse-transcribed by using the Thermoscript RT-PCR System (Life Technologies, Inc.) as previously described.41
Oligonucleotide primers used for PCR amplification: Primers for the PCR amplification were obtained by Real Time Primers LLC, according to successful approach for TLR-7,42 or as customized primers for TLR-8 obtained from Real Time Primers LLC.
PCR
PCR was performed according to the manufacturer’s recommendations, with Platinum® PCR SuperMix (Life Technologies, Inc.). Aliquots of the RT products were subjected to PCR in a total volume of 50 µl, with 100 nM adequate paired primers. PCR products were visualized on a 2% agarose gel with GelRed™ Nucleic Acid Gel Prestaining Kit (Biotium), visualized on an UV transilluminator and photographed using a Canon Powershot G10 photograph, equipped with a conversion lens 032 LA-DC58K.
Statistical analysis
ANOVA followed by the students t-test was used to compare groups, *p value of < 0.05, **p < 0.01, ***p < 0.001, considered statistically significant.
Disclosure of Potential Conflicts of Interest
All authors affiliated to Epitopoietic Research Corporation (ERC) are financially supported by ERC.
Acknowledgments
Supported in part by NIH R01 CA154256 to C.A.K.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/19068
References
- 1.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 2.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–4. [PubMed] [Google Scholar]
- 3.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 4.Nakano O, Sato M, Naito Y, Suzuki K, Orikasa S, Aizawa M, et al. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–6. [PubMed] [Google Scholar]
- 5.Vesalainen S, Lipponen P, Talja M, Syrjanen K. Histological grade, perineural infiltration, tumour-infiltrating lymphocytes and apoptosis as determinants of long-term prognosis in prostatic adenocarcinoma. Eur J Cancer. 1994;30:1797–803. doi: 10.1016/0959-8049(94)E0159-2. [DOI] [PubMed] [Google Scholar]
- 6.Marrogi AJ, Munshi A, Merogi AJ, Ohadike Y, El-Habashi A, Marrogi OL. Freeman, S. M. Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma. Int J Cancer. 1997;74:492–501. doi: 10.1002/(SICI)1097-0215(19971021)74:5<492::AID-IJC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 7.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 8.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–48. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998;95:7556–61. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckhove P, Feuerer M, Dolenc M, Schuetz F, Choi C, Sommerfeldt N, et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J Clin Invest. 2004;114:67–76. doi: 10.1172/JCI20278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofman FM, Stathopoulos A, Kruse CA, Chen TC, Schijns VEJC. Immunotherapy of Malignant Gliomas Using Autologous and Allogeneic Tissue Cells. Anticancer Agents Med Chem. 2010;10:462–70. doi: 10.2174/1871520611009060462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 13.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–33. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–80. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 15.Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res. 2007;13:4345–54. doi: 10.1158/1078-0432.CCR-07-0472. [DOI] [PubMed] [Google Scholar]
- 16.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 17.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 18.Schijns VEJC, Lavelle EC. Trends in vaccine adjuvants. Expert Rev Vaccines. 2011;10:539–50. doi: 10.1586/erv.11.21. [DOI] [PubMed] [Google Scholar]
- 19.Gollnick H, Barona CG, Frank RG, Ruzicka T, Megahed M, Tebbs V, et al. Recurrence rate of superficial basal cell carcinoma following successful treatment with imiquimod 5% cream: interim 2-year results from an ongoing 5-year follow-up study in Europe. Eur J Dermatol. 2005;15:374–81. [PubMed] [Google Scholar]
- 20.Gaitanis G, Kalogeropoulos C, Bassukas ID. Imiquimod can be combined with cryosurgery (immunocryosurgery) for locally advanced perocular basal cell carcinomas. Br J Ophthalmol. 2011;95:890–2. doi: 10.1136/bjo.2010.195800. [DOI] [PubMed] [Google Scholar]
- 21.Gaspari A, Tyring SK, Rosen T. Beyond a decade of 5% imiquimod topical therapy. J Drugs Dermatol. 2009;8:467–74. [PubMed] [Google Scholar]
- 22.Vasilakos JP, Smith RM, Gibson SJ, Lindh JM, Pederson LK, Reiter MJ, et al. Adjuvant activities of immune response modifier R-848: comparison with CpG ODN. Cell Immunol. 2000;204:64–74. doi: 10.1006/cimm.2000.1689. [DOI] [PubMed] [Google Scholar]
- 23.Risini D, Weeratna RD, Shawn R. Makinen, McCluskie MJ, Davis HL. TLR agonists as vaccine adjuvants: comparison of CpG ODN and Resiquimod (R-848) Vaccine. 2005;23:5263–70. doi: 10.1016/j.vaccine.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 24.Hamm S, Heit A, Koffler M, Huster KM, Akira S, Busch DH, et al. Immunostimulatory RNA is a potent inducer of antigen-specific cytotoxic and humoral immune response in vivo. Int Immunol. 2007;19:297–304. doi: 10.1093/intimm/dxl146. [DOI] [PubMed] [Google Scholar]
- 25.Xiong Z, Ohlfest JR. Topical imiquimod has therapeutic and immunomodulatory effects against intracranial tumors. J Immunother. 2011;34:264–9. doi: 10.1097/CJI.0b013e318209eed4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 27.Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol. 2007;85:133–48. doi: 10.1007/s11060-007-9400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berd D, Mastrangelo MJ. Effect of low dose cyclophosphamide on the immune system of cancer patients: reduction of T-suppressor function without depletion of the CD8+ subset. Cancer Res. 1987;47:3317–21. [PubMed] [Google Scholar]
- 29.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59–73. doi: 10.1038/nri2216. [DOI] [PubMed] [Google Scholar]
- 30.Kempf RA, Mitchell MS. Effects of chemotherapeutic agents on the immune response. Cancer Invest. 1984;2:459–66. doi: 10.3109/07357908409048519. [DOI] [PubMed] [Google Scholar]
- 31.Kruse CA, Molleston MC, Parks EP, Schiltz PM, Kleinschmidt-DeMasters BK, Hickey WF. A rat glioblastoma model, CNS-1, with invasive characteristics similar to those of human gliomas: a comparison to 9L gliosarcoma. J Neurooncol. 1994;22:191–200. doi: 10.1007/BF01052919. [DOI] [PubMed] [Google Scholar]
- 32.Avril T, Vauleon E, Tanguy-Royer S, Mosser J, Quillien V. Mechanisms of immunomodulation in human glioblastoma. Immunotherapy. 2011;3(Suppl):42–4. doi: 10.2217/imt.11.39. [DOI] [PubMed] [Google Scholar]
- 33.Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, et al. Immune resistence orchestrated by the tumor microenvirenment. Immunol Rev. 2006;213:131–45. doi: 10.1111/j.1600-065X.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- 34.Roszman T, Elliott L, Brooks W. Modulation of T-cell function by gliomas. Immunol Today. 1991;12:370–4. doi: 10.1016/0167-5699(91)90068-5. [DOI] [PubMed] [Google Scholar]
- 35.Ge L, Hoa N, Bota DA, Natividad J, Howat A, Jadus MR. Immunotherapy of brain cancers: the past, the present and future directions. Clin Dev Immunol. 2010; 296453; Epub doi: 10.1155/2010/296453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cherfils-Vicini J, Platonova S, Gillard M, Laurans L, Validire P, Caliandro R, et al. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest. 2010;120:1285–97. doi: 10.1172/JCI36551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kruse CA, Mitchell DH, Kleinschmidt-DeMasters BK, Bellgrau D, Eule JM, Parra JR, et al. Systemic Chemotherapy Combined with Local Adoptive Immunotherapy Cures Rats Bearing 9L Gliosarcoma. J Neurooncol. 1993;15:97–112. doi: 10.1007/BF01053931. [DOI] [PubMed] [Google Scholar]
- 38.Gomez GG, Hutchison R, Kruse CA. Chemo-immunotherapy and chemo- adoptive immunotherapy of cancer. Cancer Treat Rev. 2001;27:375–402. doi: 10.1053/ctrv.2001.0222. [DOI] [PubMed] [Google Scholar]
- 39.Stary G, Bangert C, Tauber M, Strohal R, Kopp T, Sting G. Tumoricidal activity of TLR7/8-activated inflammatory dendritic cells. J Exp Med. 2007;204:1441–51. doi: 10.1084/jem.20070021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soria I, Myhre P, Horton V, Ellefson P, McCarville S, Schmitt K, et al. Effect of food on the pharmacokinetics and bioavailability of oral imiquimod relative to a subcutaneous dose. Int J Clin Pharmacol Ther. 2000;38:476–81. doi: 10.5414/cpp38476. [DOI] [PubMed] [Google Scholar]
- 41.Westfall B, Sitaraman K, Solus J, Hughes J, Rashtchian A. High specificity and robustness of AccuPrime™ Taq provides the ideal tool for demanding miniaturized, multiplex, and high-throughput PCR. Focus. 1997;19:46. [Google Scholar]
- 42.Xiong Z, Ohlfest J. Topical Imiquimod has therapeutic and immunomodulaotory effects against intracranialt tumors. J Immunother. 2011;34:264–9. doi: 10.1097/CJI.0b013e318209eed4. [DOI] [PMC free article] [PubMed] [Google Scholar]