

Abstract
Muenke syndrome is characterized by various craniofacial deformities and is caused by an autosomal-dominant activating mutation in fibroblast growth factor receptor 3 (FGFR3P250R). Here, using mice carrying a corresponding mutation (FgfR3P244R), we determined whether the mutation affects temporomandibular joint (TMJ) development and growth. In situ hybridization showed that FgfR3 was expressed in condylar chondroprogenitors and maturing chondrocytes that also expressed the Indian hedgehog (Ihh) receptor and transcriptional target Patched 1(Ptch1). In FgfR3P244R mutants, the condyles displayed reduced levels of Ihh expression, H4C-positive proliferating chondroprogenitors, and collagen type II- and type X-expressing chondrocytes. Primary bone spongiosa formation was also disturbed and was accompanied by increased osteoclastic activity and reduced trabecular bone formation. Treatment of wild-type condylar explants with recombinant FGF2/FGF9 decreased Ptch1 and PTHrP expression in superficial/polymorphic layers and proliferation in chondroprogenitors. We also observed early degenerative changes of condylar articular cartilage, abnormal development of the articular eminence/glenoid fossa in the TMJ, and fusion of the articular disc. Analysis of our data indicates that the activating FgfR3P244R mutation disturbs TMJ developmental processes, likely by reducing hedgehog signaling and endochondral ossification. We suggest that a balance between FGF and hedgehog signaling pathways is critical for the integrity of TMJ development and for the maintenance of cellular organization.
Keywords: temporomandibular joint, craniofacial biology, mandibular condyle, articular eminence/glenoid fossa, FgfR3, Indian hedgehog
Introduction
Fibroblast growth factor receptor 3 (FGFR3), which belongs to a family of four transmembrane kinase receptors (FGFR1-4), is expressed in proliferating and differentiating chondrocytes in developing cartilage, including mandibular condyles, and plays important roles in endochondral bone development (Rice et al., 2003; Ornitz, 2005). FGFR3 consists of an extracellular ligand-binding domain bearing three immunoglobulin (Ig)-like subdomains, a single hydrophobic transmembrane domain, and an intracellular domain with a split tyrosine kinase. Targeted disruption of the FgfR3 gene in mice results in skeletal overgrowth (Colvin et al., 1996), while constitutive active FgfR3 mutations result in shortening of long bones, due mainly to inhibition of chondrocyte proliferation and maturation (Deng et al., 1996). Based on these findings, FGFR3 is considered to be a negative regulator of endochondral bone formation. A recent study suggested that constitutively active FgfR3 mutation inhibits chondrocyte proliferation through the STAT pathway and negatively regulates chondrocyte differentiation through the MAPK pathway (Murakami et al., 2004).
Muenke syndrome in humans results from an autosomal dominant mutation in FGFR3 (Pro250Arg, P250R) that is located in the linker between the extracellular IgII- and IgIII-like subdomains (Muenke et al., 1997). This mutation is shown to significantly enhance FGFR3IIIc’s affinity to FGF1, FGF2, and FGF9 (Ibrahimi et al., 2004). Patients with heterozygous mutation in FGFR3 are normal in stature, but display a wide range of craniofacial abnormalities, including unilateral or bilateral coronal synostosis and Class III dental malocclusion due to midfacial hypoplasia (Muenke et al., 1997). To study the pathophysiology of Muenke syndrome, a mouse mutant (FgfR3P244R) has been generated that carries an analogous proline-to-arginine substitution at the corresponding mouse position 244 (P244R; Twigg et al., 2009). Interestingly, in addition to the facial suture fusion and abnormal mineral density in long bones (Twigg et al., 2009), FgfR3P244R mutants displayed precocious ossification closure of cranial base synchondroses, Class III dental malocclusion, and shortening of the cranial base (Laurita et al., 2011), strongly indicating that FGF signaling activated by FgfR3P244R also affects the endochondral ossification processes.
The mandibular condyle in mammals is a major site of growth and also forms a diarthrodial joint with the articular eminence/glenoid fossa of the temporal bone. The mandibular condylar cartilage and the opposing articulating surface on the temporal bone are ontogenetically characterized as secondary cartilage. Mandibular condyle development is initiated within thickened fibrous periosteal tissues at the supralateral site of the jaw primordium. A chondrocyte mass quickly grows and acquires a growth-plate-like organization along its main axis, consisting of a fibrous superficial layer, a polymorphic layer, a flattened chondrocyte zone, and a bottom hypertrophic chondrocyte zone (Luder et al., 1988; Shibukawa et al., 2007). The polymorphic layer contains chondroprogenitors exhibiting high mitotic activity, and produces mandibular growth by apposition of chondrocytes onto the surface of the underlying condylar cartilage (Kantomaa et al., 1994). Unlike chondrocytes in developing long bones, condylar chondrocytes rapidly mature, and establish neither clear column formation nor secondary ossification centers (Shibata et al., 2006; Wadhwa and Kapila, 2008). Along with this endochondral bone ossification, bone collar formation is tightly associated with chondrocyte maturation and initiates in the adjacent perichondral tissues facing early hypertrophic chondrocytes (Yasuda et al., 2010; Kinumatsu et al., 2011). So far, little is known about the molecular mechanisms regulating the emergence and cellular organization of the articular eminence/glenoid fossa of the temporal bone.
The aim of this study was to investigate the abnormalities in temporomandibular joint (TMJ) development, morphogenesis, and growth in FgfR3P244R mutant mice. Given the rather unique developmental features of this structure and the abnormal endochondral bone ossification found in the FgfR3P244R mutant cranial base, we were particularly interested in clarifying whether activation of FGF/FGFR3 signals would alter the endochondral bone ossification process in the mandibular condyle and articulating region of the temporal bone, leading to TMJ defects.
Materials & Methods
Generation of the FgfR3P244R Mutant Mice
FgfR3P244R mice, containing a knock-in mutation (c731g) in exon 7 of the FgfR3 gene, were kindly provided by Dr. Twigg (Oxford University, Oxford, UK; Twigg et al., 2009). Animals used in this study were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Children’s Hospital of Philadelphia.
Histological, Histochemical, and in situ Hybridization Analyses
FgfR3P244R mutants and control littermates were fixed with 4% paraformaldehyde overnight, decalcified for 2 wks by 10%EDTA/2% paraformaldehyde, dehydrated, and embedded in paraffin. Serial frontal and/or parasagittal sections from mutants and control littermates were placed on the same slides and processed for histological, histochemical, and in situ hybridization analyses. Twenty-three control mice—5 from post-natal day 0 (P0), 6 from P7, 4 from P21, 2 from 3 mos, 3 from 10 mos, and 3 from 1 yr—and 32 mutant mice at identical stages (6 from P0, 6 from P7, 8 from P21, 3 from 3 mos, 3 from 10 mos, and 6 from 1 yr) were used. Cartilage and bone were stained with Alcian blue/Alizarin red. Tartrate-resistant acid phosphatase (TRAP) staining was performed with the use of a leukocyte acid phosphatase kit (Sigma, St. Louis, MO, USA). Sections were hybridized with antisense or sense 35S-labeled probes (Koyama et al., 2007).
Results
TMJ Defects and Alteration of Ihh Signaling in FgfR3P244R Mutant Mice
We first examined the expression of FgfR3 in post-natal mandibular condyles to identify possible target cells. By post-natal day 7 (P7), the condyles displayed a growth-plate-like structure consisting of flattened (fc) and hypertrophic (hc) chondrocytes overlaid by fibroblastic superficial (sf) and polymorphic (pm) cell layers along the longitudinal axis (Fig. 1A). In situ hybridization revealed that the flattened immature/early hypertrophic chondrocytes expressed collagen II (Col II), while FgfR3 transcripts were detectable in the polymorphic cells as well as in immature/early hypertrophic chondrocytes (Figs, 1B, 1C, respectively). Ptch1, a hedgehog receptor and transcriptional target, was also detected in superficial/polymorphic cells and immature chondrocytes (Fig. 1D), suggesting that FGF and hedgehog signals act on similar cell populations. Gross anatomical inspection showed that anterior-middle portions of the condyles from 12-month FgfR3P244R homozygotes (hereafter termed ‘mutant mice’; Fig. 1J, arrowhead) were deformed and became shorter along the antero-posterior axis (n = 5, p < 0.02) compared with wild-type mice (hereafter termed ‘control mice’; double arrow in Figs. 1J, 1E, respectively). Clear TMJ defects could not be detected in FgfR3P244R hemizygotes, indicating that secondary cartilage might be less susceptible than cranial base synchondroses to dosage and expression of the mutant gene.
Figure 1.
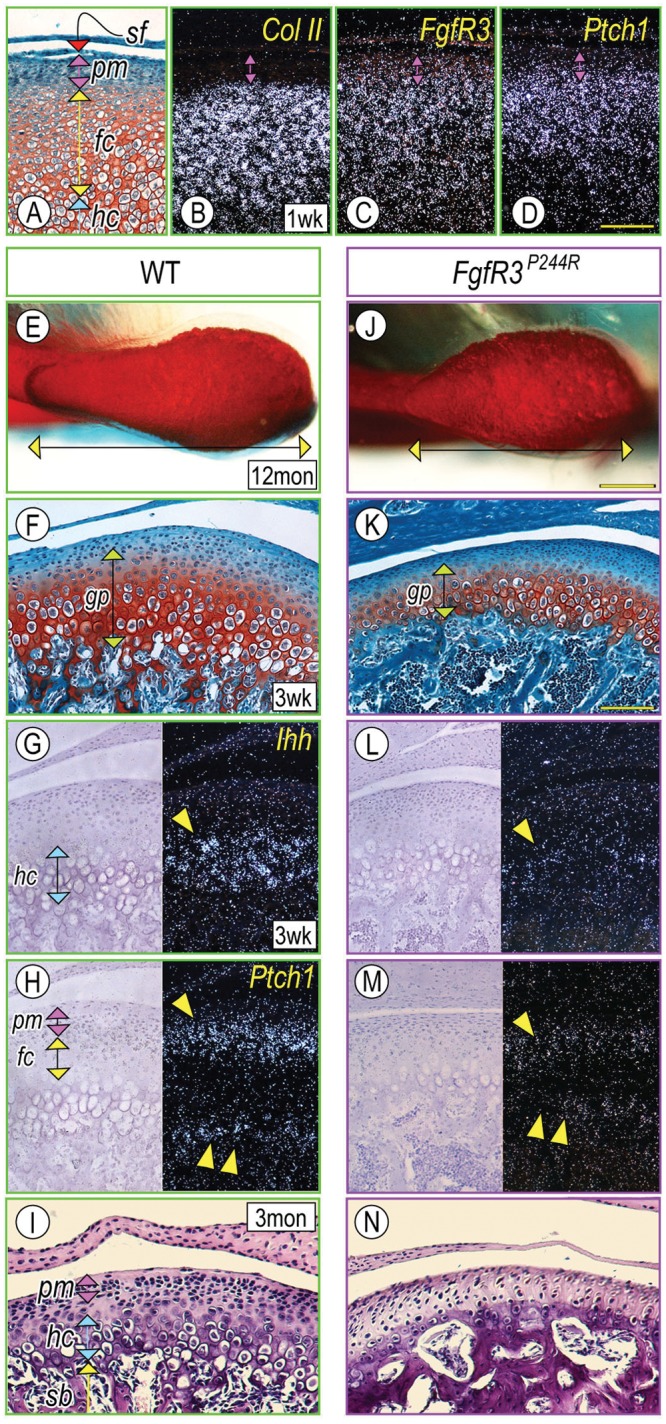
Mandibular condyle development was defective in FgfR3P244R mutant mice. Parasagittal sections from wild-type P7 (A-D), P21 (F-H), 3-month (I) and mutant P21 (K-M), and 3-month (N) condyles were processed for safranin-O/fast green staining (A, F, K), hematoxylin-eosin staining (I, N), and in situ hybridization (B-D, G-H, L-M). Twelve-month-old control (E) and mutant (J) condyles were stained with Alcian blue/alizarin red. In situ hybridization with the isotope-labeled RNA probes for collagen II (Col II) (B), Fibroblast growth factor Receptor 3 (FgfR3) (C), and Patched1 (Ptch1) (D). Note that FgfR3 expression largely overlapped with Ptch1-expressing cells in the polymorphic cell layer (pm) and flattened chondrocyte zone (fc). Superficial view of control (E) and mutant (J) condyles; mutant condyle is hypomorphic, indicated by shortening along the antero-posterior axis (a horizontal double arrow) compared with control (E). Note that P21-mutant condyles (K) exhibited the shorter growth-plate-like structure (gp, a green vertical double arrow) compared with controls (F). Ihh transcripts in the hypertrophic chondrocyte layer (hc) (G, arrowhead) and Ptch1 mRNAs in the polymorphic cell layer (pm, a purple vertical double arrow), the flattened chondrocyte layer (fc, a yellow vertical double arrow) (H, arrowhead), and the developing trabecular bones (H, a double arrowhead). Note the significant decrease of Ihh (L) and Ptch1 (M) transcripts in P21 mutant condyles, and chondroprogenitors and hypertrophic chondrocytes in 3-month mutant condyles (N). Trabecular bone with large marrow cavities underneath articular cartilage was appreciable in mutant condyles (N). sf, superficial layer; pm, polymorphic layer; fc, flattened chondrocyte layer; hc, hypertrophic chondrocyte layer. Scale bars: 120 µm for A-D (in D), 0.5 mm for E and J (in J), and 150 µm for F-I and K-N (in K).
Initially, control and mutant condyles from newborn and early post-natal mice exhibited similar cellular organization. However, by P21, mutant condylar growth-plate-like structures (gp) were significantly shorter in height than those in controls (double arrow in Figs. 1F, 1K, respectively). Ihh expression was decreased in the hypertrophic chondrocyte zone (Fig. 1L, arrowhead), and Ptch1 transcripts were decreased in the polymorphic/early flattened chondrocyte zone and primary spongiosa (Fig. 1M, arrowhead and double arrowheads, respectively) compared with controls (Figs. 1G, 1H, arrowhead and double arrowheads, respectively). By 3 mos, chondroprogenitors in the polymorphic layer and hypertrophic chondrocytes were decreased in mutant articular cartilage (Fig. 1N). In adult mutant mice, the articular disc often fused with the temporal bone and/or mandibular condyle (5/6), and the articular surface developed fissures (Appendix Fig. 1), indicating earlier onset of degenerative changes compared with wild-type mice.
Defects in Chondroprogenitor Proliferation and Endochondral Ossification in FgfR3P244R Mutant Condyles
H4C-positive proliferating cells were readily detectable in the polymorphic layer in control P7 and P21 condyles (Figs. 2A, 2B), but fewer H4C-expressing cells were observed in P21 mutant condyles (Figs. 2E, 2G; **p < 0.02). In addition, the shorter articular cartilage in P21 mutants displayed a reduction of both Col-II-expressing chondrocytes (Fig. 2H, a yellow double arrowhead; Fig. 2J, *p < 0.05) and Col-X-positive hypertrophic chondrocytes (Fig. 2I, a red double arrowhead; Fig. 2J, **p < 0.02) compared with controls (Figs. 2C, 2D, respectively), with a more drastic decrease of Col-X-expressing chondrocytes.
Figure 2.

Chondroprogentor proliferation, growth plate organization, and endochondral ossification were abnormal in FgfR3P244R mutant condyles. Parasagittal sections from control P7 (A, K, L) and P21 (B-D, M-O) condyles and mutant P7 (F, P, Q) and P21 (G-I, R-T) condyles were processed for safranin O/fast green staining (K, M, P, R), hematoxylin and eosin staining (M, R), in situ hybridization (A-D, F-I, N, O, S, T), and TRAP staining (L, Q). In situ hybridization with the isotope-labeled RNA probes for histone 4C (H4C) (A, B, F, G), and collagen type II (Col II) (C, H) and type X (Col X) (D, I). Note that, in P21 mutants, condyles displayed a significant reduction of H4C-expressing proliferating chondroprogenitors in the polymorphic layer (pm) (G), and the Col II-expressing flattened chondrocyte layer (fc) (H) and Col X-expressing hypertrophic chondrocyte layer (hc) (I) became narrower. For quantification of the proliferating chondroprogenitors (E), distinct condylar sections (approximately 100 cells/polymorphic layer, n = 6) from each of P7/P21 control and mutant mice were chosen, proliferating cells were present as the ratio/total cells, and each control was set as 100%. For quantification of the length of flattened and hypertrophic zones, Col II-expressing and Col X-expressing cells along the longitudinal axis were counted in distinct condylar sections (n = 6) from P21 control and mutant mice. The number of each control was set as 100%. p-values less than 0.05 were considered as statistically significant (*p < 0.05, **p < 0.02). Note that TRAP-positive osteoclasts were increased in the chondro-osseous border in P7 mutant condyles (E, Q; *p < 0.02) without detection of the clear histomorphogical changes of subchondral bone formation (sb) (P) compared with control (K, L, respectively). Note, in P21 mutant condyles, the significant reduction in bony spicules in the subchondral bone (sb) (R), accompanied by decreased Col I/Mmp13 (S, T)-expressing osteoblasts in the trabecular bone. pm, polymorphic layer; fc, flattened chondrocyte layer; hc, hypertrophic chondrocyte layer; sb, subchondral bone. Scale bars: 120 µm for A, B, F, G (in A); 90 µm for C, D, H, I (in C); and 90 µm for K-T (in K).
Condylar subchondral bone developing in P7 and P21 condyles displayed a typical trabecular network and harbored osteoblasts and hematopoietic cells (Figs. 2K, 2M). Developing trabeculae in subchondral bone (sb) were characterized by the presence of safranin-O-stained cartilage matrix coated by fast-green-stained bony matrix (Figs. 2K, 2M). Expression of collagen type I (Col-I), a typical bone matrix, and Matrix metalloproteinase 13 (Mmp13), an important player for cartilage in endochondral bone transition, was also evaluated (Figs. 2N, 2O). The reduced trabecular network in P21 mutant subcondral bone was appreciable by safranin-O/fast green and hematoxylin/eosin staining (Fig. 2R) and displayed fewer osteoblasts and/or marrow cells expressing Col 1 and MMP13 transcripts (Figs. 2S, 2T). Note an increase in TRAP-positive osteoclasts on the surfaces of the growing bony spicules at the cartilage-bone junction in P7 mutant condyles compared with controls (Figs. 2E, 2Q, 2L, *p < 0.02).
Abnormal Articular Eminence/Glenoid Fossa Development in FgfR3P244R Mutant Mice
To date, no study has investigated whether FGF signaling in secondary cartilage has any effect on the development of articular eminence/glenoid fossa. We first analyzed the articulating surface of the temporal bone to TMJ: the articular eminence/glenoid fossa in wild-type mice. We found evidence of condensing mesenchymal cells within a thickened periosteal tissue covering the temporal primordial at the prospective joint site by P7 (Figs. 3A, 3B), and a thin Col-II-positive secondary cartilage had developed by P21 (Figs. 3E, 3F; arrowhead). Compared with P21 mandibular condyles (Fig. 3D), the articular eminence/glenoid fossa displayed a thinner polymorphic cell layer (pm) containing fewer chondrocyte progenitors and underlying flattened/hypertrophic chondrocytes (fc/hc; Fig. 3C). The hypertrophic chondrocytes were also smaller in size, producing fast-green-stained matrix with much less Safranin-O-stained cartilage matrix, and many, if not all, hypertrophic chondrocytes appeared to be integrated into the underlying bony matrix (Fig. 3C). The articular eminence/glenoid fossa in wild-type mice contained Ihh /Osteopontin (Op)-expressing chondrocytes (fc/hc; Figs. 3G, 3H, respectively) as well as FgfR3 transcripts (Appendix Fig. 2). As expected, Col-I transcripts indicating typical secondary cartilage (Sugito et al., 2011) were also detected (Fig. 3I). In mutants, Col-II transcripts were barely detected (Fig. 3K, arrowhead), and expression of Ihh, Op, and Col-I was significantly reduced in the mutant secondary cartilage (Figs. 3L, 3M; arrowhead), while Op- and Col-I-expressing osteoblasts were detectable in the bone marrow of the developing temporal bone (tb) (Figs. 3M, 3N).
Figure 3.
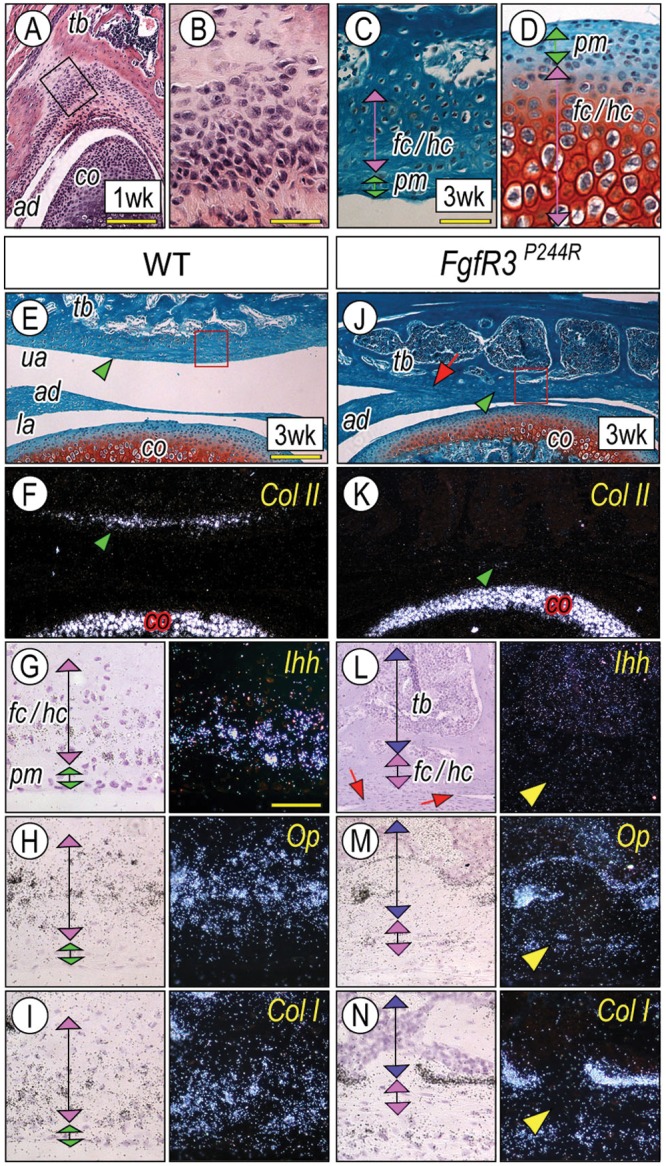
A unique process of chondrogenesis in the articular eminence/glenoid fossa and the abnormal cellular organization in FgfR3P244R-mutant mice. Serial frontal sections from wild-type P7 (A, B) and P21(C) articular eminence/glenoid fossa and P21 mandibular condyles (D) were processed for hematoxylin-eosin (A, B) and safranin O/fast green (C, D) staining. The magnified image (B) from the boxed area in (A) shows that the mesenchymal condensation of the articular eminence/glenoid fossa first emerged in the thickened periosteum of the temporal bone (tb) after the condyle (co) and articular disc (ad) formations had taken place. Note that polymorphic (pm) and differentiating chondrocyte (fc/hc) layers of the articular eminence/glenoid fossa (C) are narrower and hypertrophic chondrocytes are smaller than those in the mandibular condyles (D). Serial sections from control P21 (E-I) and mutant (J-N) TMJ were processed for safranin-O/fast green staining (E, J) and in situ hybridization with indicated probes (F-I, K-N). The magnified image (G-I, L-N) from the boxed area in E and J, respectively, shows the developing articular eminence/glenoid fossa. Note the absence of Col II transcripts (K, arrowhead) in the mutant articular eminence/glenoid fossa. Note also the articular disc fusion to the temporal bone (J, L, arrows). Scale bars: 120 µm in A, 45 µm in B, 60 µm for C, D (in C), 180 µm for E, F, J, K (in E), and 50 µm for G-I and L-N (in G).
FGF Treatment Antagonizes Hedgehog Action in Developing Condyles
Thus far, these findings indicated that activation of FGF signaling antagonistically acts on Ihh signaling. To investigate this idea further, we treated condylar explants from newborn wild-type mice with rhFGF2 or rhFGF9, both of which have been shown to activate FGFR3 (Zhang et al., 2006). In rhFGF2- and/or rhFGF9-treated condylar explants (1.0 µg/µL, n = 8, respectively), both Ptch1 and PTHrP transcripts were simultaneously decreased in the superficial/polymorphic cell layers (Figs. 4G, 4H) as compared with BSA-treated controls (Figs. 4B, 4C). EDU-labeled proliferating chondroprogenitors in the polymorphic layer were reduced compared with those in controls (Figs. 4I, 4D, respectively). Importantly, Col II expression was detectable in the flattened immature/early hypertrophic chondrocytes (Figs. 4E, 4J), signifying that this phenotypic trait was not disturbed in rhFGF-treated and control condyles.
Figure 4.
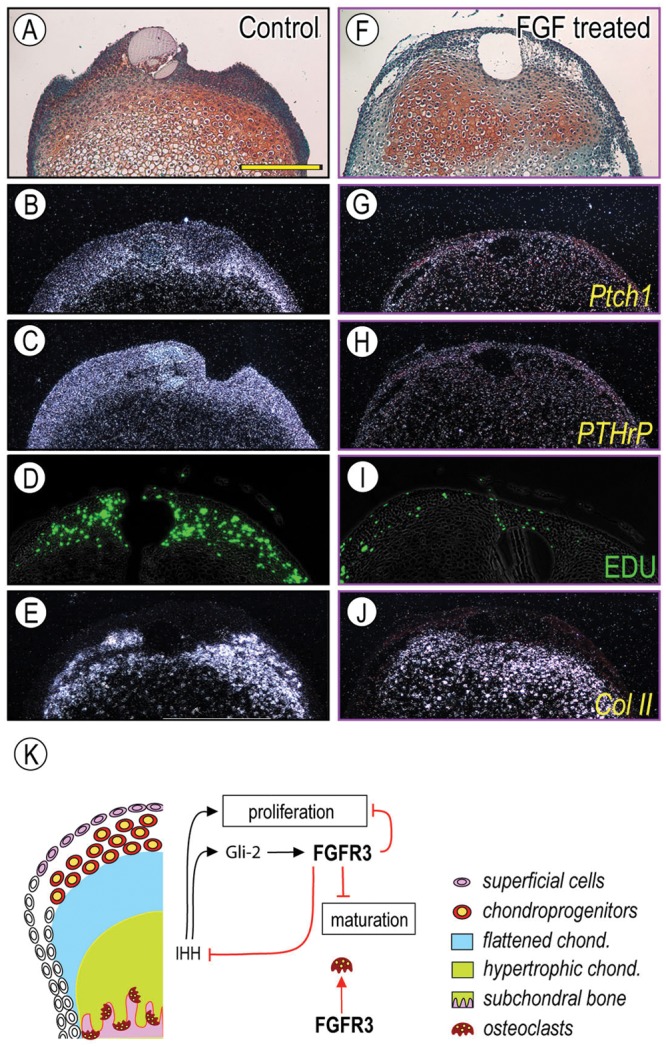
FGF signaling decreased expression of Ihh downstream targets and proliferation of chondroprogenitors in the polymorphic layer. Control BSA-treated (A-E) and rFGF9-treated (F-J) condyles were processed for in situ hybridization with isotope-labeled RNA probes for Patched1 (Ptch1) (B, G), PTHrP (C, H), and Collagen II (Col II) (E, J), and for proliferation detection (D, I). In control condyles, Ptch1 and PTHrP were expressed in the superficial/polymorphic layer (B, C), and EDU identifies proliferating chondroprogenitors in the polymorphic layers (D). Note that the rFGF9 treatment reduced Ptch1 transcripts in the superficial/polymorphic layers (G, H, respectively) and EDU incorporation (I), consistent with our observation in FgfR3P244R mutant mice and indicating that rFGF9 acted by inhibiting Ihh expression or signaling. Scale bar: 120 µm for A-J (in A). Schematic depicting our current working model for FGF-Ihh signaling interactions in post-natal TMJ is shown in (K). Ihh signaling regulated proliferation of chondroprogenitors (Shibukawa et al., 2007; Ochiai et al., 2010), and Gli2 inhibits chondrocyte hypertrophy via FgfR3 signaling (Purcell et al., 2009). FGFR3 signaling affects osteoclast differentiation/distribution. A similar molecular regulation could apply to the secondary cartilage in the articular eminence/glenoid fossa. ae/gf, articular eminence/glenoid fossa; ad, articular disc.
Discussion
Our study has demonstrated that an activating mutation of FGFR3, P244R, disturbs endochondral ossification in mandibular condyles and articular eminence/glenoid fossa in post-natal FgfR3P244R mutant mice, pointing to important roles for FgfR3 in the development and growth of secondary cartilage. The activating mutation of FGFR3 led to inhibition of both chondroprogenitor cell proliferation and chondrocyte maturation and disruption of subchondral bone formation by influencing osteoclast differentiation/distribution in the TMJ. Consistent with our findings, an earlier study has reported that FGF2 treatment inhibits cell proliferation and chondrocyte hypertrophy in mandibular condyle organ culture (Ogawa et al., 2003). Interestingly, Ihh gene expression was significantly reduced in FgfR3P244R mutant mandibular condyle as well as in FGF2-treated condylar organ cultures. We suggest that a mechanism by which FGFR3 signaling inhibits chondrocyte progenitor cell proliferation may involve down-regulation of Ihh gene expression. Purcell et al. (2009) showed that endogenous deletion of the IHH signaling mediator Gli2 (Gli2−/−) leads to decreased FgfR3 expression, along with a reduced population of immature chondrocytes, in the embryonic mouse mandibular condyle and suggested that Gli2 may be an upstream regulator of the FgfR3 gene. We thus propose that the reciprocal interaction between IHH/Gli2 and FGFR3 signaling coordinates chondrocyte proliferation and maturation in secondary cartilage of the TMJ.
These developmental defects, including abnormal endochondral ossification and defective trabecular bone formation, could be related to and/or account for earlier TMJ degenerative changes in post-natal mutants. It is also important to clarify whether mutant condyles are more susceptible to mechanical loading, given that profound phenotypic differences were appreciable at P21, when the pups naturally switched from a liquid to a hard diet.
Hedgehog signaling is also required to initiate disc development, as evident from disc agenesis in Ihh and Gli2 mutants (Shibukawa et al., 2007; Purcell et al., 2009). Given that the disc forms normally but fuses with the condyle and/or glenoid fossa in FgfR3P244R mutant mice, it is conceivable that IHH/FGFR3 signaling is also required to maintain structural integrity of the joint, including the superficial cells of the condyle/glenoid fossa. These cells secrete lubricin, which is needed for boundary lubrication, thereby protecting the articular cartilage surface from arthritic changes (Kinumatsu et al., 2011).
We have detailed secondary cartilage development that characterizes articular eminence/glenoid fossa formation. The delayed development appears to be influenced by the range of motion and masticatory action of the mandibular condyle during post-natal life (Pirttiniemi et al., 1994; Tuominen et al., 1996). Compared with the articular surface of the mandibular condyle, the articular eminence/glenoid fossa is quite distinct: Sporadically distributed chondrocyte progenitors display less proliferative activity, and hypertrophic chondrocytes synthesize very little, if any, Safranin-O-staining cartilage matrix. This finding could relate to the unique physiological and functional characteristics of the articular eminence/glenoid fossa, which exhibits distinct responses to mechanical loading compared with the mandibular condyle. Articular eminence/glenoid fossa remodeling appears to be mediated through deposition of new bone (Hinton and McNamara, 1984), indicating that activation of FGF signaling in the fibrous cell layer could be beneficial for the induction of osteoblast-like traits. A recent study reported that double knock-out of endogenous inhibitors of FGF signals, Spry 1 and Spry 2, led to defective development of the glenoid fossa due to overgrowth of lateral pterygoid and temporalis muscles in mouse embryos, in which FgfR4 signaling was likely altered (Purcell et al., 2012). Meanwhile, condylar development was not affected in Spry1−/−, Spry2−/− mutants. Thus, in the developing mandibular condyle, modulation of FGFR3-mediated signaling appears to involve distinctly different inhibitors.
In closing, it should be noted that mildly hypomorphic mandibles exhibiting increased gonial angle and decreased mandibular body length characterize patients with Muenke syndrome (Ridgway et al., 2011). Here, we unveil molecular mechanisms by which activation of FGF signaling through FgfR3P244R influences hedgehog target cells participating in the endochondral ossification of a secondary type of cartilage. Our findings point to future pharmaceutical and/or molecular-based treatments involving modulation of FGF and/or hedgehog downstream signaling in patients with skeletal abnormalities produced by disrupted FGF signaling.
Acknowledgments
We thank Drs. Steven R. Twigg and Andrew O. Wilkie (Oxford University, UK) for kindly providing FgfR3P244R mice.
Footnotes
This work was supported by NIH/NIAMS RO1AR050627 (H.-D.N) and by Departmental Funds.
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. (1996). Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet 12:390-397 [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. (1996). Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84:911-921 [DOI] [PubMed] [Google Scholar]
- Hinton RJ, McNamara JA., Jr (1984). Temporal bone adaptations in response to protrusive function in juvenile and young adult rhesus monkeys (Macaca mulatta). Eur J Orthod 6:155-174 [DOI] [PubMed] [Google Scholar]
- Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M. (2004). Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum Mol Genet 13:69-78 [DOI] [PubMed] [Google Scholar]
- Kantomaa T, Tuominen M, Pirttiniemi P. (1994). Effect of mechanical forces on chondrocyte maturation and differentiation in the mandibular condyle of the rat. J Dent Res 73:1150-1156 [DOI] [PubMed] [Google Scholar]
- Kinumatsu T, Shibukawa Y, Yasuda T, Nagayama M, Yamada S, Serra R, et al. (2011). TMJ development and growth require primary cilia function. J Dent Res 90:988-994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, et al. (2007). Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development 134:2159-2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurita J, Koyama E, Chin B, Taylor J, Lakin G, Hankenson KD, et al. (2011). The Meunke syndrome mutation (FgfR3P244R) caused cranial base shortening associated with growth plate dysfunction and premature perichondrial ossification in murine basicranial synchondroses. Dev Dyn 240:2584-2596 [DOI] [PubMed] [Google Scholar]
- Luder HU, Leblond CP, von der Mark K. (1988). Cellular stages in cartilage formation as revealed by morphometry, radioautography and type II collagen immunostaining of the mandibular condyle from weanling rats. Am J Anat 182:197-214 [DOI] [PubMed] [Google Scholar]
- Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP, et al. (1997). A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet 60:555-564 [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. (2004). Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev 18:290-305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai T, Shibukawa Y, Nagayama M, Mundy C, Yasuda T, Okabe T, et al. (2010). Indian hedgehog roles in post-natal TMJ development and organization. J Dent Res 89:349-354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Shimokawa H, Fukada K, Suzuki S, Shibata S, Ohya K, et al. (2003). Localization and inhibitory effect of basic fibroblast growth factor on chondrogenesis in cultured mouse mandibular condyle. J Bone Miner Metab 21;145-153 [DOI] [PubMed] [Google Scholar]
- Ornitz DM. (2005). FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev 16:205-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirttiniemi P, Kantomaa T, Tuominen M, Salo L. (1994). Articular disc and eminence modeling after experimental relocation of the glenoid fossa in growing rabbits. J Dent Res 73:536-543 [DOI] [PubMed] [Google Scholar]
- Purcell P, Joo BW, Hu JK, Tran PV, Calicchio ML, O’Connell DJ, et al. (2009). Temporomandibular joint formation requires two distinct hedgehog-dependent steps. Proc Natl Acad Sci USA 106:18297-18302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell P, Jheon A, Vivero MP, Rahimi H, Joo A, Klein OD. (2012). Spry1 and spry2 are essential for development of the temporomandibular joint. J Dent Res 91:387-393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP, Rice R, Thesleff I. (2003). Fgfr mRNA isoforms in craniofacial development. Bone 33:14-27 [DOI] [PubMed] [Google Scholar]
- Ridgway EB, Wu JK, Sullivan SR, Vasudavan S, Padwa BL, Rogers GF, et al. (2011). Craniofacial growth in patients with FGFR3Pro250Arg mutation after fronto-orbital advancement in infancy. J Craniofac Surg 22:455-461 [DOI] [PubMed] [Google Scholar]
- Shibata S, Suda N, Suzuki S, Fukuoka H, Yamashita Y. (2006). An in situ hybridization study of Runx2, Osterix, and Sox9 at the onset of condylar cartilage formation in fetal mouse mandible. J Anat 208:169-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibukawa Y, Young B, Wu C, Yamada S, Long F, Pacifici M, et al. (2007). Temporomandibular joint formation and condyle growth require Indian hedgehog signaling. Dev Dyn 236:426-434 [DOI] [PubMed] [Google Scholar]
- Sugito H, Shibukawa Y, Kinumatsu T, Yasuda T, Nagayama M, Yamada S, et al. (2011). Ihh signaling regulates mandibular symphysis development and growth. J Dent Res 90:625-631 [DOI] [PubMed] [Google Scholar]
- Tuominen M, Kantomaa T, Pirttiniemi P, Poikela A. (1996). Growth and type-II collagen expression in the glenoid fossa of the temporomandibular joint during altered loading: a study in the rat. Eur J Orthod 18:3-9 [DOI] [PubMed] [Google Scholar]
- Twigg SR, Healy C, Babbs C, Sharpe JA, Wood WG, Sharpe PT, et al. (2009). Skeletal analysis of the Fgfr3(P244R) mouse, a genetic model for the Muenke craniosynostosis syndrome. Dev Dyn 238:331-342 [DOI] [PubMed] [Google Scholar]
- Wadhwa S, Kapila S. (2008). TMJ disorders: future innovations in diagnostics and therapeutics. J Dent Educ 72:930-947 [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Mundy C, Kinumatsu T, Shibukawa Y, Shibutani T, Grobe K, et al. (2010). Sulfotransferase Ndst1 is needed for mandibular and TMJ development. J Dent Res 89:1111-1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. (2006). Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem 281:15694-15700 [DOI] [PMC free article] [PubMed] [Google Scholar]