


Abstract
We present a flexible, real-time-coupled transcription–translation assay that involves the continuous monitoring of fluorescent Emerald GFP formation. Along with numerical simulation of a reaction kinetics model, the assay permits quantitative estimation of the effects on full-length protein synthesis of various additions, subtractions or substitutions to the protein synthesis machinery. Since the assay uses continuous fluorescence monitoring, it is much simpler and more rapid than other assays of protein synthesis and is compatible with high-throughput formats. Straightforward alterations of the assay permit determination of (i) the fraction of ribosomes in a cell-free protein synthesis kit that is active in full-length protein synthesis and (ii) the relative activities in supporting protein synthesis of modified (e.g. mutated, fluorescent-labeled) exogenous components (ribosomes, amino acid-specific tRNAs) that replace the corresponding endogenous components. Ribosomes containing fluorescent-labeled L11 and tRNAs labeled with fluorophores in the D-loop retain substantial activity. In the latter case, the extent of activity loss correlates with a combination of steric bulk and hydrophobicity of the fluorophore.
INTRODUCTION
Protein synthesis is being studied intensively with a variety of motivations, ranging from practical needs for the development of efficient methods of cell-free protein synthesis (CFPS) for production of proteins that are difficult to express in cells (1–3), for the identification of new antibiotics (4,5), and toward achieving understanding of basic mechanisms of individual steps of the translation cycle and of overall protein synthesis (6,7). Many mechanistic studies employ fluorescent-labeled constituents of the protein synthesis machinery. Fluorescent probes permit real-time monitoring of specific reaction steps (8–15), but raise the question of whether introduction of exogenous labels unduly affects the processes under study. Assays of protein synthesis in current use are often cumbersome, involving aliquot removal and subsequent point-by-point determinations of bioluminescence (3), fluorescence (16,17), gel densitometry (16), enzymatic activity assay (18,19) or incorporation of radioactive amino acids (20–22).
Here we present a flexible, real-time coupled transcription–translation assay that, along with numerical simulation of a reaction kinetics model, permits quantitative estimation of the effects on full-length protein synthesis of introducing fluorescent probes, or other modifications, into components of the translational machinery. The assay involves the continuous monitoring of the formation of Emerald GFP (EmGFP), a rapidly maturing variant of green fluorescent protein (23), using a suitable plasmid and a commercially available CFPS kit derived from Escherichia coli (24). Straightforward alterations of the assay permit determination of (i) the fraction of ribosomes in a cell-free protein synthesis kit that is active in full-length protein synthesis and (ii) the relative activities in supporting protein synthesis of modified (e.g. mutated, fluorescent-labeled) exogenous components (ribosomes, amino acid-specific tRNAs) that replace the corresponding endogenous components.
Since the assay uses continuous fluorescence monitoring, it is much simpler and more rapid to use than other assays of protein synthesis and is compatible with high-throughput formats. It should thus find widespread application in many studies measuring protein synthesis in vitro. However, the assay does have the significant limitation that it is extremely stringent, since it treats as fully inactive those ribosomes that produce only partial or otherwise non-fluorescent EmGFP polypeptides.
MATERIALS AND METHODS
All solutions contained water of high purity generated by MilliQ deionizer equipped with pyrogen free filter (Biopak, Millipore) to ensure the absence of contaminating RNAses, DNAses and proteases. All ultracentrifugations at 110 000 r.p.m. were carried out in a S120AT2 rotor in a Sorvall M120SE ultracentrifuge. Ribosome concentrations were calculated assuming 26 pmol/A260 (25).
Exogenous component preparation
Plasmid
The pREST-EmGFP plasmid (Invitrogen, Inc.) contains a reading frame that includes a 40-residue N-terminal His-tag sequence followed by the 239-residue EmGFP sequence. We used standard mutagenesis techniques to introduce two modifications. The first, following reference (26), was to add a 31-residue C-terminal segment ending with the HindIII restriction site that, following plasmid linearization with HindIII, resulted in a reading frame devoid of a stop codon. This allowed tethering of mature, nascent EmGFPs to the ribosomes. The second change was a point mutation (S4F) near the beginning of the N-terminal His-tag sequence that was introduced for use in other studies. Mutagenesis was performed using a QuikChange Lightning site-directed mutagenesis kit (Stratagene, Inc.) with primers set according to the manufacturer's instructions.
Ribosomes
All preparative steps were carried out on ice or at 4°C. S30 fractions of E. coli strains MRE600 (wild type) and AM77 (−L11) were prepared as previously described (27) using a French Press at 5000 psi. Crude ribosome pellets were prepared by centrifuging S30 fractions at 110 000 rpm for 12 min at 4°C either directly (S100P), or by layering the S30 lysate on a sucrose cushion (S100sucP, using 1.1 M sucrose in TAM15 buffer) followed by resuspending the resulting pellet in TAM15 buffer. Small scale ribosome purification (70S-ss) was achieved by layering S100sucP ribosomes on a linear 15–30% sucrose gradient and collecting the 70S peak as described, using a VTi50 rotor (28). Large-scale ribosome purification (70S-ls) was achieved by sucrose gradient centrifugation using a zonal rotor as described (9). S100P preparations of AM77L11or AM77L11Cy3 ribosomes were prepared by incubating AM77 S30 fractions with a 2-fold excess of either unlabeled C38S/S87C-L11 (L11) or Cy3-labeled C38S/S87C-L11 (L11Cy3) at 37°C for 15 min prior to high-speed pelleting. The preparation of the L11 variant and its labeling by Cy3 is described in (11). FPLC-purified L11Cy3 contained 1.0 Cy3/protein.
tRNA
The charging and fluorescent labeling of yeast and E. coli tRNAPhes (Chemical Block, Moscow) were performed as previously described (10,11). tRNA concentrations were calculated on the basis of the amount of charged material, as determined by 14C-Phe radioactivity.
In vitro protein synthesis of EmGFP
Using the cell-free protein synthesis kit
The CFPS kit (RTS 100 E. coli HY kit – previously sold by Roche Applied Science, currently sold by 5prime) was utilized to synthesize EmGFP according to the manufacturer’s manual, with some modifications. The CFPS kit is made up of five vials, containing cell lysate (V1), reaction mixture (V2), 19 amino acids (V3), methionine (V4), reconstitution buffer (V5) and a control GFP vector that was substituted throughout these experiments with the modified pREST-EmGFP plasmid described above. In the standard reaction, the following components are pre-mixed on ice: 2.4 μl V1, 2 μl V2, 2.4 μl V3, 0.2 μl V4, 1 μl V5 and 2 μl (130 ng) plasmid in water. The total reaction volume was ∼10 μl. Fluorescence is recorded continuously at 30°C. The V3 solution was sometimes substituted by a home-made solution that contained approximately twice the original amino acid concentrations (denoted V3a). V3a, made up in V5, contained 8.33 mM tyrosine, 16.66 mM of each of the 18 other amino acids and no methionine. Final amino acid concentrations in the CFPS mixture were: 10 mM Met, 1 mM Tyr and 2 mM of the other amino acids. All other lyophilized CFPS components, were dissolved in V5 according to the manufacturer’s manual, flash-frozen and stored at −80°C in single-use aliquots.
To test the activity of various buffers (Figure 1A) the following components were mixed: 2.4 μl V1, 2 μl V2, 1.2 μl V3a, 0.2 μl V4, 9.2 μl of the indicated buffer and 0.45 μl plasmid (stock solution 430 ng/μl in water). V3a was made up in the indicated buffer rather than in V5. The total volume of the reaction was ∼15 μl. This combination yielded a reaction mixture that contained at least 70% of the indicated test buffer by volume and 30% of V5 used to reconstitute the lyophilized V1, V2 and V4. The buffers tested, included: TAM15: 50 mM Tris–HCl pH 7.5, 30 mM NH4Cl, 70 mM KCl, 15 mM Mg(Ac)2, 1 mM DTT; TAM7: 50 mM Tris–HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2, 1 mM DTT (29); Polymix Buffer: 50 mM Tris–HAc pH 7.5, 5 mM NH4Ac,100 mM KCl, 15 mM Mg(Ac)2, 0.5 mM Ca(Ac)2, 6 mM 2-mercaptoethanol, 5 mM putrescine and 1 mM spermidine (30); Polyamine Buffer: 20 mM HEPES–KOH pH 7.5, 150 mM NH4Ac, 4.5 mM MgAc2, 4 mM 2-mercaptoethanol, 0.05 mM spermine and 2 mM spermidine (31).
Figure 1.

Cell-free expression of EmGFP using the CFPS kit. (A) Buffer dependence. Activity was determined using the indicated buffers. The reaction volumes were diluted (dilution factor of 1.5) to 15 μl and measured in the fluorometer every 20 s. (B) Plasmid dependence. Activity was determined, using the buffer provided with the kit for the indicated plasmids. Anti-tmRNA oligo DNA at a concentration of 5 μM was added in addition to the linearized plasmid to prevent dissociation of the ribosome-EmGFP complex. All experiments were preformed in triplicate. Signal was acquired every 45 s, avoiding sample irradiation between consecutive measurements in order to minimize photobleaching.
Using the CFPS-ribosome kit
Ribosomes were removed from a mixture containing 2.4 μl V1, 2 μl V2, 1.2 μl V3 and 0.2 μl V4 as the pellet formed by ultracentrifugation at 110,000 rpm for 12 min at 4°C to yield the CFPS-ribosome kit. EmGFP synthesis was accomplished by adding 2.2 μl of 6.8 μM stock solution of home-made ribosomes (see above) in TAM15 buffer to the kit and initiating reaction with 130 ng of plasmid in 2 μl water. The final ribosome concentration of 1.5 μM equals the concentration found in the CFPS kit.
Using the CFPS-Phe kit
Construction of the CFPS-Phe kit required treatment of various solutions with an inhibition cocktail [100 µl of a commercial mixture of protease inhibitors (Halt, Pierce) and 0.5 µl of 4.7 M N-benzyl-2-phenylethylamine (Acros), a Phe-RS inhibitor (32)] and preparation of a V3a solution lacking Phe (denoted V3a-Phe). Additional details are found in Supplementary Information 2C and Supplementary Figure S3C. In addition, the normal ribosome concentration was sometimes reduced by half.
V1 (24 μl) was combined with 0.24 μl of an inhibition cocktail and the resulting mixture was dialyzed against 40 ml of TAM15 buffer supplemented with 5% PEG-8000 (Fisher) in 3.5 kDa cutoff micro-dialysis cups (Slide-A-Lyzer, Pierce—soaked in water for 15 min prior to first use). Following dialysis at 4°C for 30 min, 0.24 μl of fresh inhibition cocktail was added and dialysis was repeated against fresh buffer. The dialyzed V1 solution (24 μl) was combined with 20 μl V2, 12 μl V3a–Phe, 2 μl V4, 0.24 μl inhibition cocktail and 0.42 μl 10× of Polyamine buffer, generating the CFPS-Phe solution. Reduction of ribosome concentration was accomplished by combining equal volumes of a ribosome-containing CFPS-Phe kit and a ribosome-depleted CFPS-Phe kit, prepared via high-speed centrifugation as described above.
EmGFP synthesis was accomplished by adding aqueous solutions (3.8 μl) of various concentrations of Phe-tRNAPhe or Phe to the CFPS-Phe kit (6.2 µl), followed by incubating the resulting mixture at 37°C for 5 min to promote ternary complex (EF-Tu·aa-tRNA·GTP) formation, and initiating the reaction with 130 ng plasmid in water (0.3 μl). Addition of EF-Tu in amounts equimolar with added Phe-tRNAPhe results in a loss of EmGFP production (Supplementary Information 2F and Supplementary Figure S3F).
Other methods
Real-time fluorescence detection
Assays were performed at 30°C. Most assays were carried out in triplicates on a reaction volume of 10 μl dispensed in 384 well-plates (Greiner Bio-One 784076). Readings were taken every 20–45 s for 30–70 min in a plate reader (Envision 2103, Perkin-Elmer). The exposure time for individual fluorescence determinations was ∼30 ms using 10 flashes/measurement. In the absence of dye-labeled exogenous components, filters/bandpass settings of λex = 486/10 nm, λem = 535/25 nm were employed. To overcome spectral overlap of EmGFP emission with Cy3 and/or Cy5, the optimal settings were found to be λex = 450/8 nm, λem = 510/10 nm (see Supplementary Information 3 and Supplementary Figure S5). Some assays were also carried out using a reaction volume of 15 μl placed in a sub-micro fluorometer cell (Starna Cells, Inc) placed within a spectrofluorometer (Hobira Jobin Yvon) (λex = 468 nm, λem = 510 nm). Kinetic traces were recorded over 30 min at 10 s intervals, using a 1 s integration time.
EmGFP displays a fast component of photobleaching (33). To minimize photobleaching, sample irradiation was avoided except during recording, both in the fluorometer and in the plate reader. In a control experiment, no appreciable loss in EmGFP fluorescence could be detected over 70 min for purified EmGFP added to a CFPS-Phe kit using the employed plate reader configuration, although photobleaching was evident for EmGFP that was dissolved in TAM15 buffer.
Determination of the fraction of active ribosomes
Cell-free protein expression was performed for 1 h at 30°C with 130 ng circular or linearized plasmid DNA per 10 μl reaction volume. Anti-tmRNA [SsrA (34)] at a final concentration of 5 μM (6) was premixed with the reaction mixture, and reaction was initiated with linearized plasmid. The reaction mixtures were incubated either in the plate reader while following fluorescence in real time, or in a temperature controlled bath for one hour and subsequently pulled-down through a sucrose cushion. In pull-down assays, reaction volumes of 30 μl were layered onto ice-cold 1.1 M sucrose in TAM15 buffer (100 μl) and centrifuged at 110 000 rpm for 40 min at 4°C. The supernatant was collected and the pellet was suspended in 30 μl TAM15 buffer. EmGFP in the supernatant and in the pellet were quantified in the plate reader using a fluorescence calibration curve (Supplementary Information 1 and Supplementary Figure S1). The calculated ratio of EmGFP per ribosome in the pellet represented the fraction of active ribosomes.
[14C]-Phe incorporation into EmGFP
Samples were prepared with CFPS-Phe as described above, except the final sample volume was 15 μl. The [14C]-Phe-tRNAPhe variants were used at final concentrations of 10 μM. The reaction was initiated by plasmid addition and incubated at 30°C for 30 min. The reaction was quenched by the addition of 0.3 ml 5% TCA (ice-cold), heated to 95°C for 15 min, cooled on ice, filtered through a nitrocellulose filter, and washed five times with 1 ml of 5% ice-cold TCA. The filter was then dissolved in 1 ml ethyl acetate. Precipitated radioactivity, accurate to ±8%, was determined above a background equal to the radioactivity obtained with a sample that did not contain the EmGFP plasmid.
Fluorescence trace analysis
Four parameters were evaluated from the fluorescence traces that contained various Phe-tRNAPhes, using Origin software (OriginLab Corporation). Estimates of lag time, tL, maximum synthesis rate, S1 and final protein synthesis rate, S2, were obtained from the time of the maximal value of the second derivative, the maximal value of the first derivative, and a linear fit of the last eleven time points, respectively. Amplitude of the rapid synthesis period, A1, was determined by the intercept of two linear regression lines, the first centered at the point of maximal slope and the second fitted as described for S2. Before taking derivatives, the traces were smoothed using a 5-point adjacent average. All linear fits were performed using 11 time points. All values were normalized to the data obtained for 2 μM E. coli Phe-tRNAPhe.
Numerical simulations
A model of the kinetics of EmGFP fluorescence accumulation in the CFPS experiment was written in MATLAB version 7.5.0 (R2007b). Details of the algorithm are described in Supplementary Information. The MATLAB code is supplied as Supplementary File S2. A compiled executable file may also be obtained upon request to one of the corresponding authors.
RESULTS
Preliminary characterization of the cell-free coupled transcription/translation reaction
We employed a construct containing the 239-residue EmGFP sequence in between a 40-residue N-terminal His-tag extension and a 31-residue C-terminal extension. The latter sequence permitted the native EmGFP polypeptide to leave the ribosomal exit tunnel, allowing folding and maturation of EmGFP while the peptide is still attached to the ribosome. Translation of EmGFP followed by fluorophore maturation results in an accumulation of fluorescence intensity, providing a readily detectable signal that, after an initial lag phase, increases continuously in real time, reaching a near plateau after 60–90 min (Figure 1). Four different buffers were examined, with the amounts of fluorescent EmGFP formation falling in the order Polyamine (31) > TAM15 (11) > Polymix (30) ≫ TAM7 (29) (Figure 1A), with the Polyamine buffer showing similar activity to the buffer provided with the kit (unknown composition). Accordingly, only the Polyamine buffer [having a composition similar to that used previously to support transcriptional activity (35)] or the TAM15 buffer was used in subsequent translational activity assays. We also tested whether pre-transcribed mRNA would program ribosomes to produce EmGFP in the CFPS kit. Translation rates and amounts were comparable to the coupled transcription-translation beginning from the DNA plasmid (data not shown).
We next determined the fraction of active ribosomes present in a CFPS kit, as measured by the amount of ribosome-associated nascent EmGFP complex formed, adapting a known procedure (26). In this approach the EmGFP plasmid is linearized to remove the stop codon, preventing re-initiation on a new mRNA, and anti-tmRNA oligonucleotide complementary to the tmRNA sequence is added to suppress the release of EmGFP from the stalled ribosome after translation of the last codon (6,36). The results (Table 1, Figure 1B) show higher total fluorescent EmGFP production for circular plasmid than for linearized plasmid, in accord with an earlier report (22). Addition of anti-tmRNA to the linearized plasmid further reduces EmGFP production. Here the slightly higher value of total mature EmGFP synthesized (0.36/ribosome) vs. mature EmGFP cosedimenting with ribosomes (0.29/ribosome) arises from some combination of incomplete suppression of EmGFP release (i.e. a small number of ribosomes expressing multiple EmGFPs) and/or dissociation of some ribosome-bound EmGFP during either the reaction or ultracentrifugation. The fraction of active ribosomes (0.29), as measured by co-sedimenting fluorescent EmGFP, provides an important benchmark for evaluating the quality of different preparations and/or batches of CFPS kits, and of exogenous ribosomes that are substituted for endogenous ribosomes (see below). For other batches of the CFPS kit this fraction varied from 0.18 to 0.33, which is in reasonable agreement with previously reported results for SecM-stalled ribosomes with a protein of similar length (6) and somewhat higher than found in a previous single molecule experiment using similar methods (26).
Table 1.
Determination of fraction of active ribosome
| Plasmid | Circular | Linear | Linear |
|---|---|---|---|
| Anti tmRNA | − | − | + |
| Total EmGFP/ribosome | 1.62 ± 0.10 | 0.76 ± 0.05 | 0.36 ± 0.01 |
| Co-sedimenting EmGFP/ ribosome | 0.10 ± 0.01 | 0.05 ± 0.01 | 0.29 ± 0.01 |
| Co-sedimenting EmGFP/total EmGFP | 0.06 ± 0.01 | 0.06 ± 0.01 | 0.79 ± 0.02 |
All measurements were performed in triplicate and reported as means ± SD. EmGFP stoichiometry is estimated by fluorescence intensity, using the calibration curve shown in Supplementary Figure S1. Ribosome stoichiometry is estimated by 26 pmol/A260.
Substitution of exogenous ribosomes for endogenous ribosomes
High speed centrifugation can be used to remove endogenous ribosomes from the CFPS kit, resulting in a preparation, denoted the CFPS-ribosome kit, that is inactive in the EmGFP expression assay, with activity restored by addition of exogenous ribosomes. We examined the restoration of activity as a function of the extent of ribosome purification (Figure 2A and ‘Materials and Methods’ section), using the circular EmGFP plasmid, and determined that minimal purification, consisting of ribosome pelleting by high-speed centrifugation of crude S30 lysate, gave the highest restored activity. Accordingly this preparation, denoted S100P ribosomes, was used in assays comparing the activities of several exogenously added ribosomes, including wild-type MRE600 ribosomes, AM77 ribosomes lacking protein L11 (37) and AM77 ribosomes reconstituted with either wild-type L11 (AM77L11) or with an L11 variant labeled stoichiometrically at position 87 with Cy3 (AM77Cy3L11) (11) (Figure 2B). These results, showing that the lower activity of the AM77 ribosomes is partially restored by reconstitution with either wild-type or Cy3-labeled L11, indicate that derivatization of L11 with Cy3 does not interfere with translational activity, in accord with earlier results measuring other ribosomal activities (9,38,39). Addition of initiation factors improves the activity of these exogenously added ribosome preparations (Supplementary information 2B and Supplementary Figure S3A and B).
Figure 2.

Cell-free expression of EmGFP on addition of exogenous ribosomes (1.5 μM) to the CFPS-ribosome kit. Experiments were performed with ribosome preparations dissolved in TAM15 buffer mixed with CFPS reconstituted with the vendor-provided buffer, using the plate reader. (A) As a function of MRE600 ribosome purification: centrifugation of crude S30 lysate directly (S100P); through a sucrose cushion (S100sucP); small-scale (70S-ss) and large-scale (70S-ls) 70S purification via sucrose density-gradient centrifugation. A control ribosome-depleted sample (CFPS-Rib) contained no added ribosomes. (B) S100P preparations of MRE600 and AM77 ribosomes. All samples contained the CFPS-ribosome kit. Due to the spectral overlap of Cy3 with EmGFP, the AM77Cy3L11 trace exhibits slightly higher background. Experiments, repeated five times using different CFPS batches, gave reproducible results.
For the CFPS kit used for the experiments shown in Figure 2B, 0.18 ± 0.02 of the endogenous ribosomes were active (determined as described above using the linearized EmGFP plasmid in the presence of anti-tmRNA), compared with 0.32 ± 0.05 of the exogenously added MRE600 ribosomes and 0.09 ± 0.01 and 0.10 ± 0.04, respectively, for exogenously added AM77 and AM77L11 ribosomes. The lower fractions of active AM77-derived ribosomes account for the lower levels of EmGFP synthesis observed with these ribosomes (Figure 2B), although the intrinsic translation rate of active AM77-derived ribosomes is similar to that of wild-type ribosomes (see Supplementary Information 2A, and Supplementary Figure S2).
Substitution of exogenous for endogenous charged tRNAs
We next modified the EmGFP assay to permit comparison of the activities of added exogenous charged tRNA in supporting EmGFP expression. Below we detail our results with Phe-tRNAPhe, although the approach is general and has also been applied successfully to Val-tRNAVal and Lys-tRNALys. These experiments were performed using the circular EmGFP plasmid.
The principal challenge in the development of this modified assay was to prepare a CFPS-Phe kit (i.e. a kit depleted of all forms of Phe) such that synthesis of mature EmGFP was completely dependent on added exogenous Phe-tRNAPhe or Phe. This was accomplished by: (i) dialysis of the commercial CFPS lysate to remove all small molecules, including the amino acids; (ii) specific inhibition of Phe-amino-acyl tRNA synthetase (Phe-RS) with an active-site directed inhibitor; (iii) addition of a general protease inhibitor to prevent generation of Phe from protein breakdown; and (iv) adding back all of the small molecules required for protein synthesis with the exception of Phe (Supplementary Figure S3C). The ability of the resulting CFPS-Phe kit to produce detectable amounts of EmGFP was totally dependent on adding either exogenous yeast Phe-tRNAPhe (Figure 3A, Supplementary Figure S4A—similar results were obtained for E. coli Phe-tRNAPhe; Supplementary Figure S4B) or Phe (Figure 3B) with maximal EmGFP expression requiring higher concentrations of Phe (80 µM) than of Phe-tRNAPhe (8–10 μM) (Figure 3C and D).
Figure 3.

Cell-free expression of EmGFP using the CFPS-Phe kit. Experiments were performed in Polyamine buffer, using the plate reader. (A) As a function of added [Phe-tRNAPhe]. The dashed lines are derived from the numerical simulation model described in the text and in Supplementary Data. The active ribosome concentration was 0.58 ± 0.04 μM. Deviations from simulated values are attributable to various oversimplifications within the model, as well as to inhibitory effects expected at high concentrations of labeled deacylated tRNAPhe (see Supplementary Data and Figure S3E). (B) As a function of added [Phe]. (C and D) Dependence of normalized parameters tL, S1, A1 and S2 (Table 2) on [Phe-tRNAPhe] or [Phe], respectively. Solid symbols, measured values. Open symbols, values derived from the simulations of the various Phe-tRNAPhe concentrations.
The curves of EmGFP fluorescence in Figure 3A and B can be characterized by four parameters, a lag phase corresponding to all dark events preceding EmGFP maturation (tL), a region of high slope reflecting the maximum rate of fluorescent EmGFP formation (phase-1-slope, S1), the total fluorescent EmGFP synthesized in the first, rapid phase of reaction (phase-1-amplitude, A1), and a final region of lower slope (phase-2-slope, S2) corresponding to a slower, second phase. Each of the latter three parameters shows a strong Phe-tRNAPhe or Phe concentration dependence (Figure 3C and D, respectively). Both S1 and A1 increase nearly monotonically before reaching saturated values. S2 increases monotonically over the limited range of Phe-tRNAPhe concentrations employed, but goes through a maximum over the broader range of Phe concentration that was explored. In contrast, tL has only minor dependence on either Phe-tRNAPhe or Phe concentration. The overall maximal activities of the CFPS-Phe kit with added Phe-tRNAPhe (8-10 µM) or Phe (80 µM) were similar to one another (Supplementary Figure S3D), but reduced as compared with the unmodified CFPS kit, showing similar tL values but 2- to 4-fold reductions in the values of S1, S2, and A1 (Supplementary Table S1).
The CFPS-Phe kit makes very efficient use of added Phe-tRNAPhe. For a CFPS-Phe kit containing a total active ribosome concentration of 0.6 µM, saturation of Phase-1-formation was reached by ∼8 μM yeast Phe-tRNAPhe (Figure 3C). As the encoded EmGFP has 14 Phe residues (2 in the N-terminal linker sequence and 12 in the original gene) the value of 8 µM of added Phe-tRNAPhe found at saturation, in addition to the residual endogenous Phe-tRNAPhe (2 µM) that we estimate to be present in the CFPS-Phe kit (see below), is just slightly above the expected stoichiometric requirement of 8 µM Phe-tRNAPhe needed to produce one EmGFP per active ribosome. This result indicates an apparent Km for Phe-tRNAPhe incorporation at cognate codons that is <1 µM, in accord with results derived from simpler model sytems (10,40,41). The apparent Km for Phe, estimated from the concentration dependence of either S1 or A1, is ∼10–15 µM. This value, which represents the first reported estimation of Km for an amino acid in a CFPS system, is considerably higher than the value for Phe-RS (2 µM) determined in a highly purified system (42), perhaps reflecting competing binding to Phe-RS in the CFPS-Phe kit by near cognate amino acids and/or added Phe-RS inhibitor.
Numerical simulations of multi-step EmGFP synthesis
We developed a numerical simulation of a reaction kinetics model in order to gain additional understanding about the effects of exogenous Phe-tRNAPhe (see below) on the formation of EmGFP fluorescence using the CFPS-Phe kit (Figure 3A). This model includes 315 steps, which proceed in series, and include: a transcription initiation step, a transcription elongation step, a translation initiation step, 310 translation elongation steps, utilizing 296 non-Phe aminoacyl-tRNAs and 14 Phe-tRNAPhes according to the EmGFP sequence, and two fluorophore maturation steps (35). Each non-Phe translation elongation step was assigned the same pseudo-first-order rate constant (k1) and each Phe translation elongation step was assigned a second-order rate constant, k2 for endogenous Phe-tRNAPhe and 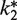 for exogenous Phe-tRNAPhe. The pseudo-first-order rate process with rate constant k1 reflects the large excess of each of the non-Phe aminoacyl-tRNAs present in the CFPS-Phe kit while the second-order process, k2, reflects the limited availability of the added Phe-tRNAPhe.
for exogenous Phe-tRNAPhe. The pseudo-first-order rate process with rate constant k1 reflects the large excess of each of the non-Phe aminoacyl-tRNAs present in the CFPS-Phe kit while the second-order process, k2, reflects the limited availability of the added Phe-tRNAPhe.
In order to account for the rather small observed threshold value of exogenously added Phe-tRNAPhe (∼1 µM) that has to be exceeded in order to detect EmGFP fluorescence (Figure 3C), the model includes endogenous Phe-tRNAPhe that is not completely removed in preparing the CFPS-Phe kit and functionally heterogeneous ribosomes. If all of the ribosomes were equally active, small amounts of added Phe-tRNAPhe would be consumed by synthesis of incomplete, non-fluorescent chains, leading to a considerably higher threshold Phe-tRNAPhe concentration (Supplementary Figure S6). A final assumption is that protease and Phe-RS activities are incompletely inhibited in the CFPS-Phe kit, allowing a continual slow regeneration of the limiting reagent, Phe-tRNAPhe, that results in the slow second phase of EmGFP formation.
The kinetic model was used to simulate actual results obtained on adding varying Phe-tRNAPhe concentrations to the CFPS-Phe kit, as described below. Further details on the simulation may be found in Supplementary Information 4A.
Assaying the activity of labeled Phe-tRNAPhe variants by EmGFP formation
We used the CFPS-Phe kit to compare the relative activities of various Phe-tRNAPhe variants in EmGFP synthesis. The Phe-tRNAPhes examined were E. coli and yeast, either unlabeled or labeled at dihydroU residues (16 and 17 in yeast or 16 and 20 in E. coli tRNAPhe) to a total stoichiometry of 0.8 – 0.9 fluorophores/tRNA (Table 2) with either Cy3, Cy5, or Cy5.5 (10). Variants were examined at one to three concentrations over a concentration range, 1–4 µM (Figure 4A), for which both S1 and A1 are highly dependent on Phe-tRNAPhe concentration (Figure 3C).
Table 2.
Phe-tRNAPhe variant parametersa
| E. coli | E. coli Cy3 | E. coli Cy5 | E. coli Cy5.5 | Yeast | Yeast Cy3 | Yeast Cy5 | |
|---|---|---|---|---|---|---|---|
| Phe-tRNAPhe preparation (%) | |||||||
| Labeling | 80 | 90 | 79 | 92 | 93 | ||
| Charging | 28 | 25 | 15 | 26 | 44 | 37 | 30 |
| Empirical parameters | |||||||
| tLb (2 µM) | 1.00 ± 0.05 | 1.47 ± 0.11 | 1.13 ± 0.22 | 1.13 ± 0.08 | 1.20 ± 0.11 | 1.27 ± 0.11 | 2.33 ± 0.15 |
| tLb (4 µM) | 1.27 ± 0.11 | 1.67 ± 0.04 | 1.07 ± 0.08 | NA | 1.07 ± 0.07 | 1.18 ± 0.11 | 2.27 ± 0.25 |
| S1c (2 µM) | 1.00 ± 0.22 | 0.48 ± 0.17 | 0.42 ± 0.04 | 0.71 ± 0.19 | 0.79 ± 0.04 | 0.42 ± 0.02 | 0.21 ± 0.02 |
| S1c (4 µM) | 2.34 ± 0.06 | 0.60 ± 0.07 | 0.43 ± 0.04 | NA | 1.43 ± 0.05 | 0.50 ± 0.03 | 0.09 ± 0.01 |
| A1d (2 µM) | 1.00 ± 0.09 | 0.87 ± 0.10 | 0.35 ± 0.05 | 0.55 ± 0.17 | 1.02 ± 0.04 | 0.68 ± 0.01 | 0.38 ± 0.02 |
| A1d (4 µM) | 2.89 ± 0.06 | 1.52 ± 0.09 | 0.81 ± 0.03 | NA | 2.41 ± 0.01 | 1.24 ± 0.06 | 0.50 ± 0.02 |
| S2e (2 µM) | 1.00 ± 0.14 | 1.17 ± 0.15 | 0.63 ± 0.07 | 0.37 ± 0.18 | 1.14 ± 0.08 | 0.71 ± 0.09 | 0.68 ± 0.04 |
| S2e (4 µM) | 2.61 ± 0.12 | 1.54 ± 0.12 | 0.78 ± 0.11 | NA ± | 1.74 ± 0.03 | 1.03 ± 0.05 | 0.73 ± 0.09 |
| Relative 14C-Phe incorporationf | |||||||
| 1.00 ± 0.02 | 0.98 ± 0.02 | 0.13 ± 0.01 | NA | 0.66 ± 0.01 | 0.43 ± 0.02 | 0.12 ± 0.01 | |
| Simulation | |||||||
Relative 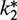
|
1g | 0.1 | 0.025 | 0.2 | 1g | 0.05 | 0.025 |
| Phase 2 steady-state Phe-tRNAPhe concentration (µM) | 0.1 | 0.1 | 0.01 | 0.01 | 0.15 | 0.025 | 0 |
aData at 2 µM added Phe-tRNAPhe are presented in Figure 4B and C. Parallel results obtained at 4 µM Phe-tRNAPhe are summarized in the table. Experiments were performed using 2–5 repeats. Data obtained with 2 µM E. coli tRNA variants was repeated with two different CFPS batches. All values were normalized to 2 µM E. coli Phe-tRNAPhe.
bLag time.
cPhase-1-slope.
dPhase-1-amplitude.
ePhase-2-slope.
fAfter 30-min incubation.
gEqual to k2.
Figure 4.

Comparisons of cell-free expression of EmGFP on addition of Phe-tRNAPhe variants to the CFPS-Phe kit. EmGFP fluorescence measurements contained an active ribosome concentration of 0.30 ± 0.02 μM. (A) Various concentrations of unlabeled E. coli and yeast Phe-tRNAPhe. (B) Escherichia coli variants (2 µM). (C) Yeast variants (2 µM). Unlabeled E. coli Phe-tRNAPhe is included in each panel for ease of comparison. The dashed lines are derived from the numerical simulation model using the parameter values listed in Table 2 and given in the text.
Although the simulation model described above adequately fits the data using identical values of 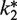 for both yeast and E. coli exogenous unlabeled Phe-tRNAPhes (equal to the k2 value for endogenous Phe-tRNAPhe), some variability in individual head-to-head comparisons caused slight differences in apparent activity, as is evident from Figure 4A. In contrast, the labeled Phe-tRNAPhes were consistently less active than the unlabeled Phe-tRNAPhes with respect to values of both S1 and A1 (Figure 4B and C, Table 2). A large increase in tL is associated with the use of Cy5-labeled yeast Phe-tRNAPhe, whereas tL is not strongly affected by the other Phe-tRNAPhes. The relative activities of labeled Phe-tRNAPhes are reduced more strongly at higher concentrations (Table 2), suggesting that high concentrations of the labeled Phe-tRNAPhe preparations are inhibitory. Such inhibition could be due, at least in part, to an increase in uncharged, labeled tRNAs in these preparations (Supplementary Figure S3E) that may offset the direct effect of raising Phe-tRNAPhe concentration.
for both yeast and E. coli exogenous unlabeled Phe-tRNAPhes (equal to the k2 value for endogenous Phe-tRNAPhe), some variability in individual head-to-head comparisons caused slight differences in apparent activity, as is evident from Figure 4A. In contrast, the labeled Phe-tRNAPhes were consistently less active than the unlabeled Phe-tRNAPhes with respect to values of both S1 and A1 (Figure 4B and C, Table 2). A large increase in tL is associated with the use of Cy5-labeled yeast Phe-tRNAPhe, whereas tL is not strongly affected by the other Phe-tRNAPhes. The relative activities of labeled Phe-tRNAPhes are reduced more strongly at higher concentrations (Table 2), suggesting that high concentrations of the labeled Phe-tRNAPhe preparations are inhibitory. Such inhibition could be due, at least in part, to an increase in uncharged, labeled tRNAs in these preparations (Supplementary Figure S3E) that may offset the direct effect of raising Phe-tRNAPhe concentration.
Because of the apparent inhibition at high labeled Phe-tRNAPhe concentrations, only the time-courses of fluorescent EmGFP formation using 2 µM exogenous labeled Phe-tRNAPhe (Figure 4B and C) were fit with the kinetic simulation model. The resulting 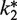 values (Table 2) show that the activities of the labeled tRNAs, relative to E. coli Phe-tRNAPhe, fall in the order Cy5.5 (5-fold reduction) > Cy3 (10-fold reduction) > Cy5 (40–fold reduction). The order of activity found with these dyes suggests that activity loss results from a combination of steric bulk and hydrophobicity, since Cy3 is both smaller and less hydrophobic than Cy5, whereas Cy5.5, a dianion at neutral pH, is both larger and less hydrophobic than Cy5.
values (Table 2) show that the activities of the labeled tRNAs, relative to E. coli Phe-tRNAPhe, fall in the order Cy5.5 (5-fold reduction) > Cy3 (10-fold reduction) > Cy5 (40–fold reduction). The order of activity found with these dyes suggests that activity loss results from a combination of steric bulk and hydrophobicity, since Cy3 is both smaller and less hydrophobic than Cy5, whereas Cy5.5, a dianion at neutral pH, is both larger and less hydrophobic than Cy5.
Assaying the activity of Phe-tRNAPhe variants by [14C]-Phe incorporation
Plasmid-dependent [14C]-Phe incorporation into polypeptide during EmGFP synthesis provides an alternative measure of the relative activities of labeled Phe-tRNAPhe in protein synthesis. [14C]-Phe incorporation is less stringent than the EmGFP fluorescence assay, since it measures synthesis of partial and full-length EmGFP chains regardless of correct folding or chromophore maturation. Indeed, for the results presented in Table 2 using unlabeled E. coli or yeast Phe-tRNAPhe, only 35–40% of the [14C]-Phe incorporated into precipitable peptide chains was incorporated in full-length fluorescent EmGFP protein in the first 30 min of the reaction, as determined by carrying out both analyses on the same sample. The relative [14C]-Phe incorporation levels are consistent with relative activities observed in ensemble measurements of polyPhe synthesis and single molecule translation of short stretches of model mRNAs using these same labeled tRNAs (10,11). Qualitatively, the relative [14C]-Phe incorporation levels showing Phe-tRNAPhe (Cy3) to be more active than Phe-tRNAPhe (Cy5) for both E. coli and yeast tRNAs parallel the order of relative activities found in the fluorescence assay measured either by 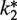 or A1 values (Table 2).
or A1 values (Table 2).
DISCUSSION
Here we describe a convenient, fluorescence-based assay that, via adaptation of a commercially available bacterial CFPS kit, permits quantitative determination of the effects on the rate and stoichiometry of fluorescent EmGFP expression of replacing endogenous with exogenous components of the protein synthesis machinery. An important adjunct to this assay is the quantification of the fraction of active ribosomes in either the CFPS kit itself, or in a sample from which endogenous ribosomes have been removed (the CFPS-ribosome kit) and replaced. Approximately 20–30% of the ribosomes in the commercial CFPS kit are active in fluorescent EmGFP expression. By comparison, wild-type MRE600 ribosomes prepared by us show ∼30% activity, whereas the fractional activities of the mutant AM77 ribosomes which lack protein L11, even when supplemented with L11, are distinctly lower (∼10%).
As described above, replacement of endogenous ribosomes is quite facile, since exogenous ribosomes prepared directly from crude cell extracts (‘S100P’ ribosomes) confer the highest activity when added to the easily prepared CFPS-ribosome kit. Replacement of endogenous Phe-tRNAPhe is somewhat more demanding, but relatively straightforward using the optimized procedures detailed in ‘Materials and Methods’ section. In order to selectively remove Phe-tRNAPhe, we elected to prepare a CFPS-Phe kit in which the endogenous tRNAPhe is retained, but Phe is depleted and Phe-RS is inhibited. With the CFPS-Phe kit, fluorescent EmGFP expression is totally dependent on either added Phe (Figure 3B) or added Phe-tRNAPhe, at much lower concentration (Figure 3A). An alternative approach for preparing a CFPS-Phe system, which we are currently pursuing, utilizes PURExpress® (New England Biolabs), a cell-free transcription/translation system reconstituted from purified E. coli components, including each of the tRNA synthetase enzymes (43). A custom-made PURExpress® mixture should essentially eliminate the residual Phe-tRNAPhe and Phe-RS content present in a CFPS-Phe kit.
Our primary goal in developing adaptations of CFPS kit assay for the expression of fluorescent EmGFP was to permit determination of the effects on translational activity of fluorescent labeling of components of the protein synthesis machinery, in particular ribosomes and tRNAs, since such labeling is used extensively in both ensemble and single molecule studies of the mechanism of protein synthesis (12–15). The results showing that dye-labeled Phe-tRNAPhes have substantially lower activity than unlabeled Phe-tRNAPhe in supporting full-length protein synthesis highlight the need to explore alternative labeling strategies to produce labeled tRNAs with higher intrinsic activities. Such strategies could include using tRNA transcripts so as to be able to direct labeling to a unique DHU position (44) as opposed to native tRNAs, for which labeling is distributed over all DHU positions (45,46); using less hydrophobic fluorophores; and introducing fluorophores at other than DHU positions (47–49).
In addition to meeting our primary goal, continuous monitoring of the formation of fluorescent EmGFP by simple or modified CFPS kits provides a general, radioactivity-free approach, amenable to high-throughput screening, for evaluating effects on the efficiency of protein synthesis of various additions, subtractions or substitutions to the protein synthesis machinery. These include (i) any kind of tRNA, rRNA or ribosomal protein variation, including mutation or enzymatic or chemical modification; (ii) addition of agents affecting transcription, translation, or protein folding (e.g. antibiotic screening effects on translation could be separated from those on transcription by initiating EmGFP synthesis with added mRNA, transcribed separately, rather than with added DNA); (iii) substituting amino acid analogues for native amino acids, whether measured by supplementing a CFPS-aminoacid kit with the amino acid analogue or with a tRNA charged with the analogue (50). In some experiments, suitable controls would be needed to exclude possible artifacts due to direct effects on EmGFP fluorescence. Finally, a CFPS-aminoacid kit, reconstituted with an isotopically labeled amino acid, could be used to prepare isotopically labeled proteins for IR and NMR studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables S1–S3, Supplementary Figures S1–S6, Supplementary Information Files S1–S2 and Supplementary Reference [51].
FUNDING
National Institutes of Health (GM080376 to B.S.C., Y.E.G.); Human Frontier Science Program fellowship (to G.R.). Funding for open access charge: National Institutes of Health (GM080376).
Conflict of interest statement. Y.E.G. and B.S.C. are paid consultants for Anima Cell Metrology.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Olga Lozynska in Professor Scott Diamond’s group at Penn for her help with the plate reader.
REFERENCES
- 1.Zubay G. In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 1973;7:267–287. doi: 10.1146/annurev.ge.07.120173.001411. [DOI] [PubMed] [Google Scholar]
- 2.Schwarz D, Junge F, Durst F, Frölich N, Schneider B, Reckel S, Sobhanifar S, Dötsch V, Bernhard F. Preparative scale expression of membrane proteins in Escherichia coli-based continuous exchange cell-free systems. Nat. Protoc. 2007;2:2945–2957. doi: 10.1038/nprot.2007.426. [DOI] [PubMed] [Google Scholar]
- 3.Langer M, Rothe M, Eck J, Mockel B, Zinke H. A nonradioactive assay for ribosome-inactivating proteins. Anal. Biochem. 1996;243:150–153. doi: 10.1006/abio.1996.0493. [DOI] [PubMed] [Google Scholar]
- 4.Brandi L, Fabbretti A, Milon P, Carotti M, Pon CL, Gualerzi CO. Methods for identifying compounds that specifically target translation. Meth. Enzymol. 2007;431:229–267. doi: 10.1016/S0076-6879(07)31012-4. [DOI] [PubMed] [Google Scholar]
- 5.Jefferson EA, Arakawa S, Blyn LB, Miyaji A, Osgood SA, Ranken R, Risen LM, Swayze EE. New inhibitors of bacterial protein synthesis from a combinatorial library of macrocycles. J. Med. Chem. 2002;45:3430–3439. doi: 10.1021/jm010437x. [DOI] [PubMed] [Google Scholar]
- 6.Evans MS, Ugrinov KG, Frese MA, Clark PL. Homogeneous stalled ribosome nascent chain complexes produced in vivo or in vitro. Nat. Methods. 2005;2:757–762. doi: 10.1038/nmeth790. [DOI] [PubMed] [Google Scholar]
- 7.Underwood KA, Swartz JR, Puglisi JD. Quantitative polysome analysis identifies limitations in bacterial cell-free protein synthesis. Biotechnol. Bioeng. 2005;91:425–435. doi: 10.1002/bit.20529. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Pan D, Pech M, Cooperman BS. Interrupted catalysis: the EF4 (LepA) effect on back-translocation. J. Mol. Biol. 2011;396:1043–1052. doi: 10.1016/j.jmb.2009.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin H, Grigoriadou C, Cooperman BS. Interaction of IF2 with the ribosomal GTPase-associated center during 70S initiation complex formation. Biochemistry. 2009;48:4699–4706. doi: 10.1021/bi900222e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan D, Qin H, Cooperman BS. Synthesis and functional activity of tRNAs labeled with fluorescent hydrazides in the D-loop. RNA. 2009;15:346–354. doi: 10.1261/rna.1257509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen C, Stevens B, Kaur J, Cabral D, Liu H, Wang Y, Zhang H, Rosenblum G, Smilansky Z, Goldman YE, et al. Single-molecule fluorescence measurements of ribosomal translocation dynamics. Mol. Cell. 2011;42:367–377. doi: 10.1016/j.molcel.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanchard SC. Single-molecule observations of ribosome function. Curr. Opin. Struct. Biol. 2009;19:103–109. doi: 10.1016/j.sbi.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frank J, Gonzalez RL., Jr Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu. Rev. Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Korostelev A, Ermolenko DN, Noller HF. Structural dynamics of the ribosome. Curr. Opin. Chem. Biol. 2008;12:674–683. doi: 10.1016/j.cbpa.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrov A, Kornberg G, O'Leary S, Tsai A, Uemura S, Puglisi JD. Dynamics of the translational machinery. Curr. Opin. Struct. Biol. 2011;21:137–145. doi: 10.1016/j.sbi.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iskakova MB, Szaflarski W, Dreyfus M, Remme J, Nierhaus KH. Troubleshooting coupled in vitro transcription-translation system derived from Escherichia coli cells: synthesis of high-yield fully active proteins. Nucleic Acids Res. 2006;34:e135. doi: 10.1093/nar/gkl462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mureev S, Kovtun O, Nguyen UT, Alexandrov K. Species-independent translational leaders facilitate cell-free expression. Nat. Biotechnol. 2009;27:747–752. doi: 10.1038/nbt.1556. [DOI] [PubMed] [Google Scholar]
- 18.Zubay G, Lederman M, DeVries JK. DNA-directed peptide synthesis. 3. Repression of beta-galactosidase synthesis and inhibition of repressor by inducer in a cell-free system. Proc. Natl Acad. Sci. USA. 1967;58:1669–1675. doi: 10.1073/pnas.58.4.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DM, Swartz JR. Regeneration of adenosine triphosphate from glycolytic intermediates for cell-free protein synthesis. Biotechnol. Bioeng. 2001;74:309–316. [PubMed] [Google Scholar]
- 20.DeVries JK, Zubay G. DNA-directed peptide synthesis. II. The synthesis of the alpha-fragment of the enzyme beta-galactosidase. Proc. Natl Acad. Sci. USA. 1967;57:1010–1012. doi: 10.1073/pnas.57.4.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jelenc PC, Kurland CG. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc. Natl Acad. Sci. USA. 1979;76:3174–3178. doi: 10.1073/pnas.76.7.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kudlicki W, Kramer G, Hardesty B. High efficiency cell-free synthesis of proteins: refinement of the coupled transcription/translation system. Anal. Biochem. 1992;206:389–393. doi: 10.1016/0003-2697(92)90383-i. [DOI] [PubMed] [Google Scholar]
- 23.Tsien RY. The green fluorescent protein. Ann. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann M, Nemetz C, Madin K, Buchberger B. Rapid translation system: a novel cell-free way from gene to protein. Biotechnol. Annu. Rev. 2004;10:1–30. doi: 10.1016/S1387-2656(04)10001-X. [DOI] [PubMed] [Google Scholar]
- 25.Cooperman BS, Jaynes EN, Brunswick DJ, Luddy MA. Photoincorporation of puromycin and N-(ethyl-2-diazomalonyl)puromycin into Escherichia coli ribosomes. Proc. Natl Acad. Sci. USA. 1975;72:2974–2978. doi: 10.1073/pnas.72.8.2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katranidis A, Atta D, Schlesinger R, Nierhaus KH, Choli-Papadopoulou T, Gregor I, Gerrits M, Büldt G, Fitter J. Fast biosynthesis of GFP molecules: a single-molecule fluorescence study. Angew. Chem. Int. Ed. Engl. 2009;48:1758–1761. doi: 10.1002/anie.200806070. [DOI] [PubMed] [Google Scholar]
- 27.Rodnina MV, Wintermeyer W. GTP consumption of elongation factor Tu during translation of heteropolymeric mRNAs. Proc. Natl Acad Sci USA. 1995;92:1945–1949. doi: 10.1073/pnas.92.6.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olah TV, Olson HM, Glitz DG, Cooperman BS. Incorporation of single dinitrophenyl-modified proteins into the 30 S subunit of Escherichia coli ribosomes by total reconstitution. J. Biol. Chem. 1988;263:4795–4800. [PubMed] [Google Scholar]
- 29.Semenkov YP, Rodnina MV, Wintermeyer W. The “allosteric three-site model” of elongation cannot be confirmed in a well-defined ribosome system from Escherichia coli. Proc. Natl Acad Sci. USA. 1996;93:12183–12188. doi: 10.1073/pnas.93.22.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fei J, Kosuri P, MacDougall DD, Gonzalez RL., Jr Coupling of ribosomal L1 stalk and tRNA dynamics during translation elongation. Mol. Cell. 2008;30:348–359. doi: 10.1016/j.molcel.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Dinos G, Kalpaxis DL, Wilson DN, Nierhaus KH. Deacylated tRNA is released from the E site upon A site occupation but before GTP is hydrolyzed by EF-Tu. Nucleic Acids Res. 2005;33:5291–5296. doi: 10.1093/nar/gki833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santi DV, Cunnion SO, Anderson RT, Webster RW., Jr In vivo inhibitors of Escherichia coli phenylalanyl-tRNA synthetase. J. Med. Chem. 1979;22:1260–1263. doi: 10.1021/jm00196a021. [DOI] [PubMed] [Google Scholar]
- 33.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat. Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 34.Tu GF, Reid GE, Zhang JG, Moritz RL, Simpson RJ. C-terminal extension of truncated recombinant proteins in Escherichia coli with a 10Sa RNA decapeptide. J. Biol. Chem. 1995;270:9322–9326. doi: 10.1074/jbc.270.16.9322. [DOI] [PubMed] [Google Scholar]
- 35.Gilmore R, Collins P, Johnson J, Kellaris K, Rapiejko P. Transcription of full-length and truncated mRNA transcripts to study protein translocation across the endoplasmic reticulum. Methods Cell Biol. 1991;34:223–239. doi: 10.1016/s0091-679x(08)61683-0. [DOI] [PubMed] [Google Scholar]
- 36.Hanes J, Pluckthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl Acad Sci. USA. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dabbs ER. Selection for Escherichia coli mutants with proteins missing from the ribosome. J. Bacteriol. 1979;140:734–737. doi: 10.1128/jb.140.2.734-737.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seo HS, Abedin S, Kamp D, Wilson DN, Nierhaus KH, Cooperman BS. EF-G-dependent GTPase on the ribosome. conformational change and fusidic acid inhibition. Biochemistry. 2006;45:2504–2514. doi: 10.1021/bi0516677. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Qin H, Kudaravalli RD, Kirillov SV, Dempsey GT, Pan D, Cooperman BS, Goldman YE. Single-molecule structural dynamics of EF-G–ribosome interaction during translocation. Biochemistry. 2007;46:10767–10775. doi: 10.1021/bi700657d. [DOI] [PubMed] [Google Scholar]
- 40.Gromadski KB, Rodnina MV. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell. 2004;13:191–200. doi: 10.1016/s1097-2765(04)00005-x. [DOI] [PubMed] [Google Scholar]
- 41.Uemura S, Aitken CE, Korlach J, Flusberg BA, Turner SW, Puglisi JD. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature. 2010;464:1012–1017. doi: 10.1038/nature08925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gabius HJ, von der Haar F, Cramer F. Evolutionary aspects of accuracy of phenylalanyl-tRNA synthetase. A comparative study with enzymes from Escherichia coli, Saccharomyces cerevisiae, Neurospora crassa, and turkey liver using phenylalanine analogues. Biochemistry. 1983;22:2331–2339. doi: 10.1021/bi00279a005. [DOI] [PubMed] [Google Scholar]
- 43.Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 44.Betteridge T, Liu H, Gamper H, Kirillov S, Cooperman BS, Hou YM. Fluorescent labeling of tRNAs for dynamics experiments. RNA. 2007;13:1594–1601. doi: 10.1261/rna.475407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaur J, Raj M, Cooperman BS. Fluorescent labeling of tRNA dihydrouridine residues: Mechanism and distribution. RNA. 2011;17:1393–1400. doi: 10.1261/rna.2670811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wintermeyer W, Zachau HG. Fluorescent derivatives of yeast tRNAPhe. Eur. J. Biochem. 1979;98:465–475. doi: 10.1111/j.1432-1033.1979.tb13207.x. [DOI] [PubMed] [Google Scholar]
- 47.Haller A, Souliére MF, Micura R. The dynamic nature of RNA as key to understanding riboswitch mechanisms. Acc. Chem. Res. 2011;44:1339–1348. doi: 10.1021/ar200035g. [DOI] [PubMed] [Google Scholar]
- 48.Motorin Y, Burhenne J, Teimer R, Koynov K, Willnow S, Weinhold E, Helm M. Expanding the chemical scope of RNA:methyltransferases to site-specific alkynylation of RNA for click labeling. Nucleic Acids Res. 2011;39:1943–1952. doi: 10.1093/nar/gkq825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333:642–646. doi: 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murakami H, Saito H, Suga H. A versatile tRNA aminoacylation catalyst based on RNA. Chem. Biol. 2003;10:655–662. doi: 10.1016/s1074-5521(03)00145-5. [DOI] [PubMed] [Google Scholar]
- 51.Teerawanichpan P, Hoffman T, Ashe P, Datla R, Selvaraj G. Investigations of combinations of mutations in the jellyfish green fluorescent protein (GFP) that afford brighter fluorescence, and use of a version (VisGreen) in plant, bacterial, and animal cells. Biochim. Biophys. Acta. 2007;1770:1360–1368. doi: 10.1016/j.bbagen.2007.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.