

Abstract
Objective:
Succinic semialdehyde dehydrogenase (SSADH) deficiency is a rare autosomal recessive disorder of GABA degradation leading to elevations in brain GABA and γ-hydroxybutyric acid (GHB). The effect of chronically elevated GABA and GHB on cortical excitability is unknown. We hypothesized that use-dependent downregulation of GABA receptor expression would promote cortical disinhibition rather than inhibition, predominantly via presynaptic GABAergic mechanisms.
Methods:
We quantified the magnitude of excitation and inhibition in primary motor cortex (M1) in patients with SSADH deficiency, their parents (obligate heterozygotes), age-matched healthy young controls, and healthy adults using single and paired pulse transcranial magnetic stimulation (TMS).
Results:
Long interval intracortical inhibition was significantly reduced and the cortical silent period was significantly shortened in patients with SSADH deficiency compared to heterozygous parents and control groups.
Conclusions:
Since long interval intracortical inhibition and cortical silent period are thought to reflect GABAB receptor–mediated inhibitory circuits, our results point to a particularly GABAB-ergic motor cortex dysfunction in patients with SSADH deficiency. This human phenotype is consistent with the proposed mechanism of use-dependent downregulation of postsynaptic GABAB receptors in SSADH deficiency animal models. Additionally, the results suggest autoinhibition of GABAergic neurons. This first demonstration of altered GABAB-ergic function in patients with SSADH deficiency may help to explain clinical features of the disease, and suggest pathophysiologic mechanisms in other neurotransmitter-related disorders. Neurology® 2012;79:47–54
Succinic semialdehyde dehydrogenase (SSADH) deficiency is a rare autosomal recessive neurologic disorder, in which an enzyme defect in the GABA degradation pathway causes a consecutive elevation of γ-hydroxybutyric acid (GHB) and GABA.1 The clinical symptoms include developmental delay, hypotonia, mental retardation, ataxia, seizures, hyperkinetic behavior, aggression, and sleep disturbances.2–4 GHB is known to activate GHB receptors5–8 and in higher concentration GABAB receptors, without substantial binding to GABAA receptors.9,10 In SSADH knockout mice, use-dependent downregulation of (predominantly postsynaptic) GABAB but not GHB receptor expression, likely due to increased synaptic GABA and GHB, has been described.11,12 Similarly, downregulation of GABAA receptor expression has been observed in the transgenic mouse model using binding studies with a selective GABAA receptor antagonist.13 We have also reported widespread reduction in flumazenil binding potential on [11C]flumazenil PET in patients with SSADH deficiency, supporting downregulation of binding sites at the GABA-benzodiazepine receptor complex.14
Increased levels of GABAergic transmitters in SSADH deficiency could lead to functionally increased inhibition. Alternatively, both animal and human data provide some support for the hypothesis that reduced postsynaptic GABA receptor availability could lead to reduced cortical inhibition due to a predominantly presynaptic mechanism of action of GABA. Hence, human evidence for the influence of GABAA vs GABAB-ergic mechanisms for this disorder is missing. Here, we used transcranial magnetic stimulation (TMS) paradigms that are susceptible to changes in GABAA-ergic and GABAB-ergic neurotransmission to quantify excitation and inhibition in the primary motor cortex (M1) of patients with SSADH deficiency, their parents (obligate heterozygotes), and age-matched healthy controls.
METHODS
Subjects.
Seven families with at least one child or young adult with SSADH deficiency were recruited to participate in this study. Affected patients and family members were screened for inclusion by the Children's National Medical Center Department of Neurology, followed by referral to NIH. Inclusion criteria were clinical characteristics consistent with SSADH deficiency, persistent 4-hydroxybutyric aciduria (γ-hydroxybutyric aciduria), and confirmed leukocyte extract succinic semialdehyde dehydrogenase enzyme deficiency or identification of a pathogenic mutation in DNA samples.15,16 Eight affected patients (age 10–27 years; mean, 15.4 ± 1.5 years; 4 male) and at least 1 parent per family (age 30–58 years; mean, 42.8 ± 2.2 years; 7 male, 6 female) were assessed with TMS. Furthermore, 2 groups of healthy controls (11 adults: age 23–48 years; mean, 36.5 ± 2.7 years, 4 male; 8 age-matched “young controls”: age 9–27 years; mean, 14.4 ± 2.3 years, 6 male) were enrolled, matching the mean age of the affected patients and of parents, respectively. Some of the patients had participated in a previous study.14 All subjects were right-handed according to the Edinburgh Handedness Inventory.17 Healthy participants had never been treated with neuroleptic drugs and had no history of neuropsychiatric disorders, neurosurgery, or metal or electronic implants. One SSADH deficiency patient was treated with carbamazepine for seizures; another 2 patients had a history of seizures but were medically untreated. All patients had been seizure-free for several months at the time of the study (for details, see tables 1 and 2).
Table 1.
Demographics
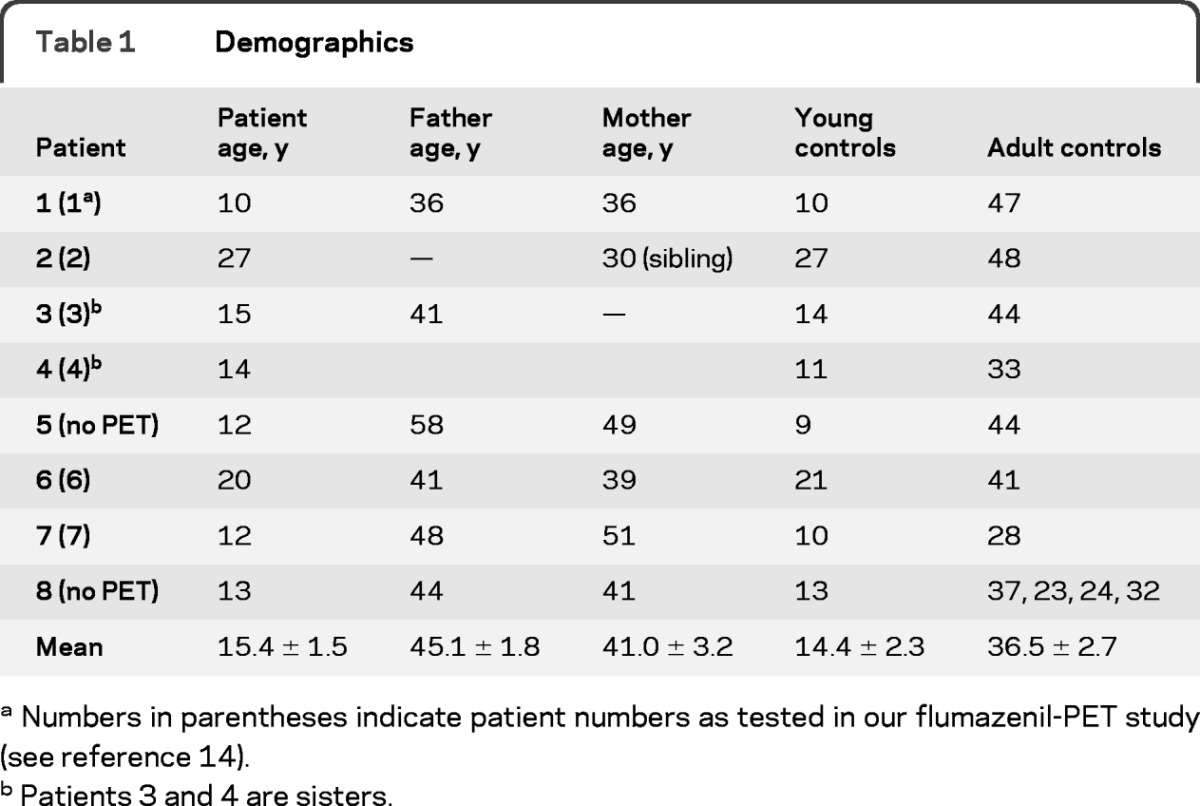
Numbers in parentheses indicate patient numbers as tested in our flumazenil-PET study (see reference 14).
Patients 3 and 4 are sisters.
Table 2.
Clinical features of 8 patients with SSADH deficiencya
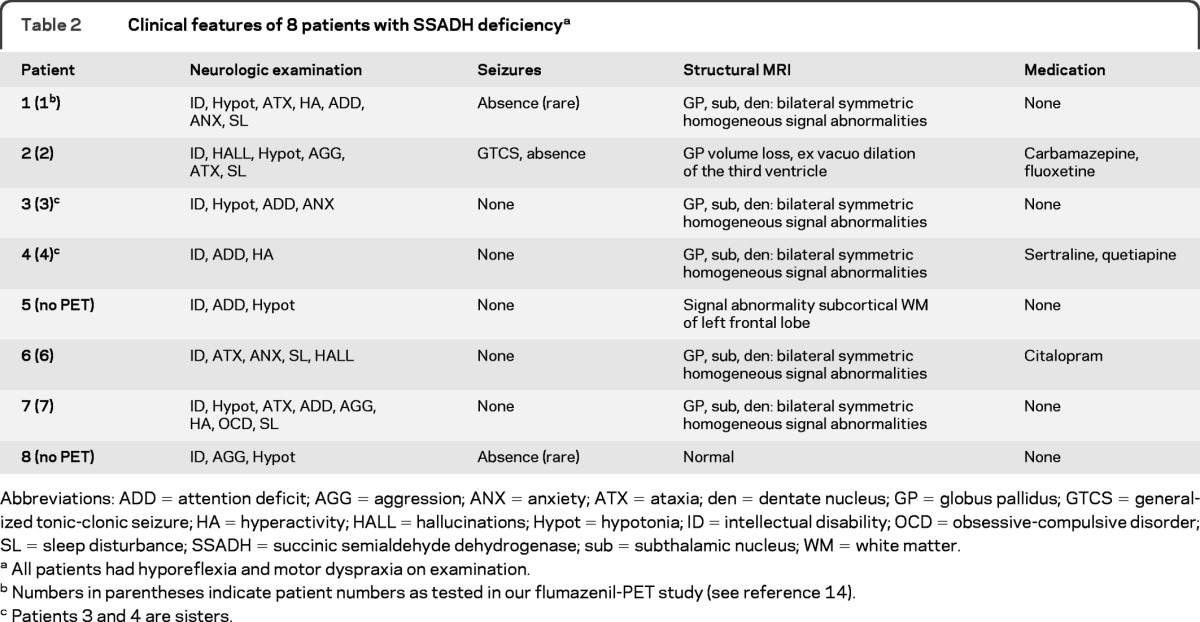
Abbreviations: ADD = attention deficit; AGG = aggression; ANX = anxiety; ATX = ataxia; den = dentate nucleus; GP = globus pallidus; GTCS = generalized tonic-clonic seizure; HA = hyperactivity; HALL = hallucinations; Hypot = hypotonia; ID = intellectual disability; OCD = obsessive-compulsive disorder; SL = sleep disturbance; SSADH = succinic semialdehyde dehydrogenase; sub = subthalamic nucleus; WM = white matter.
All patients had hyporeflexia and motor dyspraxia on examination.
Numbers in parentheses indicate patient numbers as tested in our flumazenil-PET study (see reference 14).
Patients 3 and 4 are sisters.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the National Institute of Neurological Disorders and Stroke Institutional Review Board. All subjects gave their written informed consent or assent before the experiments. Parents consented for minors or patients unable to give informed consent due to cognitive dysfunction. Four young control subjects were tested under an associated protocol approved by the Institutional Review Board of the University of Freiburg.
Transcranial magnetic stimulation.
Transcranial magnetic stimulation was delivered through a figure-of-eight shaped magnetic coil (90 mm external loop diameter) connected to 2 Magstim 2002 magnetic stimulators via a BiStim-module (Magstim, USA). The coil was placed over the left motor cortex “hot spot,” optimal for eliciting motor evoked potentials (MEPs) from the contralateral first dorsal interosseus muscle (FDI). Coil orientation was tangential to the scalp with the handle pointing backwards and laterally at a 45° angle away from the midline, inducing a posterior-anterior current in the brain. This is thought to be the most effective way to transsynaptically activate the corticospinal system.18 In all paired pulse TMS procedures the intertrial interval was fixed at 0.2 Hz; in all single pulse procedures the intertrial interval was randomly changed between 0.1 and 0.12 Hz.
Figure 1. Age-dependent differences in M1 excitability.

(A) Young subjects show higher resting motor thresholds (RMT, in % of maximum stimulator output) compared to parents and adult controls. (B) Single subject data for motor thresholds. Filled circles (gray) indicate patients with medication. (C) Recruitment curves for the several groups show age-dependent differences between young subjects (including patients with succinic semialdehyde dehydrogenase [SSADH] deficiency) and adults. The color code is identical to that of A and E. (D) Single subject data for recruitment curve slopes. Normalization could only be performed in 4 patients. Filled circles (gray) indicate patients with medication. (E) The slope of the recruitment curve is significantly steeper in adults than in patients with SSADH deficiency or young controls. All group data are presented as mean ± SEM. Level of significance: *p < 0.05; **p < 0.01.
EMG recording.
Subjects were seated in a comfortable chair with their arms resting on a pillow. Disposable surface Ag-AgCl EMG electrodes were placed on the FDI of the right hand in a belly-tendon montage. Impedance was reduced to <5 kΩ. The raw EMG signal was amplified using a conventional EMG machine (Viking Nicolet 4), bandpass filtered (20 Hz to 10 kHz), digitized (analog/digital rate 40 kHz), and recorded onto a PC using a data collection and averaging program (Signal version 4, Cambridge Electronics, UK) for offline analysis. Muscle activity was continuously monitored by audio-visual feedback. MEP size was generally determined by averaging peak-to-peak amplitudes. The resting motor threshold (RMT) was defined as the lowest stimulator output intensity required to evoke MEPs of at least 50 μV in 5 out of 10 consecutive trials, using a step-by-step intensity resolution of 1% of the maximal stimulator output. Trials with a background EMG of >20 μV in the FDI (assessed as root mean square) within 200 msec before the onset of the MEP were rejected. Furthermore, the first evoked response per testing block was excluded to avoid data contamination by startle responses.
Experimental paradigms.
All patients were severely affected, and many of them showed symptoms of attention deficit, anxiety, or hyperactivity (table 2). Thus, in order to examine a sufficient set of TMS parameters within 45 minutes, we tested only one interstimulus interval (ISI) and the commonly used stimulation intensity for each paired-pulse TMS parameter. Furthermore, suprathreshold electrical stimulation of the median nerve to measure the maximum compound muscle action potential (Mmax) for recruitment curve (REC) normalization was explored only in patients who tolerated this uncomfortable procedure.
Ten single TMS stimuli at intensities of 100, 120, 140, 160, and 180% RMT were applied to obtain RECs and the averaged MEP amplitude per intensity was calculated. In those participants (4 patients) in whom Mmax was obtained, REC was also expressed as % of Mmax. The cortical stimulation–induced silent period (CSP) was averaged over 20 trials at a stimulus intensity of 150% RMT in the moderately contracted FDI muscle (30% of maximal voluntary contraction [MVC[rsq]). MVC during a lateral pinch grip was measured using a custom-made Honeywell force transducer connected to a signal conditioner. The 30% MVC was visualized on a screen monitor as horizontal bar and presented online to the subjects for force control during CSP measurement. CSP duration was defined as the time from TMS stimulus artifact to the first reoccurrence of voluntary EMG activity exceeding prestimulus muscle activity. Short intracortical inhibition (SICI) was investigated at an ISI of 3 msec, and intracortical facilitation (ICF) was investigated at an ISI of 10 msec.19,20 The conditioning stimulus was set to an intensity of 75% of RMT to exclude changes of excitability in the spinal cord.18 The intensity of the test stimulus was adjusted to produce MEPs of approximately 0.8 mV peak-to-peak amplitude, since children younger than 12 years typically show saturation of MEP amplitude below 1 mV when probed by recruitment curves, reflecting maturation level of the pyramidal tract.21 Other rationales for using lower amplitude test MEP than those utilized in adults was to account for muscle hypotonia and to reduce the discomfort associated with TMS application in patients with SSADH deficiency, with higher motor thresholds than adults. Late intracortical inhibition (LICI) was tested at an ISI of 150 msec, but in contrast to SICI and ICF 2 equal stimuli of 150% RMT were delivered.20,22 Fifteen trials of the control single test stimuli and 15 paired stimuli of each ISI were recorded, delivered 4–8 seconds apart in random order generated by the software. The conditioned MEP response per ISI was expressed as percentage of the mean amplitude of the unconditioned test MEP to obtain values for inhibition and facilitation. For a detailed review of the assessed parameters, see reference 23.
Electrical stimulation of the median nerve.
An electrical stimulator (Digitimer DS7) was used to generate single square-wave pulses of 1 msec duration. The maximal amplitude of the M response (Mmax) was determined by supramaximal electrical stimulation.
Statistical analysis.
TMS variables (RMT, REC, SICI, ICF, LICI, CSP) were subjected to separate one-way analyses of variance (ANOVA) with the factor “group” as the independent variable (patients with SSADH deficiency, parents, adult controls, young controls) and “MEP amplitude” as the dependent variable. For ANOVAs and Bonferroni post hoc tests significance was assumed at p < 0.05. Fathers and mothers were treated as one “Parent” group (see also figure e-1 on the Neurology® Web site at www.neurology.org for separated data); groups were not separated by gender due to the few patients. SPSS 16.0 and Excel 2003 were used for statistical testing. For graphical illustration Excel 2003 and Adobe Illustrator CS 4 were used. Data are presented as mean ± SEM.
RESULTS
All subjects tolerated the experimental procedures well; none of them reported any side effects after TMS.
Resting motor threshold and recruitment curve.
The one-way ANOVAs for the factor resting motor threshold and recruitment curve revealed a significant difference among the 4 groups (F = 4.359, p = 0.010 and F = 3.729, p = 0.022, respectively). Resting motor thresholds were significantly higher in patients with SSADH deficiency (57 ± 4.8% MSO) than in parents or adult controls (46.5 ± 2.0% MSO, p = 0.008, and 44.0 ± 3.2% MSO, p = 0.002, respectively). RMT in patients with SSADH deficiency was not significantly different from young control subjects (52.2 ± 2.7% MSO). There was a trend toward higher RMT in young controls compared to adult controls (p = 0.09) and no difference compared to parents (p = 0.2, figure 1, A and B). Additionally, we found a significantly lower slope of the recruitment curve in patients with SSADH deficiency (0.26 ± 0.10) and young control subjects (0.33 ± 0.11) compared to adult controls (0.80 ± 0.12; p = 0.013 and 0.010, respectively); there was a trend toward lower slopes in both groups compared to parents (0.61 ± 0.10; p = 0.09 and 0.10, respectively, figure 1, C–E).
Late interval inhibition.
The one-way ANOVA for the factor LICI revealed a highly significant difference among the 4 groups (F = 10.39, p = 0.000053). LICI was virtually absent in patients with SSADH deficiency (% unconditioned MEP: 101.16 ± 8.7), but present in all other groups (% unconditioned MEP: parents: 46.7 ± 5.8, p < 0.0001; adult controls: 37.95 ± 8.5, p < 0.0001; young controls: 61.0 ± 10.5, p = 0.004 vs patients with SSADH deficiency; figure 2, A–C).
Figure 2. Late intracortical inhibition (LICI) is reduced in succinic semialdehyde dehydrogenase (SSADH) deficient patients.

(A) In patients with SSADH deficiency, LICI was virtually absent, as illustrated by a lack of suppression in the conditioned motor evoked potential (MEP) response. (B) Single subject data for LICI. (C) Characteristic comparison between a young control subject and a SSADH deficiency patient. (D) In patients with SSADH deficiency, cortical stimulation–induced silent period (CSP) was significantly shortened relative to parents and controls. (E) Single subject data for CSP. (F) EMG traces with lack of EMG activity following the MEP in a young control subject and a SSADH deficiency patient. The control has a 2-fold longer CSP compared to the patient. All data are presented as mean ± SEM. Level of significance: *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.000001.
Cortical silent period.
In accordance with changes in LICI, we also found significant group differences for the cortical silent period. The one-way ANOVA for the factor CSP revealed a significant difference among the 4 groups (F = 4.052, p = 0.014). CSP was shortened in patients with SSADH deficiency (98.57 ± 15.8 msec) relative to CSP in parents (154.23 ± 9.0 msec, p = 0.003), adult controls (135.34 ± 6.4 msec, p = 0.049), and young controls (155.37 ± 20.3 msec, p = 0.006) (figure 2, D–F). In general, there was no correlation between the amount of LICI and CSP duration.
Short intracortical inhibition and facilitation.
We also found a slightly decreased short intracortical inhibition in patients with SSADH deficiency (% unconditioned MEP: 46.4 ± 11.5) compared to the other experimental groups (% unconditioned MEP: parents: 30.6 ± 6.0; adult controls: 41.2 ± 8.6; young controls: 36.9 ± 9.6, figure 3, A and B). However, this difference was not statistically significant (F = 0.61; p = 0.61). Intracortical facilitation was slightly lower in patients with SSADH deficiency and their parents compared to the 2 control groups, but we did not observe any statistically significant difference (F = 0.83, p = 0.49; % unconditioned MEP: SSADH: 131.1 ± 25.1; parents: 123.2 ± 8.3; adult controls: 162.5 ± 24.3; young controls: 160.6 ± 36.9, figure 3, C and D).
Figure 3. Short interval intracortical inhibition (SICI) and intracortical facilitation.

(A) Conditioned motor evoked potential (MEP) amplitudes in % of unconditioned MEP. There was a slight trend toward less SICI in patients with succinic semialdehyde dehydrogenase (SSADH) deficiency compared to all other groups. (B) Single subject data for SICI. Filled circles (gray) indicate patients with medication. (C) There were no significant group differences for the conditioned MEP amplitudes tested at an ISI of 10 msec (intracortical facilitation [ICF]). (D) Single subject data for ICF. Filled circles (gray) indicate patients with medication. All group data are presented as mean ± SEM. Data from 1 patient and 1 healthy young control could not be analyzed due to technical reasons (group size n = 7).
DISCUSSION
We demonstrate that young patients with SSADH deficiency show specific alterations of motor cortical excitability that are not found in heterozygous carriers (parents) or healthy age-matched controls. TMS measures of cortical inhibition, namely LICI and CSP, point toward impaired GABAB-ergic function in these patients. Surprisingly, these changes were much more robust than changes in SICI, the parameter assumed to reflect GABAA-ergic function.
In young subjects (patients with SSADH deficiency and young controls), measures of gross motor excitability (RMT and recruitment curve) differed from those observed in adults. Previous studies in preschool children showed higher motor thresholds24 and reduced MEP recruitment with increasing stimulus intensity21: with increasing age, there is an increase in the steepness of the slope; MEP output size plateaus approaching adolescence. Since we observed significant differences between the 2 age groups tested in our study (young vs adults), but not between young patients with SSADH deficiency and young controls, these differences most likely reflect pyramidal tract maturation.
LICI and CSP are TMS measures of intracortical inhibition, which are susceptible to modifications in GABAB-ergic neurotransmission.23 LICI was consistently present in parents and both control groups but virtually absent in patients with SSADH deficiency. In general, LICI decreases with higher test MEP amplitudes, suggesting higher susceptibility of M1 neurons mediating LICI at low test stimulus intensities.25 However, in our study test stimuli were given at stimulation intensities usually inducing strong LICI. Moreover, we excluded the possibility that differences in MEP output between experimental groups were responsible for the LICI group differences observed. While LICI is tested at rest, CSP is apparent as a period of EMG inhibition following the MEP in a voluntarily contracted muscle.26 In accordance with group differences in LICI, CSP was significantly shortened in patients with SSADH deficiency compared to parents and controls. The later part of the CSP is thought to result from suppression of interneurons at the cortical level.26,27 Thus, it is likely that CSP shortening in patients with SSADH deficiency is mediated by cortical rather than spinal mechanisms. CSP duration correlates strongly with the magnitude of voluntary contraction, and MEP amplitude, respectively.28 We excluded this confounding factor using matched 30% of maximum voluntary contraction for CSP elicitation. CSP and LICI are susceptible to alterations by CNS-active drugs targeting GABAB-ergic populations, i.e., tiagabine and baclofen.22,29,30 Thus, it is conceivable that in the patients concomitant changes of LICI and CSP reflect a dysfunction of GABAB-ergic inhibition. These results represent the first demonstration of an abnormality in GABAB-ergic function in human SSADH deficiency. Our data are consistent with results derived from an SSADH deficiency animal model, in which reduced postsynaptic GABAB receptor expression by accumulated GHB or GABA has been shown.12 Despite the increased constitutive GABA levels resulting in increased tonic inhibition,31 we found evidence for a reduction of GABAergic inhibition in patients with SSADH deficiency. If postsynaptic GABAB receptor downregulation is evident, it is possible that elevated GABA binds predominantly to presynaptic GABAB receptors, leading to reduced activity-dependent secretion of GABA into the synaptic cleft. In this scenario, functional changes observable by TMS would be more related to activity-dependent transsynaptic (phasic) inhibition than to constitutive (tonic) inhibition.
In contrast to the highly significant changes of LICI and CSP, we observed only a tendency toward reduced short intracortical inhibition, a TMS parameter that is most likely mediated by GABAA receptors.32,33 At first sight, it appears less pronounced than the results of an earlier study using [11C]flumazenil PET in patients with SSADH deficiency suggesting a downregulation of benzodiazepine binding sites at the GABAA receptor. However, in animal studies effects of elevated GHB have been strongly associated with activation of GABAB receptors rather than GABAA receptors,10 which might also explain the contrasting result observed in our study. In addition, in a familial generalized epilepsy syndrome a mutation in the GABAA receptor γ2 subunit led to reduced [11C]flumazenil binding.34 Affected patients showed reduced short-interval intracortical inhibition and increased intracortical facilitation on paired-pulse TMS, but no differences from controls in motor threshold or CSP.35 The contrast with our results reinforces the importance of GABAB receptor dysfunction in SSADH deficiency, as opposed to a disorder confined to GABAA receptors. Of note, SICI was measured at test stimulus intensities evoking relatively small MEPs. SICI increases with higher test MEP amplitudes.25 Thus, it is also possible that we missed significant results, because we did not assess SICI with higher test MEP amplitudes. Finally, we did not observe significant changes in intracortical facilitation. ICF is a TMS parameter responding predominantly to glutamatergic interventions, i.e., NMDA receptor antagonists have been shown to reduce ICF.36–38 Since SSADH deficiency is a disorder of the GABAergic pathway the lack of difference in patients with SSADH deficiency compared to other experimental groups is expected.
In the developing brain, GABA has depolarizing properties due to a higher intracellular chloride concentration in immature neurons.39 Since GABA is the key excitatory player in neurodevelopment preceding the action of glutamate, elevated cerebral GABA and GHB levels may cause both functional and structural alterations during this critical period. In other words, chronically elevated cerebral GABA and GHB may have different effects on neurotransmission than acute increases in neurotransmitters, e.g., by drug intake. It is striking that a noninherited, immunologically mediated disease such as stiff-person syndrome, in which antibodies against glutamic acid decarboxylase lead to a reduction in cerebral GABA levels, causes a TMS phenotype of disinhibition (shortening of CSP, decreased SICI40) similar to SSADH deficiency. Hence, developmental factors and chronic compensatory mechanisms may play a critical role for the direction of the GABAergic dysbalance in SSADH deficiency.
To date, a particular diagnostic problem in SSADH deficiency is the heterogeneity of clinical manifestations, featuring intellectual disability, epilepsy, ataxia, sleep disorders, and neuropsychiatric disturbances. Nevertheless, all patients are severely affected. Furthermore, no consistently successful therapy has emerged for the treatment of SSADH deficiency. Since excitability of the motor cortex was specifically altered in these patients (not in obligate heterozygous parents), TMS could be helpful to detect the homozygous carrier status, facilitating diagnosis of the disease, but also to monitor novel treatment strategies. Our study has implications for the understanding of the pathophysiology of neurotransmitter disorders in general, by showing how neurophysiologic techniques can help explain the clinical effects of alterations in transmitter levels and receptor expression.
Supplementary Material
GLOSSARY
- ANOVA
analysis of variance
- CSP
cortical stimulation–induced silent period
- FDI
first dorsal interosseus
- GHB
γ-hydroxybutyric acid
- ICF
intracortical facilitation
- ISI
interstimulus interval
- LICI
late intracortical inhibition
- M1
primary motor cortex
- MEP
motor evoked potential
- MVC
maximal voluntary contraction
- REC
recruitment curve
- RMT
resting motor threshold
- SICI
short intracortical inhibition
- SSADH
succinic semialdehyde dehydrogenase
- TMS
transcranial magnetic stimulation
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
J.R. designed and performed the study, analyzed the data, and wrote the paper. L.G.C. designed the study and wrote the paper. P.L.P. recruited and evaluated patients. B.F. performed parts of the study, analyzed data, and wrote the paper. N.J. performed parts of the study. I.D. was involved in patient recruitment, care, and logistics. W.H.T. designed the study, performed patient care, and wrote the paper.
DISCLOSURE
J. Reis received personal funding (postdoctoral fellowship) from the Alexander von Humboldt Foundation Germany and the Intramural Research Program, NINDS, NIH. L. Cohen received funding from the Intramural Research Program, NINDS, NIH. P. Pearl, B. Fritsch, and N. Jung report no disclosures. I. Dustin received funding from the Intramural Research Program, NINDS, NIH. W. Theodore received funding from the Intramural Research Program, NINDS, NIH, serves as the Co-Editor in Chief of Epilepsy Research, and holds stock in General Electric. Go to Neurology.org for full disclosures.
REFERENCES
- 1. Akaboshi S, Hogema BM, Novelletto A, et al. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum Mutat 2003;22:442–450 [DOI] [PubMed] [Google Scholar]
- 2. Pearl PL, Novotny EJ, Acosta MT, et al. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol 2003;54(suppl 6):S73–S80 [DOI] [PubMed] [Google Scholar]
- 3. Pearl PL, Shamim S, Theodore WH, et al. Polysomnographic abnormalities in succinic semialdehyde dehydrogenase (SSADH) deficiency. Sleep 2009;32:1645–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis 2009;32:343–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benavides J, Rumigny JF, Bourguignon JJ, et al. High affinity binding sites for gamma-hydroxybutyric acid in rat brain. Life Sci 1982;30:953–961 [DOI] [PubMed] [Google Scholar]
- 6. Snead OC, 3rd, Liu CC. Gamma-hydroxybutyric acid binding sites in rat and human brain synaptosomal membranes. Biochem Pharmacol 1984;33:2587–2590 [DOI] [PubMed] [Google Scholar]
- 7. Lingenhoehl K, Brom R, Heid J, et al. Gamma-hydroxybutyrate is a weak agonist at recombinant GABA(B) receptors. Neuropharmacology 1999;38:1667–1673 [DOI] [PubMed] [Google Scholar]
- 8. Murphy TC, Poppe C, Porter JE, et al. 4-Hydroxy-trans-2-nonenoic acid is a gamma-hydroxybutyrate receptor ligand in the cerebral cortex and hippocampus. J Neurochem 2004;89:1462–1470 [DOI] [PubMed] [Google Scholar]
- 9. Carter LP, Koek W, France CP. Behavioral analyses of GHB: receptor mechanisms. Pharmacol Ther 2009;121:100–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Serra M, Sanna E, Foddi C, et al. Failure of gamma-hydroxybutyrate to alter the function of the GABAA receptor complex in the rat cerebral cortex. Psychopharmacology 1991;104:351–355 [DOI] [PubMed] [Google Scholar]
- 11. Buzzi A, Wu Y, Frantseva MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res 2006;1090:15–22 [DOI] [PubMed] [Google Scholar]
- 12. Vardya I, Drasbek KR, Gibson KM, Jensen K. Plasticity of postsynaptic, but not presynaptic, GABAB receptors in SSADH deficient mice Exp Neurol 2011;225:114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol 2006;59:42–52 [DOI] [PubMed] [Google Scholar]
- 14. Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology 2009;73:423–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gibson KM, Aramaki S, Sweetman L, et al. Stable isotope dilution analysis of 4-hydroxybutyric acid: an accurate method for quantification in physiological fluids and the prenatal diagnosis of 4-hydroxybutyric aciduria. Biomed Environ Mass Spectrom 1990;19:89–93 [DOI] [PubMed] [Google Scholar]
- 16. Gibson KM, Lee CF, Chambliss KL, et al. 4-Hydroxybutyric aciduria: application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin Chim Acta 1991;196:219–221 [DOI] [PubMed] [Google Scholar]
- 17. Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 1971;9:97–113 [DOI] [PubMed] [Google Scholar]
- 18. DiLazzaro V, Oliviero A, Profice P, et al. Comparison of descending volleys evoked by transcranial magnetic and electric stimulation in conscious humans. Electroencephalogr Clin Neurophysiol 1998;109:397–401 [DOI] [PubMed] [Google Scholar]
- 19. Kujirai T, Caramia MD, Rothwell JC, et al. Corticocortical inhibition in human motor cortex. J Physiol 1993;471:501–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakamura H, Kitagawa H, Kawaguchi Y, Tsuji H. Intracortical facilitation and inhibition after transcranial magnetic stimulation in conscious humans. J Physiol 1997;498:817–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garvey MA, Gilbert DL. Transcranial magnetic stimulation in children. Eur J Paediatr Neurol 2004;8:7–19 [DOI] [PubMed] [Google Scholar]
- 22. McDonnell MN, Orekhov Y, Ziemann U. The role of GABA(B) receptors in intracortical inhibition in the human motor cortex. Exp Brain Res 2006;173:86–93 [DOI] [PubMed] [Google Scholar]
- 23. Reis J, Swayne OB, Vandermeeren Y, et al. Contribution of transcranial magnetic stimulation to the understanding of cortical mechanisms involved in motor control. J Physiol 2008;586:325–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heinen F, Glocker FX, Fietzek U, et al. Absence of transcallosal inhibition following focal magnetic stimulation in preschool children. Ann Neurol 1998;43:608–612 [DOI] [PubMed] [Google Scholar]
- 25. Chen R, Curra A. Measures of cortical inhibition in health and disease. Suppl Clin Neurophysiol 2004;57:691–701 [DOI] [PubMed] [Google Scholar]
- 26. Fuhr P, Agostino R, Hallett M. Spinal motor neuron excitability during the silent period after cortical stimulation. Electroencephalogr Clin Neurophysiol 1991;81:257–262 [DOI] [PubMed] [Google Scholar]
- 27. Tergau F, Wanschura V, Canelo M, et al. Complete suppression of voluntary motor drive during the silent period after transcranial magnetic stimulation. Exp Brain Res 1999;124:447–454 [DOI] [PubMed] [Google Scholar]
- 28. Orth M, Rothwell JC. The cortical silent period: intrinsic variability and relation to the waveform of the transcranial magnetic stimulation pulse. Clin Neurophysiol 2004;115:1076–1082 [DOI] [PubMed] [Google Scholar]
- 29. Werhahn KJ, Kunesch E, Noachtar S, et al. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J Physiol 1999;517:591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siebner HR, Dressnandt J, Auer C, Conrad B. Continuous intrathecal baclofen infusions induced a marked increase of the transcranially evoked silent period in a patient with generalized dystonia. Muscle Nerve 1998;21:1209–1212 [DOI] [PubMed] [Google Scholar]
- 31. Drasbek KR, Vardya I, Delenclos M, et al. SSADH deficiency leads to elevated extracellular GABA levels and increased GABAergic neurotransmission in the mouse cerebral cortex. J Inherit Metab Dis 2008;31:662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Lazzaro V, Pilato F, Dileone M, et al. GABAA receptor subtype specific enhancement of inhibition in human motor cortex. J Physiol 2006;575:721–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziemann U. Pharmacology of TMS. Suppl Clin Neurophysiol 2003;56:226–231 [PubMed] [Google Scholar]
- 34. Fedi M, Berkovic SF, Marini C, et al. A GABAA receptor mutation causing generalized epilepsy reduces benzodiazepine receptor binding. Neuroimage 2006;32:995–1000 [DOI] [PubMed] [Google Scholar]
- 35. Fedi M, Berkovic SF, Macdonell RA, et al. Intracortical hyperexcitability in humans with a GABAA receptor mutation. Cereb Cortex 2008;18:664–669 [DOI] [PubMed] [Google Scholar]
- 36. Ziemann U, Chen R, Cohen LG, Hallett M. Dextromethorphan decreases the excitability of the human motor cortex. Neurology 1998;51:1320–1324 [DOI] [PubMed] [Google Scholar]
- 37. Schwenkreis P, Witscher K, Janssen F, et al. Influence of the N-methyl-D-aspartate antagonist memantine on human motor cortex excitability. Neurosci Lett 1999;270:137–140 [DOI] [PubMed] [Google Scholar]
- 38. Reis J, Tergau F, Hamer HM, et al. Topiramate selectively decreases intracortical excitability in human motor cortex. Epilepsia 2002;43:1149–1156 [DOI] [PubMed] [Google Scholar]
- 39. Ben-Ari Y. Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 2002;3:728–739 [DOI] [PubMed] [Google Scholar]
- 40. Sandbrink F, Syed NA, Fujii MD, et al. Motor cortex excitability in stiff-person syndrome. Brain 2000;123:2231–2239 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.