

Abstract
The role of the dual specificity protein phosphatase, Cdc25, in activating the cyclin-dependent kinase-cyclin B complex (Cdk1-CycB) by overcoming the inhibitory Wee1 kinase is a long-established principle for mitotic entry. Recently, however, evidence has emerged of a regulatory network that facilitates Cdk1-CycB activity by inhibiting the form of protein phosphatase 2A having a B55 regulatory subunit (PP2A-B55). Here, I review the genetic and biochemical evidence for Greatwall kinase and its substrate Endosulphine as the key components of this previously obscure regulatory network. Not only is the inhibition of PP2A-B55 by phospho-endosulphine required to prevent dephosphorylation of Cdk1-CycB substrates until mitotic exit, but it is also required to promote Cdc25 activity and inhibit Wee1 at mitotic entry. I discuss how these alternating states of preferential PP2A-B55 or Cdk1-CycB activity can have an impact upon the regulation of Polo kinase and its ability to bind different partner proteins as mitosis progresses.
Keywords: mitosis, Greatwall kinase, Endos, PP2A
2. A short history of the protein kinases regulating mitosis
In this paper, I would like to review some recent studies of the regulation of protein dephosphorylation as a counter to the activity of the Cdk1 mitotic kinase. I will try and place some of these recent findings into a historical context in order to view the broader roles of these proteins in mitotic regulation. This recent work, like many of the pioneering studies of the cell cycle, has relied upon embryos that undertake rapid cleavage divisions. Such embryos, from insects, echinoderms, molluscs and amphibians, have been good friends of the cell cycle research community because they all come fully loaded with a maternal dowry of proteins needed for the cleavage division cycles, thus making them amenable for biochemical and, in some cases, genetic studies. Much has been written about such biochemical approaches that have their origins in the work of Yoshio Masui & Clement Markert [1] and James Reynhout & Dennis Smith [2] of maturation promoting factor (MPF), a cytoplasmic entity that could be withdrawn from unfertilized frog eggs and injected into oocytes, causing them to ‘mature’ (i.e. undertake meiosis I and arrest in meiosis II). An entity with similar properties was soon found to be active in the starfish oocyte by Takeo Kishimoto & Haruo Kanatani [3], where the involvement of increased protein kinase activity in meiotic maturation was quickly appreciated by Marcel Doree and co-workers [4]. Together Fred Lohka & Yoshio Masui established a cell-free system from activated frog eggs in which sperm nuclei would undergo decondensation and DNA synthesis, and condense to form mitotic chromosomes [5]. This, together with work from Marc Kirschner's laboratory in the early 1980s, was instrumental in developing Xenopus embryos as a system in which to study the oscillations of MPF activity in the cleavage cycles that are dependent on protein synthesis [6]. Over the intervening decades, we have realized what a fantastic system this is, not only in providing cell-free extracts in which the oscillatory behaviour of MPF activity could be examined in the test tube, but also in providing an in vitro system that is able to recapitulate the complex dynamics of spindle formation and function. However, it was the cleavage embryos of sea-urchins that allowed Tim Hunt and co-workers to first observe the mitotic cyclins, proteins undergoing periodic cycles of destruction and re-synthesis in phase with mitotic progression [7]. Of course, all eventually became crystal clear when Fred Lohka, Marianne Hayes and Jim Maller got to work to purify MPF from Xenopus [8]. MPF proved to be a complex of a mitotic cyclin and a protein kinase, now known as Cdk1, whose genes, CDC28 and cdc2+, had been described in the respective budding and fission yeasts by Lee Hartwell [9] and Paul Nurse [10]. Nurse's work revealed Cdc2 kinase's conserved function in mitotic entry [11] and also identified the gene for its activating protein Cdc25p [12], later discovered to be a dual-specificity protein phosphatase. The protein kinase opposing Cdc25p, which phosphorylates a critical tyrosine residue in Cdc2p's active site to inhibit the kinase, was the product of the wee1+ gene [13]. This regulatory wiring turned out to be shared by all eukaryotic organisms. Collectively, therefore, these findings provided the field with an explosive burst of activity in the late 1980s that established the key facets of mitotic regulation and a broad understanding of how the conserved mitotic kinase, Cdk1, was regulated. As we shall see in this review, we can now add a new dimension to this regulatory process.
Genetic studies of cell cycle regulation in Drosophila also got under way in the 1980s. However, by and large, the fly community followed independent genetic routes that lagged a little behind their yeast colleagues in identifying mitotic regulatory genes. These exploited the fact that two stages of the Drosophila life cycle are particularly dependent on the cell division cycle: the embryo, whose early cycles are driven by maternally supplied proteins; and the late larval/pupal stages, when the imaginal tissues and central nervous system proliferate to form the adult structures. Because much of the earlier stages of larval development involves the growth of tissue whose cells undergo endoreduplication, repeated rounds of S-phase in the absence of mitosis, there is little demand for the mitotic machinery during these stages. Consequently, the maternal provision of many wild-type proteins from a heterozygous mutant mother is sufficient to allow her homozygous mutant offspring to survive to third instar larval or pupal stages. Mitotic defects then begin to accumulate in the proliferating diploid tissues that will form the adult structures. Nevertheless, for some gene products, the transition from maternal to zygotic provision occurs very much earlier—in the embryo at cycle 14 immediately after cellularization of the nuclei of the synctium. Bruce Edgar & Pat O'Farrell [14] showed that one such gene that has to be expressed zygotically at this stage is string, which encodes one of the two Drosophila cdc25 homologues. It was Maurizio Gatti & Bruce Baker [15], however, who classically exploited the transition between maternal and zygotic control of cell division by screening mutants exhibiting late larval/pupal lethality in a search for genes required for cell proliferation at these stages. Roger Karess in my laboratory followed their example by carrying out one of the first P-transposon screens for such mutants that was instrumental in defining several of our favourite genes. However, we also adopted a complementary genetic approach to study the maternal contribution to cell cycle regulation. Encouraged by Janni Nuesslein-Volhard, we first screened Janni's own collection of Drosophila maternal effect lethal mutants generated by EMS mutagenesis. Notably, this led to the characterization of gnu [16], a gene now known to participate in regulating the translation of maternal mRNA for Cyclin B [17]. Strikingly, Janni's collection harboured mutant genes for two interesting protein kinases, which we named polo [18] (figure 1a) and aurora [19]. We chose these names because their phenotypes, defective spindle poles, reminded us of phenomena at the geomagnetic poles of the Earth. Although these protein kinases were in the shade of Cdk1-cycB for many years, we now know that they have key roles in mitotic progression and these have been reviewed elsewhere [20,21]. Polo, which will later become a lead character in this essay, plays multiple roles in co-ordinating mitotic progression. In so doing, it moves about the cell from centrosomes to kinetochores, and finally to the central spindle, to act out its part in a multitude of processes from centrosome maturation to cytokinesis.
Figure 1.

Origins of some of the alleles of polo and greatwall. (a) The orginal polo allele was identified as a mutation that, when homozygous in the mother, led to mitotic abnormalities and death of syncytial Drosophila embryos. Scant was isolated as a mutation that, when trans-heterozygous with polo (one mutant copy of Scant and one mutant copy of polo), caused females to produce embryos that died owing to a specific mitotic defect—loss of centrosomes from one pole (see text and figure 2). Scheme for the identification of mutations that would suppress the + Scant/polo + phenotype. These were polo+ duplications; revertants of Scant to its recessive alleles, Sr; and a second-site suppressor, su. Sr was identified as greatwall, su as endos. We may also refer to Scant as gwlScant.
3. The genetical trail: from Arctic to the Greatwall via the Antarctic
It was to be genetic studies with polo that set our group on a trail to the then-unknown destination of Greatwall kinase and its substrate Endos. This began in the late 1980s, when we had just discovered the polo gene and were still unaware that it encoded a protein kinase. We drew inspiration from the ideas of Minx Fuller that second-site mutations that failed to complement mutations in the male-specific tubulin gene represented genes encoding interacting proteins within protein complexes [22] (box 1). This led Tano Gonzalez and myself to write a grant application in which we proposed to take the polo1 mutation, a hypomorphic allele with reduced activity, together with mutations in several other mitotic genes and screen for non-complementing mutations at other sites. We got the grant and I am sure we somehow put it to good use, but the project was not brought to fruition until Helen White-Cooper took on the project for her PhD studies in the early 1990s [23]. Helen actually recombined five mitotic mutants onto the same chromosome and carried out a screen to isolate non-complementing mutations following EMS mutagenesis [24]. One of these five genes was represented by the polo1 allele, and indeed one of many new mutants discovered failed to complement polo in this test (figure 1b). This turned out to be the first allele of greatwall; it was not a straightforward recessive allele but rather a gene with a dominant phenotype that only became apparent when the levels of functional Polo were reduced, and then only in the embryos derived from mothers of such a genotype. We named the gene Scant (after Scott of the Antarctic) because of its mutant phenoptype, which led us to suspect that the gene would be interesting (see figure 2 and legend): embryos derived from polo1+/+Scant mothers developed mitotic spindles from which centrosomes were lost from one of the poles. When this first happened, most centrosomes popped right back into place later in the cycle, but eventually the outcome was mitotic mayhem and embryonic lethality.
Box 1. Second-site non-complementing mutations.

(a) Non-complementing second-site mutations lead to mutant phenotypes when the two genes under study are each present as one wild-type copy and one mutant copy. The resulting mutant phenotype can be accounted for in a number of ways. In this example, it is imagined that the second-site gene encodes a partner of Polo kinase essential for its function as a heterodimer and that both mutant genes carry recessive loss-of-function mutations. Thus, of the four possible combinations of heterodimeric protein that can be formed between wild-type and mutant proteins, only one will be functional. This 75 per cent reduction of functional heterodimer can lead to a mutant phenotype.
(b) The Scant mutation, identifying the first mutant allele of greatwall, was identified as a second-site non-complementing mutation of polo1, but it is a gain-of-function mutant that we now know to encode a hyperactive (i.e. gain-of-function) form of Greatwall kinase. Thus, Scant exerts a dominant mitotic phenotype in the presence of mutations showing reduced Polo activity. This repressive effect of greatwallScant upon polo is discussed in the text.
Figure 2.
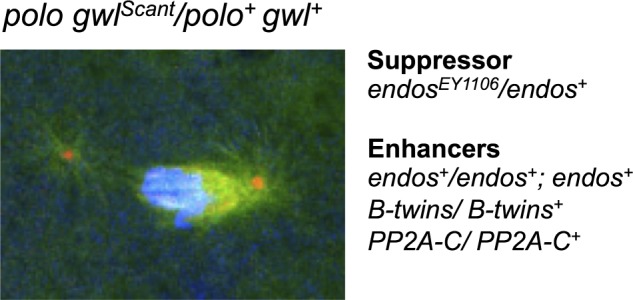
The ‘Scant’ phenotype; its suppressor and enhancers. Typical mitotic figure from embryos derived from a mother with one mutant copy of polo and one mutant copy of gwlScant. We first named the gene Scant after Scott of the Antarctic, the British explorer who set out to find the mysterious southern geomagnetic pole of the Earth in a ship, the Discovery, which is now anchored in full view of the University of Dundee campus. Reducing the wild-type gene dosage of endos in the mother suppresses, whereas increasing the endos gene dosage enhances the phenotype.
Recessive alleles of greatwall were later to be isolated by Mike Goldberg's group, who gave the gene its popular name [25]. These recessive mutations resulted in cell cycle delay at the G2-to-M transition and mitotic chromosomes showing unusual states of condensation. Contemporaneously, we had found Greatwall kinase through an RNAi screen to downregulate every protein kinase in the Drosophila genome in cultured Drosophila cells, in a search for protein kinases regulating cell cycle progression [26]. It was some time before we recognized greatwall as an allele of Scant. This was really the result of Adelaide Carpenter's inspiration to set up a screen to identify Scant revertants (figure 1c). As polo1 +/+ Scant mothers produce no viable offspring, it was relatively straightforward to mutagenize chromosomes carrying Scant, this time by X-irradiation, place the mutagenized chromosome against one carrying polo1 and search for mothers who produced viable progeny. At last, in the mid-2000s, we had isolated several Scant revertants and grouped them into an allelic series of greatwall mutations [27]. An amorphic allele displayed pupal lethality, indicating that, in common with many other cell cycle genes of Drosophila, it was essential for the proliferation of diploid tissues to form the adult. Only once we had these recessive alleles were we able to map the Scant locus using a combination of classical and molecular genetic approaches, and identify it on the genome as encoding the Greatwall kinase. We found that the Scant mutation corresponded to a single amino acid change, K97M, which we could show, when introduced into Greatwall kinase to be expressed in cultured cells, resulted in dramatically increased activity towards artificial substrates.
Depletion of Greatwall from cultured cells led to mitotic delays and a characteristic phenotype of conjoined chromatids scattered upon mitotic spindles that were elongated as if in anaphase B [26]. Of our own recessive gwl alleles isolated as Scant revertants, several showed mutant phenotypes in larval neuroblasts similar to those previously described by the Goldberg laboratory, and one allele, a female-specific germ-line splicing mutant, showed only female sterility [27]. It turned out that flies have two isoforms of Greatwall, one of which is the only form produced in the female germ-line and is absolutely essential for normal progression through female meiosis. The oocytes of females hemizygous for this allele, gwlSr18, fail to arrest in metaphase of meiosis I, and both homologues and sister chromatids separate on elongated, often highly branched meiotic spindles.
Our screen for Scant revertants also produced two other interesting types of mutant (figure 1c). The first of these were duplications of the polo locus that rescued the maternal effect lethality by restoring polo to the wild-type gene dosage. This was in accord with the need for reduced polo function in order to see reduced fertility in the presence of gwlScant. The second was exemplified by a ‘third site’ suppressor of polo1 +/+ Scant that turned out to be a mutation in the gene for the Greatwall kinase substrate Endos [28].
endos encodes a small phospho-protein, α-endosulphine, originally (but probably erroneously) suggested in vertebrates to be the ligand of sulphonylurea receptor K+ channels [29]. Drosophila endos mutants showed female sterility [30], later shown to be due to a failure of oocytes to progress properly to metaphase I and subsequently undertake aberrant meiosis [31]. The meiotic phenotype was said to resemble that of females mutant for the Drosophila germ-line-specific form of Cdc25, twine [32–34], and, interestingly, levels of both Twine and Polo kinase proteins were reported to be reduced. However, the latter observation is curious because genetic experiments predict that Greatwall/Endos should antagonize Polo function (see below). Indeed why specific protein levels should be reduced is not clear because, as we shall see below, the majority of the mutant phenotypes of endos can be accounted for by the ability of its phosphorylated form to inhibit PP2A with a B55 regulatory subunit.
Subsequently, we have found mutations in a number of other cell cycle regulatory genes that either suppress or enhance polo1 gwlScant + (figure 2). Of these, we focused upon genes for subunits of PP2A because of the findings from biochemical studies that this phosphatase was regulated by Greatwall kinase (see below) and because of our earlier findings in flies that the regulatory B subunit of PP2A encoded by twins/abnormal anaphase resolved (aar) was required for anaphase progression, and this form of PP2A preferentially dephosphorylated substrates of Cdk1-cycB [35–37]. Moreover, a separate screen carried out by Vince Archambault and his co-workers also identified mutations in PP2A as enhancers of either a strong hypomorphic polo mutant or of Scant [38]. Our study showed that lowering the dosage of endos suppressed polo1 gwlScant, allowing many embryos to survive [28]. In contrast, lowering the dosage of either the catalytic C subunit or the B55/Twins regulatory subunit of PP2A enhanced the maternal-dominant effect of polo1 gwlScant (figure 2). Increasing the gene dosage of wild-type endos also acted as an enhancer. Thus, Endos and PP2A-twins appeared to be acting antagonistically in this genetic test.
A second set of experiments using RNAi to knockdown gene function in cultured Drosophila cells also suggested that Endos was required to antagonize the function of PP2A-B55Twins [28]. The effects of either endos or greatwall knockdown in cultured Drosophila cells are very similar; mitotic progression into anaphase is delayed and in fixed preparations scattered chromosomes are seen on unusually elongated spindles (figure 3a). The phenotype of the double knockdown was not significantly more severe, suggesting that the two genes work in the same pathway. The endos knockdown phenotypes could be suppressed by co-depleting the catalytic (C), structural (A) or the regulatory B55-subunit of PP2A encoded by twins. There are genes for four different regulatory B subunits of PP2A in the Drosophila genome, and co-depletion of the other three, Widerborst, B′ or B″, had no effect. The phenotype of lagging and bridging anaphase chromosomes in endos RNAi-treated cells could also be suppressed by knocking down PP2A-Btwins (figure 3b). Thus, Endos appeared to counteract PP2A-B55Twins functions; a finding entirely consistent with parallel biochemical studies in Xenopus that I shall now review. These showed that inhibition or depletion of PP2A-B55 from mitotic extracts rescues the inability of Gwl-depleted extracts to enter M phase [39,40] and that Endos becomes a potent inhibitor of PP2A after phosphorylation by Greatwall [41,42].
Figure 3.

Suppression of the endos knockdown phenotype in cultured cells by simultaneous knockdown of PP2A-B55twins. (a) RNAi-mediated depletion of either Greatwall or Endos results in prolonged mitoses in which chromosomes remain scattered on elongated spindles before attempting anaphase. This phenotype is suppressed by the simultaneous depletion of either the catalytic subunit of PP2A (encoded by mts—microtubule star), the structural A subunit (encoded by PP2A 29B) or the B subunit (encoded by twins also known as aar-abnormal anaphase resolved). It is not suppressed by knocking down the three other regulatory B subunits in Drosophila (wdb,widerborst; B′; or B″). (b) Cells depleted of Endos display lagging chromosomes at anaphase. This phenotype is rescued by simultaneous depletion of the PP2A B-subunit, Twins.
4. The biochemical trail: do not fantasize about the poles, stay in the coldroom
After identifying the first recessive greatwall alleles in Drosophila, Mike Goldberg returned to his biochemical roots and switched to studying the Xenopus counterpart of Greatwall kinase [43]. His group showed the kinase was activated in mitosis, probably by Cdk1, and that the depletion of Greatwall from mitotic extracts led to the accumulation of inhibitory phosphorylations on Cdc2 kinase. As Greatwall depletion would also prevent cycling extracts from entering M phase, and this could be rescued by constitutively active Cdk1, they concluded that Greatwall participates in the autoregulatory activation loop for Cdk1. This notion found support from experiments in which they showed that activated Greatwall could induce phosphorylations of Cdc25 in the absence of the activity of kinases of the activation loop or in the presence of an activator of protein kinase A that normally blocks mitotic entry [44]. These effects are all very similar to those of the phosphatase inhibitor okadaic acid, and indeed okadaic acid could drive cycling extracts into M phase in the absence of Greatwall. This led them to the idea that Greatwall negatively regulates a crucial phosphatase that inhibits Cdc25 activation and M phase entry.
Meanwhile, the party was joined by a group in Montpellier, originally assembled by Marcel Dorée and with considerable expertise in biochemical studies on Xenopus egg extracts. This group, now led by Anna Castro and Thierry Lorca, showed that depletion of Greatwall also promoted mitotic exit, even in the presence of a high Cdk1 activity, by inducing dephosphorylation of mitotic substrates [39]. Two findings led to the idea that Greatwall activity was inhibiting PP2A. First, the depletion of PP2A from mitotic extracts rescued the phenotype induced by loss of Greatwall; and second, the PP2A-dependent dephosphorylation of Cdk1-cycB substrates was increased in Greatwall-depleted Xenopus egg extracts. This idea that Greatwall inactivates ‘antimitotic’ phosphatases also found support from the Goldberg group [40], who showed that, once activated, Gwl promotes inhibition of the PP2A trimer with the B55δ subunit (counterpart of B55twins of Drosophila). In the absence of Greatwall, PP2A-B55δ remained active even when Cdk1 activity was high. Moreover, the removal of PP2A-B55δ corrected the inability of Greatwall-depleted extracts to enter M phase. Thus, there appeared to be two components to Greatwall function: one to inhibit PP2A to promote the Cdk1 activation loop and a second to suppress the PP2A activity that would otherwise remove Cdk1-driven phosphorylations [45]. Thus, there is some ambiguity in interpreting the requirements for Greatwall in inhibiting PP2A; some experiments emphasized its role to promote in mitotic entry and others to maintain the mitotic state.
Clarity into the biochemical mode of action of Greatwall kinase came from the identification of its principal substrates [41,42]. Tim Hunt's laboratory had been studying the roles of protein phosphatases in mitotic progression for some time and also had convincing evidence that in Xenopus, PP2A-B55δ was indeed the major phosphatase for Cdk1 substrates; depletion of this form of PP2A accelerated mitotic progression in mitoic extracts [46]. As they were not able to detect phosphorylated forms of PP2A, they were led to suspect a role for Greatwall in phosphorylating some intermediate protein to achieve PP2A-B55δ inhibition. This led them to screen for Greatwall substrates in interphase egg extracts and identify α-endosulphine (Ensa) and a related protein, Arpp-19, a substrate of cyclic AMP-activated protein kinase in post-synaptic neurons [47]. The Montpellier group reached similar findings in screening interphase extracts for Greatwall substrates [42] to find the same two proteins. Once phosphorylated by Greatwall these proteins became inhibitors of PP2A-B55δ. In the absence of Gwl activity, Arpp19 and α-Endosulphine were dephosphorylated, and lost their capacity to bind and inhibit PP2A. The London and Montpellier groups disagree about the relative importance of ARPP19 or Ensa in frogs, but as the two proteins are so highly similar, it may be questionable whether any distinction is of biological importance. Endos, the single orthologue of these proteins in Drosophila, is phosphorylated on the equivalent serine residue by the fly Greatwall kinase. Mutations at this site abolish the ability of the protein to rescue Endos depletion in cultured Drosophila cells [28]. Thus, genetic and biochemical approaches converged to identify this novel form of mitotic regulation by a protein kinase. Greatwall promotes mitotic progression not by phosphorylating a particular protein to directly promote its mitotic activity but rather to enable the inhibitor of an anti-mitotic phosphatase (figure 4).
Figure 4.
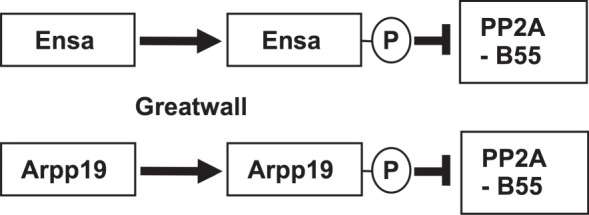
The paralogues Ensa (Endos) and Arpp19 are phosphorylated by Greatwall kinase to become inhibitors of PP2A-B55.
Aside from their importance in opposing the activity of PP2A to reverse the phosphorylation of Cdk1 substrates in mitotic exit, Greatwall kinase and its Endos substrate now emerge as key components of the regulatory loop that governs mitotic entry. This is because PP2A inhibition results in the accumulation of the phosphorylated, active form of Cdc25 and the phosphorylated, inhibited form of the Wee1 kinases. The consequence—full activation of Cdk1 even at low levels of Cyclin B—accounts for the long-standing observation that the phosphatase inhibitor okadaic acid can promote mitotic entry [48]. This has led Bela Novak and has collaborators to point out that the inhibition of PP2a by Greatwall/Endos contributes additional amplification loops to an inherently bistable mitotic switch that governs mitotic entry. This reflects the antagonistic interaction between one group of proteins promoting M phase (Cdk1-cyclin B, Cdc25, Gwl and Endos) and another that promotes interphase (Wee1, PP2A-B55; figure 5) [49,50]. As might be expected, such a central tenet for the regulation of mitotic entry is highly conserved, and the human counterpart of Greatwall, MASTL kinase (microtubule-associated serine threonine kinase-like protein), has similar roles in mitotic progression [51–53]. Indeed, the mitotic defects resulting from the depletion of MASTL can be rescued by simultaneous knockdown of PP2A or treatment with okadaic acid, once again indicating the importance of regulating the balance between Cdk1 and PP2A activity [51]. Interestingly, downregulation of MASTL can even overcome the mitotic arrest resulting from failure to activate the anaphase-promoting complex/cyclosome (APC/C) in cells ablated for Cdc20 [53]. Thus, by relieving the inhibition of PP2A-B55, it is possible to overcome even this block to the natural progression through the metaphase–anaphase transition and exit mitosis.
Figure 5.

Greatwall–Endos regulates mitotic entry and stabilizes the mitotic state by inhibiting PP2A-B55. Mitotic entry is regulated by a positive-amplification loop in which the dual-specificity phosphatase Cdc25 dephosphorylates and thereby activates Cdk1-cycB kinase. Cdk1-cycB phosphorylates and activates Cdc25. Cdc25 is opposed by the Wee1 kinase that is inhibited by Cdk1-cycB phosphorylation. Thus, PP2A dephosphorylates Cdc25-P and Wee1-P to oppose Cdk1-cycB. This accounts for the long-known fact that inhibition of PP2A (by okadaic acid) promotes mitotic entry. Endos, phosphorylated by Greatwall, acts in an analogous way. By inhibiting PP2A, phospho-Endos also maintains the mitotic state by enabling the multiple mitotic substrates of Cdk1-cycB to retain their phosphate groups.
5. Greatwall antagonizes some key functions of Polo
In spite of the clarity of understanding we now have of this regulatory loop, the relationship between Polo and Greatwall functions is confusing. Paradoxically, both Polo and Greatwall kinases promote progression through mitosis, and yet the genetic interactions outlined above suggest that the gain-of-function mutation gwlScant negatively regulates the function of Polo or one of its targets. So what is the evidence that the mitotic kinases Polo and Greatwall can act antagonistically, and how can we account for this? In considering this conundrum, it is important to note that this antagonism is observed specifically with respect to what appears to be a sensitive threshold requirement for Polo kinase activity to maintain centrosomes at the nuclear envelope in the division cycles of syncytial embryos. This phenotype can also be seen in other situations in which Polo kinase activity is reduced; for example, following the over-expression of Map205, a known interphase-binding partner of Polo that sequesters the kinase onto microtubules [54].
Two other lines of evidence suggest that the polo1+/+ Scant phenotype represents an enhancement of the polo phenotype as a result of the gain-of-function mutation in the Greatwall kinase. First, polo1+/+ Scant maternal effect lethality can be rescued by increasing the activity of Polo kinase, for example, as a result of polo+ duplications we obtained in the screen for revertants [27]. Second, the degree of embryo lethality resulting from the cumulative effects of centrosome loss covaries with strength of polo allele. The weak hypomorphic allele, polo1, shows only moderate embryonic defects with Scant, whereas the amorphic allele, polo11, shows centrosome loss defects that prevent any embryonic survival. Because the function of Scant can be ascribed to a mutation that we demonstrated to result in hyperactive Greatwall kinase [27], these experiments suggest either that Greatwall kinase might decrease the level of active Polo via PP2A or that Greatwall is independently inhibiting a pathway that is positively regulated by Polo.
Further evidence supporting a role for Greatwall in antagonising Polo has come from a recent study from Daniela Drummond-Barbosa's laboratory to search for second-site non-complementing mutants of endos [55]. Mutation in matrimony (mtrm), which encodes a known Polo kinase inhibitor [56], resulted in mitotic abnormalities in syncytial embryos when transheterozygous with an endos mutant in mothers (i.e. mtrm +/+ endos females). This sterility could be rescued by removing one wild-type copy of polo. Thus, in the absence of sufficient Matrimony protein to depress Polo activity, 50 per cent of functional Endos is unable to correctly exert mitotic control over a Polo-regulated function. These observations are thus consistent with the Greatwall–Endos pathway negatively regulating Polo.
There are a number of possible ways to account for this negative regulatory relationship between Greatwall–Endos and Polo that need not necessarily be exclusive and certainly still need to be clarified. First, both Polo and PP2A have been shown to be required for the centrosome maturation [57], and thus by promoting inhibition of PP2A, Greatwall would essentially antagonize a pathway promoted by Polo. Second, it has been proposed that loss of PP2A function synergizes with loss of Polo function because both activities are required to maintain the association of centrosomes to the nucleus or spindle, albeit at different stages of the nuclear division cycle [58]. A third possibility is that at some stages of the cycle, Greatwall and Polo together promote mitotic progression, whereas at other stages they act antagonistically. This is certainly possible because of the multiple ways in which Polo can interact, through its Polo-box domains, with partner proteins. The Polo-box domain generally binds phosphorylated sequences on a partner protein that has either been primed by another kinase or self-primed by Polo itself [58]. Thus, as the cell cycle proceeds we see a progression from Polo interactions at mitotic exit and in interphase that can be not only independent of Cdk1 but can also be actively disrupted by Cdk1-cycB phosphorylation to ones from late G2 until anaphase that are totally dependent on priming phosphorylation by Cdk1-cycB (box 2). We have argued that it is the alternation of Polo's functional interactions between a Cdk1-cycB dependency and independency that might account for the paradoxical relationship between Greatwall and Polo evidenced by centrosome detachment in the syncytial cycles of polo +/+ Scant-derived embryos [27]. We proposed that in prophase and prometaphase Greatwall, activated by Cdk1-cycB, inhibits PP2A via Endos, and this sustains the association of Polo with its Cdk1-cycB-phosphorylated partners. Once cyclin B is degraded at the onset of anaphase, Cdk1 activity falls and Polo begins to associate with proteins dephosphorylated at their Cdk1 sites by PP2A-B55Twins. We postulate at least one such of these latter proteins, which undergo Cdk1 independent interactions with Polo, to be required for the maintenance of centrosome attachment to the nuclear envelope. In syncytial embryos, hyperactive GreatwallScant kinase would lead to reduced interphase activity of PP2A, so lowering levels of functional complexes between Polo and dephosphorylated partners below some critical level (figure 6). At present, however, we do not know the molecular players participating in this process, nor indeed whether it reflects a single molecular interaction or a rather a readout of the effect of disrupting the Cdk1-PP2A balance on this stage of mitotic progression.
Box 2. Diverse interactions of the Polo-box domain of Polo/Plk1 kinases with their partner proteins.

The C-terminal part of the polo-like kinases has two Polo-box motifs that form interaction sites with partner proteins. Typically, a priming phosphorylation on the partner protein mediated by another protein kinase generates a docking site for Polo/Plk1. (a) In mitosis, the priming kinase is often Cdk1-cycB itself, thus ensuring that targeting of Polo to specific sequences occurs only when mitosis is underway. The binding of Plk1 to, and its subsequent phosphorylation of, the checkpoint protein BubR1 exemplifies such an interaction, likely to mediate the association of Plk1 with the kinetochore [59–61]. (b) Plk1 also interacts with other partners when Cdk1 is inactivated following cyclin B degradation. Perhaps the best examples of these interactors and substrates are the microtubule PRC1 protein [62] and the central-spindlin subunit CYK-4 that each participate in mediating Polo functions in the early stages of cytokinesis [63,64]. It is postulated that the initial phase of binding of Plk1 to such proteins may be phosphorylation-independent, but that subsequent phsophorylation of the target protein by Plk1 may effectively act as a self-priming event and accentuate the interaction [65]. We recently described an extreme case of an interaction of this type in syncytial Drosophila embryos, where a microtubule-associated protein Map205 sequesters Polo kinase onto microtubules during interphase (shown here). This interaction, which also takes place via the Polo-box domain, is actually disrupted by Cdk1-cycB phosphorylation of Map205 at an adjacent site (figure 6b) [54].
Figure 6.

Hypothesis for how Greatwall might act antagonistically to Polo late in mitosis in the syncytial nuclear division cycles of the Drosophila embryo. As both Greatwall and Polo are ‘mitotic kinases’, it seems counterintuitive that Greatwall might inhibit some Polo functions as suggested by the interactions between the gwlScant and polo mutations. Several explanations for this are possible and are discussed in the text. This schematic presents one of these potential explanations. It postulates that because Polo can interact in mitosis with proteins (X-P) that have been phosphorylated by Cdk1-cycB, and at mitotic exit and interphase with proteins that do not have such mitotic phosphorylations (Y), downregulating PP2A in Greatwall-Scant-derived embryos can prolong Polo's interactions with its mitotic partners and deny its interactions with interphase partners. In the context of this scheme, the consequence of the latter would be to favour retention of high Y-P levels and thereby lead to loss of centrosomes from nuclei, postulated to be an interphase process requiring dephosphorylated protein Y.
6. Regulation of Greatwall activity
Although available evidence supports the idea that Greatwall is activated by Cdk1, the precise mechanism for this is not clear and the possibility still exists that other mechanisms are involved. Greatwall is a member of the AGC family of kinases that includes enzymes such as PKA, PKC and RSK. Typically, the activation of this group of enzymes requires phosphorylation of an activation loop in the C-lobe of the enzyme, together with interactions between N- and C-terminal tails. The latter interaction involves phosphorylation of a hydrophobic motif that appears to be absent from Greatwall and it has been suggested that another AGC kinase might interact with Greatwall to provide this [66]. Much remains to be done in order to understand how exactly the activity of Greatwall is regulated. Greatwall is activated by Cdk1 or a Cdk1-dependent protein kinase, but although Greatwall's phosphorylation sites have been mapped [66], the significance of each site needs further investigation. Equally little is known of why the K97M mutation results in the activation of GreatwallScant in Drosophila [27], although introducing the equivalent mutation into Xenopus Greatwall kinase (K71M) also results in a hyperactive enzyme [67]. In fact, this mutant form of Greatwall is able to induce oocytes to enter M phase even in the absence of progesterone, the normal hormonal stimulus for this process. We are even more ignorant of the protein phosphatases required to inactivate Greatwall on mitotic exit, or indeed to inactivate its substrate Endos. It has, however, been suggested that Endos might be dephosphorylated at mitotic exit by PP1 [50]. If so, this may contribute to another interesting regulatory loop because PP1 has itself been shown to be inhibited by Cdk1s [68].
7. What other roles might Greatwall have?
While the focus has naturally been upon the role of Greatwall in mitosis, other studies raise the possibility of its role in different processes. It has been reported, for example, that Greatwall promotes recovery from the DNA damage checkpoint [69,70]. Thus, an increased DNA damage response was seen in Xenopus extracts depleted of Greatwall, whereas active Greatwall kinase inhibited the response. Greatwall was itself inhibited by the DNA damage response in a caffeine-sensitive manner, indicating a response to ATM (ataxia telangiectasia mutated)/ATR (ATM-related) signalling. The mechanisms of neither this inhibitory effect nor of the interphase activation of Greatwall in response to DNA damage are clear. However, it is of interest that Greatwall and Plk1 appear to associate and that the two kinases appear to show mutual dependency in promoting recovery from the damage checkpoint. Plk1 appears able to phosphorylate Greatwall directly, whereas it is suggested that Greatwall activates Aurora A, which in turn activates Plk1 [70]. These interactions and the precise roles of the phosphorylation reactions resulting from them demand more detailed study in different systems before we have a true understanding of Greatwall activation both in the damage checkpoint recovery and in mitotic entry.
We might also expect Greatwall and Endos or their counterparts to function outside of the cell cycle given that PP2A-B55twins functions in a wide range of biological processes. In Drosophila, there is evidence for involvement of the twins gene in the maintenance of neuroblast polarity, pattern formation in imaginal discs and in sensory organ development. Thus, as a PP2A-B55twins inhibitor, Endos could also participate in these processes. Moreover, it also seems that the Endos family of proteins might have other functions beyond the regulation of PP2A-B55. The budding yeast counterpart of Greatwall and Endos, the respective Rim5 protein kinase and its substrates Igo1 and Igo2, participate in the response to limiting amounts of nitrogen and/or carbon sources. Such starvation leads to downregulation of the conserved TORC1 and PKA signalling pathways and the consequential activation of Rim15 kinase, which in turn controls expression of specific downstream genes by regulating both transcription and mRNA stability. Once phosphorylated by Rim15, the Igo1 and Igo2 proteins associate with the mRNA decapping activator Dhh1 and protect newly expressed mRNAs from the mRNA decay pathway [71]. If these results extend into analogous pathways in higher eukaryotes, then this could identify a whole new range of functions for the Greatwall kinase. The participation of Arpp19 in stabilizing GAP-43 mRNA in response to nerve growth factor treatment could perhaps turn out to work through a similar mechanism [72]. Indeed, there are also some hints that Endos might be involved in mRNA stability, which come from the finding indicated above that in females heteroallelic or hemizygous for endos, where its protein levels are reduced by more than 95 per cent, levels of Polo and Cdc25twine protein are drastically reduced [31]. Thus, Endos could have an additional role in the post-transcriptional regulation of gene expression leading up to meiosis in the female germ-line of Drosophila. What is undoubtedly clear is that it is truly difficult to study protein phosphatase functions because of the need to relate them to those of the counteracting protein kinase. If the protein phosphatase regulatory proteins are as pleiotropic as it seems, then we must look forward to some very interesting times.
8. Acknowledgements
Work in my laboratory on Greatwall kinase and the Endos phosphoprotein is supported by a Programme Grant from the Medical Research Council. I would like to thank Elvan Boke, Iain Hagan and Tim Hunt for their very helpful comments on the manuscript.
References
- 1.Masui Y, Markert CL. 1971. Cytoplasmic control of nuclear behavior during meiotic maturation of frog oocytes. J. Exp. Zool. 177, 129–145 10.1002/jez.1401770202 (doi:10.1002/jez.1401770202) [DOI] [PubMed] [Google Scholar]
- 2.Reynhout JK, Smith LD. 1974. Studies on the appearance and nature of a maturation-inducing factor in the cytoplasm of amphibian oocytes exposed to progesterone. Dev. Biol. 38, 394–400 10.1016/0012-1606(74)90016-5 (doi:10.1016/0012-1606(74)90016-5) [DOI] [PubMed] [Google Scholar]
- 3.Kishimoto T, Kanatani H. 1976. Cytoplasmic factor responsible for germinal vesicle breakdown and meiotic maturation in starfish oocyte. Nature 260, 321–322 10.1038/260321a0 (doi:10.1038/260321a0) [DOI] [PubMed] [Google Scholar]
- 4.Guerrier P, Doree M, Freyssimet G. 1975. Early stimulation of protein kinase activity during hormonal meiosis reinitiation in starfish ovocytes. C R. Acad. Sci. Hebd. Seances Acad. Sci. D. 281, 1475–1478 [PubMed] [Google Scholar]
- 5.Lohka M, Masui Y. 1983. Formation in vitro of sperm pronuclei and mitotic chromosomes induced by amphibian ooplasmic components. Science 220, 719–721 10.1126/science.6601299 (doi:10.1126/science.6601299) [DOI] [PubMed] [Google Scholar]
- 6.Gerhart J, Wu M, Kirschner M. 1984. Cell cycle dynamics of an M-phase-specific cytoplasmic factor in Xenopus laevis oocytes and eggs. J. Cell. Biol. 98, 1247–1255 10.1083/jcb.98.4.1247 (doi:10.1083/jcb.98.4.1247) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans T, Rosenthal ET, Youngblom J, Distel D, Hunt T. 1983. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389–396 10.1016/0092-8674(83)90420-8 (doi:10.1016/0092-8674(83)90420-8) [DOI] [PubMed] [Google Scholar]
- 8.Lohka MJ, Hayes MK, Maller JL. 1988. Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl Acad. Sci. USA. 85, 3009–3013 10.1073/pnas.85.9.3009 (doi:10.1073/pnas.85.9.3009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hartwell LH, Culotti J, Pringle JR, Reid BJ. 1974. Genetic control of the cell division cycle in yeast. Science 183, 46–51 10.1126/science.183.4120.46 (doi:10.1126/science.183.4120.46) [DOI] [PubMed] [Google Scholar]
- 10.Nurse P, Thuriaux P, Nasmyth K. 1976. Genetic control of the cell division cycle in the fission yeast Schizosaccharomyces pombe. Mol. Gen. Genet. 146, 167–178 10.1007/BF00268085 (doi:10.1007/BF00268085) [DOI] [PubMed] [Google Scholar]
- 11.Lee MG, Nurse P. 1987. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327, 31–35 10.1038/327031a0 (doi:10.1038/327031a0) [DOI] [PubMed] [Google Scholar]
- 12.Russell P, Nurse P. 1986. cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell 45, 145–153 10.1016/0092-8674(86)90546-5 (doi:10.1016/0092-8674(86)90546-5) [DOI] [PubMed] [Google Scholar]
- 13.Russell P, Nurse P. 1987. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 49, 559–567 10.1016/0092-8674(87)90458-2 (doi:10.1016/0092-8674(87)90458-2) [DOI] [PubMed] [Google Scholar]
- 14.Edgar BA, O'Farrell PH. 1990. The three postblastoderm cell cycles of Drosophila embryogenesis are regulated in G2 by string. Cell 62, 469–480 10.1016/0092-8674(90)90012-4 (doi:10.1016/0092-8674(90)90012-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatti M, Baker BS. 1989. Genes controlling essential cell-cycle functions in Drosophila melanogaster. Genes Dev. 3, 438–453 10.1101/gad.3.4.438 (doi:10.1101/gad.3.4.438) [DOI] [PubMed] [Google Scholar]
- 16.Freeman M, Glover DM. 1987. The gnu mutation of Drosophila causes inappropriate DNA synthesis in unfertilized and fertilized eggs. Genes Dev. 1, 924–930 10.1101/gad.1.9.924 (doi:10.1101/gad.1.9.924) [DOI] [PubMed] [Google Scholar]
- 17.Vardy L, Orr-Weaver TL. 2007. The Drosophila PNG kinase complex regulates the translation of cyclin B. Dev. Cell. 12, 157–166 10.1016/j.devcel.2006.10.017 (doi:10.1016/j.devcel.2006.10.017) [DOI] [PubMed] [Google Scholar]
- 18.Sunkel CE, Glover DM. 1988. polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell. Sci. 89, 25–38 [DOI] [PubMed] [Google Scholar]
- 19.Glover DM, Leibowitz MH, McLean DA, Parry H. 1995. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 81, 95–105 10.1016/0092-8674(95)90374-7 (doi:10.1016/0092-8674(95)90374-7) [DOI] [PubMed] [Google Scholar]
- 20.Archambault V, Glover DM. 2009. Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell. Biol. 10, 265–275 10.1038/nrm2653 (doi:10.1038/nrm2653) [DOI] [PubMed] [Google Scholar]
- 21.Ruchaud S, Carmena M, Earnshaw WC. 2007. Chromosomal passengers: conducting cell division. Nat. Rev. Mol. Cell. Biol. 8, 798–812 10.1038/nrm2257 (doi:10.1038/nrm2257) [DOI] [PubMed] [Google Scholar]
- 22.Regan CL, Fuller MT. 1988. Interacting genes that affect microtubule function: the nc2 allele of the haywire locus fails to complement mutations in the testis-specific beta-tubulin gene of Drosophila. Genes Dev. 2, 82–92 10.1101/gad.2.1.82 (doi:10.1101/gad.2.1.82) [DOI] [PubMed] [Google Scholar]
- 23.White-Cooper HM. 1994. Drosophila cell cycle mutants, and their interactions. PhD thesis, University of Dundee, Dundee, UK. [Google Scholar]
- 24.White-Cooper H, Carmena M, Gonzalez C, Glover DM. 1996. Mutations in new cell cycle genes that fail to complement a multiply mutant third chromosome of Drosophila. Genetics 144, 1097–1111 [DOI] [PubMed] [Google Scholar]
- 25.Yu J, Fleming SL, Williams B, Williams EV, Li Z, Somma P, Rieder CL, Goldberg ML. 2004. Greatwall kinase: a nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J. Cell. Biol. 164, 487–492 10.1083/jcb.200310059 (doi:10.1083/jcb.200310059) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bettencourt-Dias M, et al. 2004. Genome-wide survey of protein kinases required for cell cycle progression. Nature 432, 980–987 10.1038/nature03160 (doi:10.1038/nature03160) [DOI] [PubMed] [Google Scholar]
- 27.Archambault V, Zhao X, White-Cooper H, Carpenter AT, Glover DM. 2007. Mutations in Drosophila Greatwall/Scant reveal its roles in mitosis and meiosis and interdependence with Polo kinase. PLoS Genet. 3, e200. 10.1371/journal.pgen.0030200 (doi:10.1371/journal.pgen.0030200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rangone H, et al. 2011. Suppression of scant identifies Endos as a substrate of greatwall kinase and a negative regulator of protein phosphatase 2A in mitosis. PLoS Genet. 7, e1002225. 10.1371/journal.pgen.1002225 (doi:10.1371/journal.pgen.1002225) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Virsolvy-Vergine A, Leray H, Kuroki S, Lupo B, Dufour M, Bataille D. 1992. Endosulfine, an endogenous peptidic ligand for the sulfonylurea receptor: purification and partial characterization from ovine brain. Proc. Natl Acad. Sci. USA 89, 6629–6633 10.1073/pnas.89.14.6629 (doi:10.1073/pnas.89.14.6629) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drummond-Barbosa D, Spradling AC. 2004. Alpha-endosulfine, a potential regulator of insulin secretion, is required for adult tissue growth control in Drosophila. Dev. Biol. 266, 310–321 10.1016/j.ydbio.2003.10.028 (doi:10.1016/j.ydbio.2003.10.028) [DOI] [PubMed] [Google Scholar]
- 31.Von Stetina JR, Tranguch S, Dey SK, Lee LA, Cha B, Drummond-Barbosa D. 2008. α-Endosulfine is a conserved protein required for oocyte meiotic maturation in Drosophila. Development 135, 3697–3706 10.1242/dev.025114 (doi:10.1242/dev.025114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alphey L, Jimenez J, White-Cooper H, Dawson I, Nurse P, Glover DM. 1992. twine, a cdc25 homolog that functions in the male and female germline of Drosophila. Cell 69, 977–988 10.1016/0092-8674(92)90616-K (doi:10.1016/0092-8674(92)90616-K) [DOI] [PubMed] [Google Scholar]
- 33.White-Cooper H, Alphey L, Glover DM. 1993. The cdc25 homologue twine is required for only some aspects of the entry into meiosis in Drosophila. J. Cell. Sci. 106, 1035–1044 [DOI] [PubMed] [Google Scholar]
- 34.Courtot C, Fankhauser C, Simanis V, Lehner CF. 1992. The Drosophila cdc25 homolog twine is required for meiosis. Development 116, 405–416 [DOI] [PubMed] [Google Scholar]
- 35.Gomes R, Karess RE, Ohkura H, Glover DM, Sunkel CE. 1993. Abnormal anaphase resolution (aar): a locus required for progression through mitosis in Drosophila. J. Cell. Sci. 104, 583–593 [DOI] [PubMed] [Google Scholar]
- 36.Mayer-Jaekel RE, Ohkura H, Gomes R, Sunkel CE, Baumgartner S, Hemmings BA, Glover DM. 1993. The 55 kd regulatory subunit of Drosophila protein phosphatase 2A is required for anaphase. Cell 72, 621–633 10.1016/0092-8674(93)90080-A (doi:10.1016/0092-8674(93)90080-A) [DOI] [PubMed] [Google Scholar]
- 37.Mayer-Jaekel RE, Ohkura H, Ferrigno P, Andjelkovic N, Shiomi K, Uemura T, Glover DM, Hemmings BA. 1994. Drosophila mutants in the 55 kDa regulatory subunit of protein phosphatase 2A show strongly reduced ability to dephosphorylate substrates of p34cdc2. J. Cell. Sci. 107, 2609–2616 [DOI] [PubMed] [Google Scholar]
- 38.Wang P, Pinson X, Archambault V. 2011. PP2A-twins is antagonized by greatwall and collaborates with polo for cell cycle progression and centrosome attachment to nuclei in Drosophila embryos. PLoS Genet. 7, e1002227. 10.1371/journal.pgen.1002227 (doi:10.1371/journal.pgen.1002227) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vigneron S, Brioudes E, Burgess A, Labbé J. C., Lorca T, Castro A. 2009. Greatwall maintains mitosis through regulation of PP2A. EMBO J. 28, 2786–2793 10.1038/emboj.2009.228 (doi:10.1038/emboj.2009.228) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castilho PV, Williams BC, Mochida S, Zhao Y, Goldberg ML. 2009. The M phase kinase Greatwall (Gwl) promotes inactivation of PP2A/B55delta, a phosphatase directed against CDK phosphosites. Mol. Biol. Cell 20, 4777–4789 10.1091/mbc.E09-07-0643 (doi:10.1091/mbc.E09-07-0643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mochida S, Maslen SL, Skehel M, Hunt T. 2010. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330, 1670–1673 10.1126/science.1195689 (doi:10.1126/science.1195689) [DOI] [PubMed] [Google Scholar]
- 42.Gharbi-Ayachi A, Labbé J. C., Burgess A, Vigneron S, Strub JM, Brioudes E, Van-Dorsselaer A, Castro A, Lorca T. 2010. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330, 1673–1677 10.1126/science.1197048 (doi:10.1126/science.1197048) [DOI] [PubMed] [Google Scholar]
- 43.Yu J, Zhao Y, Li Z, Galas S, Goldberg ML. 2006. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol. Cell. 7, 83–91 10.1016/j.molcel.2006.02.022 (doi:10.1016/j.molcel.2006.02.022) [DOI] [PubMed] [Google Scholar]
- 44.Zhao Y, Haccard O, Wang R, Yu J, Kuang J, Jessus C, Goldberg ML. 2008. Roles of Greatwall kinase in the regulation of cdc25 phosphatase. Mol. Biol. Cell. 19, 1317–1327 10.1091/mbc.E07-11-1099 (doi:10.1091/mbc.E07-11-1099) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldberg ML. 2010. Greatwall kinase protects mitotic phosphosites from barbarian phosphatases. Proc. Natl Acad. Sci. USA 107, 12 409–12 410 10.1073/pnas.1006046107 (doi:10.1073/pnas.1006046107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mochida S, Ikeo S, Gannon J, Hunt T. 2009. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 28, 2777–2785 10.1038/emboj.2009.238 (doi:10.1038/emboj.2009.238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Girault JA, Horiuchi A, Gustafson EL, Rosen NL, Greengard P. 1990. Differential expression of ARPP-16 and ARPP-19, two highly related cAMP-regulated phosphoproteins, one of which is specifically associated with dopamine-innervated brain regions. J. Neurosci. 10, 1124–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picard A, Capony JP, Brautigan DL, Dorée M. 1989. Involvement of protein phosphatases 1 and 2A in the control of M phase-promoting factor activity in starfish. J. Cell. Biol. 109, 3347–3354 10.1083/jcb.109.6.3347 (doi:10.1083/jcb.109.6.3347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krasinska L, et al. 2011. Protein phosphatase 2A controls the order and dynamics of cell-cycle transitions. Mol. Cell. 44, 437–450 10.1016/j.molcel.2011.10.007 (doi:10.1016/j.molcel.2011.10.007) [DOI] [PubMed] [Google Scholar]
- 50.Domingo-Sananes MR, Kapuy O, Hunt T., Novak B. 2011. Switches and latches: a biochemical tug-of-war between the kinases and phosphatases that control mitosis. Phil. Trans. R. Soc. Lond. B 366, 3584–3594 10.1098/rstb.2011.0087 (doi:10.1098/rstb.2011.0087) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burgess A, Vigneron S, Brioudes E, Labbé J. C., Lorca T, Castro A. 2010. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc. Natl Acad. Sci. USA 107, 12 564–12 569 10.1073/pnas.0914191107 (doi:10.1073/pnas.0914191107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voets E, Wolthuis RM. 2010. MASTL is the human orthologue of Greatwall kinase that facilitates mitotic entry, anaphase and cytokinesis. Cell Cycle 9, 3591–3601 10.4161/cc.9.17.12832 (doi:10.4161/cc.9.17.12832) [DOI] [PubMed] [Google Scholar]
- 53.Manchado E, et al. 2010. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55α,δ phosphatase. Cancer Cell 18, 641–654 10.1016/j.ccr.2010.10.028 (doi:10.1016/j.ccr.2010.10.028) [DOI] [PubMed] [Google Scholar]
- 54.Archambault V, D'Avino PP, Deery MJ, Lilley KS, Glover DM. 2008. Sequestration of Polo kinase to microtubules by phosphopriming-independent binding to Map205 is relieved by phosphorylation at a CDK site in mitosis. Genes Dev. 22, 2707–2720 10.1101/gad.486808 (doi:10.1101/gad.486808) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Von Stetina JR, LaFever KS, Rubin M, Drummond-Barbosa D. 2011. A genetic screen for dominant enhancers of the cell-cycle regulator α-Endosulfine identifies matrimony as a strong functional interactor in Drosophila. G3 1, 607–613 10.1534/g3.111.001438 (doi:10.1534/g3.111.001438) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiang Y, et al. 2007. The inhibition of polo kinase by matrimony maintains G2 arrest in the meiotic cell cycle. PLoS Biol. 5, e323. 10.1371/journal.pbio.0050323 (doi:10.1371/journal.pbio.0050323) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dobbelaere J, Josué F., Suijkerbuijk S, Baum B, Tapon N, Raff J. 2008. A genome-wide RNAi screen to dissect centriole duplication and centrosome maturation in Drosophila. PLoS Biol. 16, e224. 10.1371/journal.pbio.0060224 (doi:10.1371/journal.pbio.0060224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elia AE, et al. 2003. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115, 83–95 10.1016/S0092-8674(03)00725-6 (doi:10.1016/S0092-8674(03)00725-6) [DOI] [PubMed] [Google Scholar]
- 59.Wong OK, Fang G. 2007. Cdk1 phosphorylation of BubR1 controls spindle checkpoint arrest and Plk1-mediated formation of the 3F3/2 epitope. J. Cell. Biol. 179, 611–617 10.1083/jcb.200708044 (doi:10.1083/jcb.200708044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsumura S, Toyoshima F, Nishida E. 2007. Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J. Biol. Chem. 282, 15 217–15 227 10.1074/jbc.M611053200 (doi:10.1074/jbc.M611053200) [DOI] [PubMed] [Google Scholar]
- 61.Elowe S, Hümmer S, Uldschmid A, Li X., Nigg EA. 2007. Tension-sensitive Plk1 phosphorylation on BubR1 regulates the stability of kinetochore microtubule interactions. Genes Dev. 21, 2205–2219 10.1101/gad.436007 (doi:10.1101/gad.436007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neef R, Gruneberg U, Kopajtich R, Li X, Nigg EA, Sillje H, Barr FA. 2007. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat. Cell. Biol. 9, 436–444 10.1038/ncb1557 (doi:10.1038/ncb1557) [DOI] [PubMed] [Google Scholar]
- 63.Burkard ME, et al. 2009. Plk1 self-organization and priming phosphorylation of HsCYK-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol. 7, e1000111. 10.1371/journal.pbio.1000111 (doi:10.1371/journal.pbio.1000111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolfe BA, Takaki T, Petronczki M, Glotzer M. 2009. Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol. 7, e1000110. 10.1371/journal.pbio.1000110 (doi:10.1371/journal.pbio.1000110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park JE, Soung NK, Johmura Y, Kang YH, Liao C, Lee KH, Park CH, Nicklaus MC, Lee KS. 2010. Polo-box domain: a versatile mediator of polo-like kinase function. Cell. Mol. Life Sci. 67, 1957–1970 Review 10.1007/s00018-010-0279-9 (doi:10.1007/s00018-010-0279-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vigneron S, Gharbi-Ayachi A, Raymond AA, Burgess A, Labbé J. C., Labesse G, Monsarrat B, Lorca T, Castro A. 2011. Characterization of the mechanisms controlling Greatwall activity. Mol. Cell. Biol. 31, 2262–2275 10.1128/MCB.00753-10 (doi:10.1128/MCB.00753-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto TM, Blake-Hodek K, Williams BC, Lewellyn AL, Goldberg ML, Maller JL. 2011. Regulation of Greatwall kinase during Xenopus oocyte maturation. Mol. Biol. Cell. 22, 2157–2164 10.1091/mbc.E11-01-0008 (doi:10.1091/mbc.E11-01-0008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dohadwala M, da Cruz e Silva EF, Hall FL, Williams RT, Carbonaro-Hall DA, Nairn AC, Greengard P, Berndt N. 1994. Phosphorylation and inactivation of protein phosphatase 1 by cyclin-dependent kinases. Proc. Natl Acad. Sci. USA 91, 6408–6412 10.1073/pnas.91.14.6408 (doi:10.1073/pnas.91.14.6408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peng A, Yamamoto TM, Goldberg ML, Maller JL. 2010. A novel role for greatwall kinase in recovery from DNA damage. Cell Cycle 9, 4364–4369 10.4161/cc.9.21.13632 (doi:10.4161/cc.9.21.13632) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peng A, Wang L, Fisher LA. 2011. Greatwall and Polo-like kinase 1 coordinate to promote checkpoint recovery. J. Biol. Chem. 286, 28 996–29 004 10.1074/jbc.M111.257121 (doi:10.1074/jbc.M111.257121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Talarek N, et al. 2010. Initiation of the TORC1-regulated G0 program requires Igo1/2, which license specific mRNAs to evade degradation via the 5′-3′ mRNA decay pathway. Mol. Cell. 38, 345–355 10.1016/j.molcel.2010.02.039 (doi:10.1016/j.molcel.2010.02.039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Irwin N, Chao S, Goritchenko L, Horiuchi A, Greengard P, Nairn AC, Benowitz LI. 2002. Nerve growth factor controls GAP-43 mRNA stability via the phosphoprotein ARPP-19. Proc. Natl Acad. Sci. USA 99, 12 427–12 431 10.1073/pnas.152457399 (doi:10.1073/pnas.152457399) [DOI] [PMC free article] [PubMed] [Google Scholar]