

Abstract
Hydration water is vital for various macromolecular biological activities, such as specific ligand recognition, enzyme activity, response to receptor binding, and energy transduction. Without hydration water, proteins would not fold correctly and would lack the conformational flexibility that animates their three-dimensional structures. Motions in globular, soluble proteins are thought to be governed to a certain extent by hydration-water dynamics, yet it is not known whether this relationship holds true for other protein classes in general and whether, in turn, the structural nature of a protein also influences water motions. Here, we provide insight into the coupling between hydration-water dynamics and atomic motions in intrinsically disordered proteins (IDP), a largely unexplored class of proteins that, in contrast to folded proteins, lack a well-defined three-dimensional structure. We investigated the human IDP tau, which is involved in the pathogenic processes accompanying Alzheimer disease. Combining neutron scattering and protein perdeuteration, we found similar atomic mean-square displacements over a large temperature range for the tau protein and its hydration water, indicating intimate coupling between them. This is in contrast to the behavior of folded proteins of similar molecular weight, such as the globular, soluble maltose-binding protein and the membrane protein bacteriorhodopsin, which display moderate to weak coupling, respectively. The extracted mean square displacements also reveal a greater motional flexibility of IDP compared with globular, folded proteins and more restricted water motions on the IDP surface. The results provide evidence that protein and hydration-water motions mutually affect and shape each other, and that there is a gradient of coupling across different protein classes that may play a functional role in macromolecular activity in a cellular context.
Introduction
Water is an integral part of protein structures, mediates macromolecular recognition, modulates ligand binding and allosteric effects, reacts in biochemical processes, and participates in electron and proton transfer (1). The formation of a productive enzyme-substrate complex, for instance, is preceded by a slowing of water motions in the protein hydration layer (2), providing strong evidence that water is indeed actively involved in macromolecular biological activity (3). Furthermore, water plays essential roles via its unusual hydrogen-bond dynamics, which contributes to the hydrophobic effect in protein folding and modulates macromolecular flexibility through formation and breaking of hydrogen bonds at the protein-water interface (4). Consequently, it has been proposed that macromolecular dynamics is controlled by hydration-water dynamics (5) not only at the macromolecule's surface but also in its interior (6). Whereas the influence of hydration water on the dynamics of folded, soluble proteins has been studied extensively (4,7,8), little is known about the coupling of hydration water in the case of IDPs. The corollary aspect of dynamical coupling, viz., the influence of protein motions on water motions, also remains largely unstudied, mostly because of experimental difficulties in directly accessing water dynamics. Before we can draw a more general picture of protein-water dynamical coupling, we need to study different protein classes and protein effects on hydration-water dynamics. Here, we focused on the largely unexplored class of IDPs and directly assessed their hydration-water dynamics by studying perdeuterated proteins using elastic incoherent neutron scattering (EINS).
IDPs are, by definition, proteins that function without the requirement of a well-defined, unique three-dimensional (3D) structure in isolation (9,10). Approximately 30% of eukaryotic proteins are thought to be either fully or partially disordered (11), and it seems that evolution selected disorder to go with species complexity. IDPs fulfill specific biological roles in cells (12,13) that are often dependent on their partial folding upon interaction with a functional partner (14). This is the case for the microtubule-associated tau protein, which folds partially upon binding to tubulin. Tau has attracted considerable attention because its abnormal intracellular deposition as so-called paired-helical filaments is one of the main hallmarks of Alzheimer disease (15). Other IDPs are also prone to aggregate into amyloid fibrils, and as such are involved in many neurodegenerative diseases (e.g., the β-amyloid peptide and α-synuclein in Alzheimer disease and Parkinson disease, respectively (16)). Tau has six isoforms, ranging from 352 to 441 residues in length, that are generated through alternative splicing in the central nervous system (17). The proposed biological role of tau involves stabilization of microtubules, which is regulated through specific phosphorylation (15). If aberrant hyperphosphorylation of tau occurs, the protein's repeat domains, which have a propensity for β-sheets (18) and β-turns (19), form cross-β-structures and aggregate into paired helical filaments (20). As is generally the case for IDPs (21), tau has a large content of polar and charged residues compared with that of hydrophophic residues, which prevents the protein's hydrophobic collapse into a folded structure. As a consequence, tau likely features a much larger solvent-accessible surface compared with a globular protein of similar molecular weight. One might suspect, therefore, that the coupling of hydration-water dynamics with tau dynamics, and with IDP dynamics in general, would differ in nature and strength from that observed for a folded, globular protein. Yet, no experimental evidence directly addressing this issue has been reported.
Neutron scattering is a unique experimental method that provides direct access to molecular dynamics on a nano- to picosecond timescale and angstrom (Å) length scale (22). The incoherent scattering of neutrons by a biological sample is largely dominated by hydrogen nuclei, whose incoherent scattering cross section is ∼40 times larger than those of other nuclei, including deuterium, the stable isotope of hydrogen. Because hydrogen atoms are homogeneously distributed throughout a protein and located mainly in amino acid side chains (∼84% for tau), the mean-square displacement (MSD) extracted from EINS experiments yields information concerning averaged side-chain motions and thus provides a measure of a protein's global flexibility (23). Because the INS cross section of deuterium is much lower than that of hydrogen, one can employ specific deuteration to mask the contribution of the labeled part in a sample. Perdeuterating a protein and then hydrating it in H2O minimizes its contribution to the INS signal, and therefore it is mostly the water dynamics that is measured, despite the limited deuterium-hydrogen exchange (7,24–26). In contrast to x-ray crystallography, which provides atomic-resolution structural information on hydration water (27), and to NMR (28) and time-resolved fluorescence spectroscopy (29), which provide site-resolved dynamical information on hydration water on the nano- to picosecond timescale, neutron scattering yields an average but model-free amplitude of nano- to picosecond water motions on the angstrom length scale. Conversely, when a natural-abundance protein is hydrated in D2O, the protein dynamics can be directly measured by INS. To focus on hydration water in our neutron scattering experiments, we examined hydrated powders that were devoid of bulk water. A hydrated powder of a medium-sized globular, folded protein at 0.4 g water/g protein is sufficient to ensure macromolecular biological activity (30,31) and is thought to correspond to monolayer water coverage of the protein surface (30). This level of hydration is six times lower than that of a typical animal cell containing ∼75% water by weight (32). Previous 17O magnetic relaxation dispersion experiments suggested that the dynamic perturbation factor of water in hydrated protein powders is increased 1–2 orders of magnitude compared with the free hydration layer of a protein in solution (33). Therefore, it is important to examine samples at the same hydration level if protein and hydration-water dynamics are to be compared among different classes of proteins, as was the case in this study.
Here, we employed EINS to examine powders of hydrogenated and deuterated tau, hydrated in D2O and H2O, respectively, to study hydration water and IDP motions separately but on the same protein. The extracted MSDs reveal a greater motional flexibility of IDP compared with globular, folded proteins in powders hydrated at the same level and more restricted water motions on the IDP surface. The comparison of MSDs across different protein classes reveals a gradient of coupling between hydration-water and protein motions, which is tightest in IDP and loosest in membrane proteins.
Materials and Methods
Expression and purification of unlabeled and perdeuterated htau40 protein
The htau40 isoform of the tau protein was expressed in the Institut Laue-Langevin/European Molecular Biology Laboratory (ILL-EMBL) Deuteration Laboratory (Grenoble, France) as a histidine-tagged fusion protein in Escherichia coli BL21(DE3) following protocols that will be published in detail elsewhere (A. Laganowsky, J.-P. Colletier, and D. Eisenberg., unpublished results) in its hydrogenated (H-tau) and perdeuterated (D-tau) forms. Briefly, a high-cell-density fermentation process with Enfors minimal medium (34) was used to grow bacteria to an OD600 of 12–14, followed by induction of protein expression by isopropyl-β-D-thio-galactoside to a final concentration of 0.5 mM. Bacteria were harvested when an OD600 of 20 was reached. To obtain deuterated tau, E. coli cells were grown in D2O minimal medium with d8-glycerol (fully deuterated glycerol) as the carbon source and D2O as solvent. The expressed protein was purified by immobilized-metal-ion-affinity chromatography on a nickel affinity column, followed by size-exclusion chromatography. For purification of both D-tau and H-tau samples, H2O-based buffer systems were used. The crude protein extract was incubated with the nickel resin in 20 mM Tris pH 8, 500 mM NaCl, washed three times with 10 mM imidazole, 5 mM β-mercaptoethanol (BME), and 1 M, 500 mM and 250 mM sodium chloride, respectively. Protein was eluted with 250 mM imidazole, 5 mM BME, and 300 mM sodium chloride. Eluted fractions containing tau were pooled and dialyzed against 500 mM ammonium acetate and loaded on a Sephacryl S200 size-exclusion column that was previously equilibrated with the same buffer. The eluted fractions were pooled. Protein purity was assessed by 12% Tris-Tricine SDS-PAGE.
Small-angle x-ray scattering
Small-angle x-ray scattering (SAXS) experiments on H-tau (4.7 mg/ml) in H2O, 20 mM Tris pH 8.0, 150 mM NaCl were performed at 15°C in a glass capillary on the ID14-3 BioSAXS beamline at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The sample-detector distance was 2.6 m, yielding Q-range from 0.005 to 0.56 Å−1 for an x-ray wavelength of 0.931 Å (13.32 keV). The absence of radiation damage was verified by 10 successive exposures, each of 10 s duration. The final exposure time was 100 s for all samples and buffers. The 2D diffraction patterns were normalized to an absolute scale and azimuthally averaged to obtain the intensity profiles I(Q) (see Fig. S1 in the Supporting Material) within BSxCuBE (ESRF beamline data collection software). Solvent contributions (buffer backgrounds collected before and after every protein sample) were averaged and subtracted from the corresponding protein sample using the program PRIMUS (35). The same program was used to extract the radius of gyration (Rg) in a Q-range with QmaxRg = 1.
Multi-angle laser light scattering
We performed size-exclusion chromatography coupled with multi-angle laser light scattering (MALLS) to assess the monodispersity, oligomerization state, and average molecular weight of the purified H-tau. For each point on the size exclusion chromatogram, the amount of light at various angles was recorded and converted directly into a molecular weight value, because the size of the particle was much lower than the wavelength. We then obtained the average molecular weight from the weight distribution across the elution peak. We used a 690 nm laser and a DAWNEOS detector (Wyatt Technology, Santa Barbara, CA), and processed the data with the use of ASTRA software (Wyatt Technology). The size-exclusion chromatography column was equilibrated in 150 mM NaCl, 20 mM Tris, pH 8.
Electron microscopy
We verified the absence of paired helical filaments in the H-tau sample in solution before lyophilization by electron microscopy employing negative staining with uranyl acetate and 0.5 mg/ml H-tau in H2O.
Sample preparation for EINS and neutron diffraction experiments
The tau solutions were flash-cooled in liquid nitrogen and then lyophilized. Because ammonium acetate is volatile, the samples did not contain any residual salt. The lyophilized protein powders were dried over P2O5 for 4 days on a 4×3 cm2 flat aluminum sample holder. Dolman and co-workers (36) reported that after such a procedure, lysozyme retains only four tightly bound structural water molecules. The resulting hydration level was defined as corresponding to 0 g water/g tau. Dry H-tau (149 mg) and D-tau (204 mg) powders were then rehydrated over pure D2O and H2O, respectively, up to a hydration level of 0.44 g D2O/g H-tau (H-tau-D2O sample) and 0.38 g H2O/g D-tau (D-tau-H2O sample) as determined by weighting, corresponding in both cases to 1040 water molecules per tau molecule. Samples were then sealed by an aluminum cover (0.3 mm neutron path length) and a 1 mm indium seal. The two closed samples were used for all neutron (spectroscopy and diffraction) experiments described herein.
If we assume that all exchangeable hydrogens in the H-tau and all exchangeable deuterons in the D-tau protein (i.e., 24% of all hydrogens/deuterons in the protein) exchange, we can calculate the following contributions to the total incoherent scattering cross section of the samples:
D-tau-H2O sample: 29% protein (8% from deuterated protein and 92% from the deuterons exchanged against hydrogens) and 71% from hydration water (H2O)
H-tau-D2O sample: 97% protein (99% from hydrogenated protein and 1% from the hydrogen atoms exchanged against deuterons) and 3% from hydration water (D2O)
Similar contributions were previously calculated for hydrogenated and deuterated maltose-binding protein (MBP) powders hydrated at a level of ∼0.4 g water/ g protein in D2O (H-MBP-D2O sample) and H2O (D-MBP-H2O sample), respectively (7).
To determine the MSD of dry tau powder, 104 mg of H-tau was placed in a 4×3 cm2 aluminum sample holder and dried over P2O5 for 4 days (H-tau-dry sample). The sample holder was then sealed with a 1 mm indium seal and closed with an aluminum cover.
Natural-abundance (hydrogenated) horse-heart myoglobin (Mb) was purchased as a lyophilized powder (Sigma, St. Louis, MO) and 125 mg were dried over P2O5 for 24 h on a 4×3 cm2 flat aluminum sample holder. The resulting hydration level was defined as corresponding to 0 g water/g Mb. The dry Mb powder was then rehydrated through the vapor phase over 100% D2O. When a final weight corresponding to a hydration level of 0.43 g D2O/g Mb (corresponding to 0.4 g H2O/g hydrogenated protein or 386 water molecules) was reached, the sample (hereafter termed H-Mb-D2O) was sealed by an aluminum cover (0.3 mm neutron path length) and a 1 mm indium seal.
EINS experiments on the IN16 spectrometer
EINS experiments were carried out on the backscattering IN16 spectrometer (37) at the ILL (Grenoble, France). The instrumental energy resolution of 0.9 μeV (full width at half-maximum) allowed motions faster than 1 ns to be probed. The instrument uses neutrons with a wavelength of 6.27 Å and scattering vectors Q in the range of 0.02–1.9 Å−1. Each sample was inserted at room temperature into an orange ILL cryostat at 135° with respect to the incoming neutron beam. The temperature was lowered to 20 K over 2 h. Elastically scattered neutrons were then counted while the temperature was continuously increased from 20 to 300 K at a rate of 0.12 K/min for H-tau-D2O, 0.10 K/min for D-tau-H2O, 0.21 K/min for H-tau-dry, and 0.20 K/min for H-Mb-D2O (elastic scans). The elastically scattered intensity of the empty cell was measured at 20, 150, and 290 K. The elastically scattered signal of the empty cell at intermediate temperatures was obtained by linear interpolation. The elastically scattered signals of the protein samples were then calculated according to the following formula:
| (1) |
where Isample,T(Q) is the normalized intensity scattered from the sample at the temperature T, corrected for instrument effects and empty-cell scattering; Itotal,T(Q) is the measured sample intensity; and Iempty,T(Q) is the scattered intensity from the empty cell. The brackets represent the elastic intensity averaged in the temperature window from 20 to 24 K, and t is the measured sample transmission (0.89 for H-tau-D2O and 0.77 for D-tau-H2O). Isample,T(Q) is presented at selected temperatures in Fig. S2 for H-tau-D2O (panel A) and D-tau-H2O (panel B).
We extracted atomic MSDs (〈u2〉) using the Large Array Manipulation Program of ILL from the Q dependence of the elastic intensity, which can be described in the Gaussian approximation by
| (2) |
This expression remains valid as long as Q2〈u2〉 ≤ 2. MSDs were extracted in the range of 0.19 Å−2 < Q2 < 1.13 Å−2 for the D-tau-H2O sample (Fig. S2 B), and in the range of 0.19 Å−2 < Q2 < 1.32 Å−2 for the H-tau-D2O (Fig. S2 A) and H-tau-dry samples. We verified that the MSD of H-tau-D2O did not change significantly when extracted from data in a range reduced to 0.19 Å−2 < Q2 < 1.13 Å−2 (not shown). Published data on MBP (7) and RNase (38) were reexamined for MSD extraction in the same Q2-ranges as for tau (i.e., 0.19 Å−2 < Q2 < 1.13 Å−2 for the D-MBP-H2O, and 0.19 Å−2 < Q2 < 1.32 Å−2 for H-MBP-D2O, H-RNase-D2O, and H-RNase-dry). MSDs were extracted in the range of 0.20 Å−2 < Q2 < 1.40 Å−2 for the H-Mb-D2O sample.
The slope of MSD versus temperature decreased suddenly at 263 K for both D-MBP-H2O (7) and D-tau-H2O. Such a change in slope indicates that motions partly leave the experimental time-space window covered by the spectrometer, resulting in an apparent reduction in MSD (39). Therefore, only MSDs below 263 K were meaningful for D-MBP-H2O (7) and D-tau-H2O, respectively (Fig. 1 B; also see Fig. 4).
Figure 1.
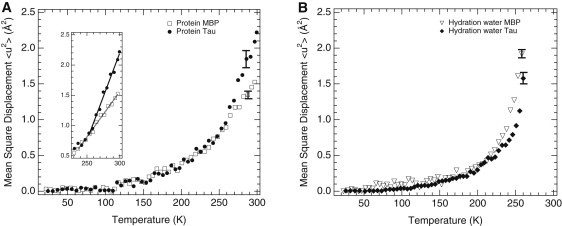
(A) MSDs of side-chain motions as a function of temperature for the IDP tau (solid circles, H-tau-D2O) and the folded MBP (open squares, H-MBP-D2O) as determined by neutron spectroscopy on hydrogenated protein powders hydrated in D2O to ∼0.4 g water/g protein. Effective force constants were 0.185 Nm−1 (MBP) and 0.096 Nm−1 (tau) as extracted from the slopes above 250 K (inset). (B) MSDs of hydration-water motions on the surfaces of tau (solid diamonds; D-tau-H2O) and MBP (open triangles; D-MBP-H2O) as determined on deuterated protein powders hydrated in H2O at ∼0.4 g water/g protein. The MBP data (7) were reexamined for MSD extraction in the same Q2-ranges as for tau. Errors are shown for selected high-temperature data points and are smaller at lower temperatures (not shown). Data were measured on the IN16 spectrometer at the ILL, Grenoble, France.
Figure 4.
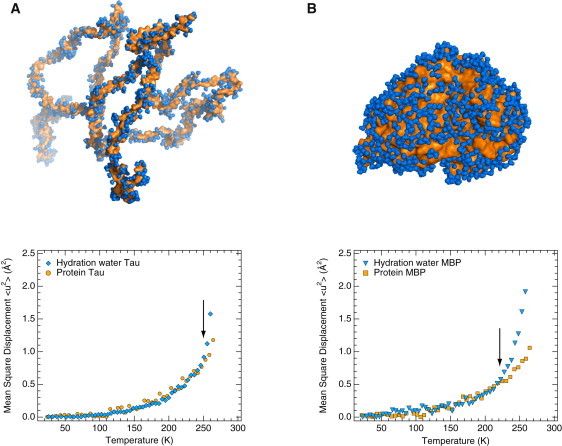
Dynamical coupling of protein dynamics with hydration-water motions for different protein classes. Temperature-dependent atomic MSDs of proteins (orange data points) are compared with those of hydration water (blue data points) and are identical to the ones shown in Fig. 1. (A) IDP tau. (B) Folded, globular MBP (7). An ensemble of 100 tau structures was generated using a coil library (58) and a structure with a calculated radius of gyration (63 Å) identical to that measured by SAXS (63 Å) was chosen for the upper half of panel A. Approximately 1000 water molecules forming the first hydration shell are shown in panel A, corresponding to the experimental hydration level of 0.4 g water/g protein. Data were obtained on the IN16 spectrometer at the ILL, Grenoble, France. Arrows indicate the temperature at which water and protein MSD diverge (250 K (A) and 220 K (B) for tau and MBP, respectively). MSDs > 260 K were omitted for the protein (tau and MBP) experiments for the sake of comparison with hydration-water MSDs (tau and MBP), which could be determined only up to 260 K (see Materials and Methods section).
Neutron diffraction on H-tau-D2O as a function of the hydration level
We determined the hydration threshold above which the presence of crystalline ice at 200 K indicates the presence of bulk-like water in addition to hydration water with a second H-tau-D2O sample by making use of the neutron diffraction option on IN16 (40). The presence of hexagonal crystalline ice (Ih; space group P63/mmc) was indicated by its 1 0 0 reflection at 2θ = 107° (λ = 6.27 Å). Ih was present at 0.8, 0.7, and 0.6 but not at 0.5 and 0.4 g D2O/g H-tau.
Neutron diffraction on D-tau-H2O to exclude the formation of paired helical filaments
We examined the same D-tau-H2O and H-tau-D2O samples as used for neutron spectroscopy on the D16 diffractometer at the ILL with a wavelength of 4.75 Å. The samples were placed perpendicularly to the incident neutron beam, and the diffraction pattern was measured at room temperature in a Q-range of 0.35–1.42 Å−1. Cross-β-structures, such as those found in paired helical filaments of neurofibrillary tangles, are characterized by peaks at 1.33 Å−1 (0.47 nm) and 0.59 Å−1 (1.07 nm) (20). The absence of any such diffraction peak indicated that no neurofibrillary tangles had formed in the samples.
Results
Biophysical characterization of H-tau and D-tau
The human htau40 isoform (the longest and most commonly found isoform of human tau with 441 residues; 45 kDa; UniProt entry code P10636-8 (Isoform Tau-F); referred to as tau throughout this work) was expressed and purified in its hydrogenated (H-tau) and perdeuterated (D-tau) forms. H-tau in solution was analyzed by SAXS and found to be monodisperse, with a radius of gyration of 63 Å in accordance with published data (41). MALLS provided evidence for the absence of tau oligomers in the H-tau solution. Negative-staining electron microscopy on H-tau in solution showed that no fiber structures were present. For neutron scattering experiments, lyophilized powders of hydrogenated and perdeuterated tau were hydrated to a level of 0.44 g D2O/g H-tau (H-tau-D2O sample) and 0.38 g H2O/g D-tau (D-tau-H2O sample), corresponding to 0.4 g H2O/g H-tau or 1040 water molecules per tau molecule, as determined by weighing. A neutron diffraction pattern of the hydrated D-tau-H2O powder sample was collected at room temperature and excluded the presence of the diffraction peaks at 0.47 and 1.07 nm that are characteristic of cross-β-structures, such as in paired helical filaments (20). A neutron diffraction pattern at 200 K of the H-tau-D2O sample also indicated the absence of crystalline ice. However, crystalline ice was present at 200 K when an H-tau-D2O sample was hydrated to levels of ≥0.6 g water/g protein. The solvent-accessible surface area of tau was estimated to be 47,000 Å2 by the protSA server (42), which employs the Flexible-Meccano algorithm to generate ensembles of IDP (43). This value compares with 15,000 Å2 computed for folded MBP, a folded globular protein of similar molecular mass (43 kDa) to tau (45 kDa).
EINS experiments
To examine the protein and water dynamics separately, we conducted EINS experiments as a function of temperature (ranging from 20 to 300 K) on H-tau-D2O and D-tau-H2O powders, respectively. Atomic MSDs were extracted and compared with values based on previously reported data (7) for deuterated and hydrogenated MBP powders hydrated in H2O and D2O, respectively (Fig. 1).
We monitored the protein dynamics by using the temperature-dependent MSD of side-chain motions as determined for H-proteins hydrated in D2O (Fig. 1 A). In an empirical approach based on a quasi-harmonic approximation, an effective force constant can be extracted from the slope of the MSD versus temperature (23). This force constant corresponds to the average structural resilience of the protein in the respective temperature range. Above 250 K, this constant is two times lower for tau than for MBP. At room temperature, MSDs are 50% larger for tau than for MBP. Interestingly, the differences in MSD between tau and MBP (Fig. 1 A) by far exceed the small differences observed between the MSDs of folded, globular proteins of different sizes, viz., MBP (387 amino acid residues; 43 kDa), Mb (153 residues; 17 kDa), and RNase (124 residues; 14 kDa), all hydrated at a level of ∼0.4 g D2O/g hydrogenated protein (Fig. 2). EINS experiments were also carried out on a dry tau powder (H-tau-dry sample), and the MSDs compared with that of dry RNase (38). Above 220 K, the MSDs of H-tau-dry are larger than those of a dry RNase powder (Fig. 3).
Figure 2.
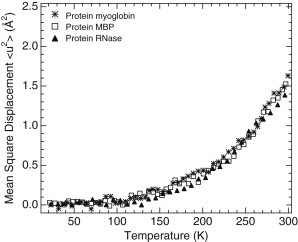
MSDs of hydrogenated powders of three folded, globular proteins of different sizes, viz., MBP (open squares; H-MBP-D2O), RNase (solid triangles; H-RNase-D2O), and Mb (crosses; H-Mb-D2O), hydrated at ∼0.4 g D2O/g protein. Published neutron spectroscopy data for H-RNase-D2O (38) and H-MBP-D2O (7) as measured on IN16 were reexamined for MSD extraction in the same Q2-range (0.19–1.32 Å−2) used for the H-tau-D2O sample.
Figure 3.
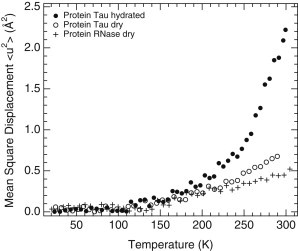
MSDs of dry tau (H-tau-dry, open circles), hydrated tau (H-tau-D2O, solid circles), and dry RNase (H-RNase-dry, crosses) powders. Published neutron spectroscopy data for H-RNase-dry (38), obtained on IN16, were reexamined for MSD extraction in the same Q2-range (0.19–1.32 Å−2) as for the H-tau-dry sample. MSDs of H-tau-D2O are the same as those displayed in Figs. 1A and 4A.
By examining the deuterated proteins hydrated in H2O, we were able to obtain direct information about the hydration-water dynamics, because the protein signal was largely masked (see Materials and Methods section for details). Below 230 K, the hydration water of tau and MBP displayed similar MSDs (Fig. 1 B). Above 230 K, the MSDs of hydration water were smaller for tau than for MBP. The MSDs of tau and its hydration water were determined in several independent experiments and on two different neutron backscattering spectrometers (IN16 (37) at the ILL, and SPHERES (44) at the Jülich Center for Neutron Science at FRM II, Garching, Germany), both of which monitor motions on a nano- to picosecond timescale. Almost identical MSDs were obtained (see Fig. S3 and Fig. S4), indicating the accuracy with which MSD can be determined by EINS.
One can assess the degree of coupling between protein and hydration-water dynamics by comparing the temperature dependences of their MSDs (Fig. 4). For the disordered tau, the protein and water MSDs largely follow each other up to 250 K (Fig. 4 A), whereas they diverge above 220 K for the folded MBP (Fig. 4 B).
Discussion
Increased side-chain flexibility of intrinsically disordered tau
At temperatures above 260 K, the MSDs of side-chain motions in the hydrated IDP tau are larger than those in the folded, globular protein MBP (Fig. 1 A). At room temperature, they differ by 50%. The associated effective force constant, which expresses the average structural resilience of the protein, is two times lower for tau than for MBP. Thus, the IDP is characterized by increased flexibility and reduced resilience compared with folded proteins of various sizes (Figs. 1 A and 2). The increased flexibility may be due to differences in the amino acid composition of IDP and folded proteins (IDPs have a large content of polar and charged residues compared with that of hydrophophic residues), because different types of amino acids display different nano- to picosecond dynamics (45). An additional or alternative explanation for increased IDP flexibility above 260 K could be increased conformational freedom of the backbone that propagates to increased side-chain flexibility in EINS experiments. Our results are in line with quasi-elastic neutron scattering experiments on hydrated powders of intrinsically disordered casein proteins, which showed that they display higher flexibility than folded proteins (46). However, hydrated powders of folded wild-type and partially unfolded mutant staphylococcal nuclease displayed similar MSDs (47), as did hydrated powders of native and denatured lysozyme (48). Interestingly, unfolded tRNA showed smaller MSDs than folded tRNA (49), in contrast to the opposite behavior of proteins. Even in the absence of hydration water, tau is characterized at room temperature by larger MSDs than are dry folded proteins (Fig. 3), in line with results obtained on dry powders of folded wild-type and partially unfolded mutant staphylococcal nuclease (47). Whether disordered (Fig. 1 A) or denatured, proteins thus display a much broader dynamical spectrum than do their folded counterparts (Fig. 2).
Tau adopts different partially folded conformations when it interacts with microtubules than when it interacts with biological membranes (50). The energy landscape of IDPs in general is described as being rather flat (51), which would rationalize the rapid structural adaptation to their targets or binding partners in a cellular context. The greater side-chain flexibility of tau, on the nano- to picosecond timescale, relative to the folded proteins MBP, Mb, and RNase, may reflect a rapid interconversion between the different conformational substates (structural rearrangements) that are required for tau to bind to microtubules.
Restricted hydration-water motions on the surface of IDPs
The MSDs of the hydration water of tau are smaller than those of MBP above 230 K (Fig. 1 B), indicating that the disordered nature of the tau protein surface hinders and confines water motions, in contrast to the behavior of folded globular proteins. Most probably, the side chains of IDP interact predominantly with their surrounding aqueous environment, and not with each other. Consequently, the lower water mobility of tau can be explained by geometrical constraints imposed by side chains being involved in more interactions with hydration-water molecules in IDPs than in folded proteins. It is interesting to note, however, that crystalline ice formed at 200 K when tau powders were hydrated at levels above 0.5 g water/g tau, indicating that the first hydration layer does not seem to involve more water molecules than are found around MBP, even though the solvent-accessible surface of tau is three times larger than that of MBP.
A recent 17O magnetic relaxation dispersion study of a small folded protein of 76 amino acids and its unfolded mutant in solution indicated that the rotational mobility of hydration water is greater in unfolded protein than in folded protein (52). In contrast, chemically denatured proteins have been shown to display reduced water mobility with respect to the folded state, and the hypothesis has been put forward that they are in a compact conformation that hinders water motion (53). Taken together (52,53), these results may indicate that the more compact the unfolded state of a protein is, the more its water dynamics will be perturbed. In analogy, the reduction in the hydration-water MSD of tau compared with MBP suggests that in the tau sample, water is confined, possibly by a rather compact conformation of the unfolded protein.
In agreement with the reduced MSD of tau hydration water found in this study, previous solid-state NMR relaxation experiments showed the activation energy of hydration water motions to be 50% larger for IDPs than for globular proteins (54). Intrinsic protein disorder thus shapes and modifies protein hydration, leading to hydration-water properties that differ among different classes of proteins. For example, in a study by Awile et al. (55), molecular-dynamics simulations of nudix hydrolase from Deinococcus radiodurans showed a lower hydration free energy for the disordered segments of the protein. Because the organism contains several IDPs that are thought to be involved in recovery after desiccation and are not present in nonextremophile homologs, the authors hypothesized that disordered regions may allow the protein to stay in intracellular patches of residual water, and suggested a new function for disordered regions, viz., to serve as hydrators (55).
Gradient of coupling with hydration-water dynamics across different protein classes
One can assess the coupling between dynamics of hydration water and proteins by comparing the temperature dependence of their respective MSDs (Fig. 4). With increasing temperature, they largely follow each other up to 250 K in the case of tau, but separate at 220 K in the case of MBP. The coupling with hydration-water dynamics is thus tighter for an IDP than for a folded, globular protein. In the case of membrane proteins, the onset of hydration-water translational motions at 200 K does not affect the nano- to picosecond dynamics of the underlying membrane, as evidenced by the temperature-dependent water and protein MSDs (24), and membrane-protein motions have been shown to be coupled to lipid rather than to water motions (56). Thus, there exists a gradient of coupling with hydration-water motions across different protein classes. The coupling decreases in strength from IDPs to globular-folded and finally membrane proteins. The slowing down of DNA dynamics compared with RNA dynamics has been suggested to be accompanied by a slowing down of water dynamics (57). A coherent picture thus emerges in which hydration water, rather than being a mere epiphenomenon, is an integral part of the biologically active protein. It is the delicately balanced give-and-take that occurs between a biological macromolecule's structural dynamics and those of its hydration water, rather than a master-slave relationship (5), that governs macromolecular function in a cellular context.
Acknowledgments
We thank Martin Field for help in the preparation of Fig. 4 A; Dan Tawfik, Israel Silman, and Colin Jackson for critical readings of the manuscript; Daphna Fenel for carrying out electron microscopy experiments; and Marc Jamin, Bruno Démé, and Louiza Zerrad for assistance during the MALLS, D16, and ID14-3 experiments, respectively. We thank the ILL and ESRF for allocating beam time and Bernhard Frick for his continuous support related to IN16 experiments.
This work was supported by the Commissariat à l'Énergie Atomique et aux Énergies Alternatives, Centre National de la Recherche Scientifique, Université Joseph Fourier, and Agence Nationale de la Recherche (project number ANR-11-BSV5-027 to M.W.). This work benefited from the activities of the DLAB consortium funded by the European Union under contracts HPRI-2001-50065 and RII3-CT-2003-505925, and from UK Engineering and Physical Sciences Research Council-funded activity within the ILL-EMBL Deuteration Laboratory under grants GR/R99393/01 and EP/C015452/1. The study was also supported by the European Commission under the 7th Framework Programme through the Research Infrastructures action of the Capacities Programme, contract CP-CSA_INFRA-2008-1.1.1 number 226507-NMI3. K.W. acknowledges funding from the Access to Major Research Facilities Program, supported by the Commonwealth of Australia under the International Science Linkages Program.
Footnotes
L. van Eijck's present address is Reactor Institute Delft, Delft University of Technology, Delft, The Netherlands.
A. Laganowsky's present address is Department of Chemistry, Chemistry Research Laboratory, University of Oxford, Oxford, UK.
Supporting Material
References
- 1.Ball P. Water as an active constituent in cell biology. Chem. Rev. 2008;108:74–108. doi: 10.1021/cr068037a. [DOI] [PubMed] [Google Scholar]
- 2.Grossman M., Born B., Havenith M. Correlated structural kinetics and retarded solvent dynamics at the metalloprotease active site. Nat. Struct. Mol. Biol. 2011;18:1102–1108. doi: 10.1038/nsmb.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ball P. Biophysics: more than a bystander. Nature. 2011;478:467–468. doi: 10.1038/478467a. [DOI] [PubMed] [Google Scholar]
- 4.Doster W., Settles M. Protein-water displacement distributions. Biochim. Biophys. Acta. 2005;1749:173–186. doi: 10.1016/j.bbapap.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Fenimore P.W., Frauenfelder H., Young R.D. Bulk-solvent and hydration-shell fluctuations, similar to α- and β-fluctuations in glasses, control protein motions and functions. Proc. Natl. Acad. Sci. USA. 2004;101:14408–14413. doi: 10.1073/pnas.0405573101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vitkup D., Ringe D., Karplus M. Solvent mobility and the protein ‘glass’ transition. Nat. Struct. Biol. 2000;7:34–38. doi: 10.1038/71231. [DOI] [PubMed] [Google Scholar]
- 7.Wood K., Frölich A., Weik M. Coincidence of dynamical transitions in a soluble protein and its hydration water: direct measurements by neutron scattering and MD simulations. J. Am. Chem. Soc. 2008;130:4586–4587. doi: 10.1021/ja710526r. [DOI] [PubMed] [Google Scholar]
- 8.Jansson H., Bergman R., Swenson J. Role of solvent for the dynamics and the glass transition of proteins. J. Phys. Chem. B. 2011;115:4099–4109. doi: 10.1021/jp1089867. [DOI] [PubMed] [Google Scholar]
- 9.Dyson H.J. Expanding the proteome: disordered and alternatively folded proteins. Q. Rev. Biophys. 2011;44:467–518. doi: 10.1017/S0033583511000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chouard T. Structural biology: breaking the protein rules. Nature. 2011;471:151–153. doi: 10.1038/471151a. [DOI] [PubMed] [Google Scholar]
- 11.Fink A.L. Natively unfolded proteins. Curr. Opin. Struct. Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Dunker A.K., Silman I., Uversky V.N., Sussman J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008;18:756–764. doi: 10.1016/j.sbi.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 14.Sugase K., Dyson H.J., Wright P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 15.Mandelkow E.M., Mandelkow E. Tau in Alzheimer's disease. Trends Cell Biol. 1998;8:425–427. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Y., LeGall T., Dunker A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Goedert M., Spillantini M.G., Crowther R.A. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 18.Mukrasch M.D., Biernat J., Zweckstetter M. Sites of tau important for aggregation populate β-structure and bind to microtubules and polyanions. J. Biol. Chem. 2005;280:24978–24986. doi: 10.1074/jbc.M501565200. [DOI] [PubMed] [Google Scholar]
- 19.Mukrasch M.D., Markwick P., Blackledge M. Highly populated turn conformations in natively unfolded tau protein identified from residual dipolar couplings and molecular simulation. J. Am. Chem. Soc. 2007;129:5235–5243. doi: 10.1021/ja0690159. [DOI] [PubMed] [Google Scholar]
- 20.von Bergen M., Barghorn S., Mandelkow E. 2005. Tau aggregation is driven by a transition from random coil to β-sheet structure. Biochim. Biophys. Acta. 2012;1739:158–166. doi: 10.1016/j.bbadis.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Mao A.H., Crick S.L., Vitalis A., Chicoine C.L., Pappu R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA. 2010;107:8183–8188. doi: 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabel F., Bicout D., Zaccai G. Protein dynamics studied by neutron scattering. Q. Rev. Biophys. 2002;35:327–367. doi: 10.1017/s0033583502003840. [DOI] [PubMed] [Google Scholar]
- 23.Zaccai G. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science. 2000;288:1604–1607. doi: 10.1126/science.288.5471.1604. [DOI] [PubMed] [Google Scholar]
- 24.Wood K., Plazanet M., Weik M. Coupling of protein and hydration-water dynamics in biological membranes. Proc. Natl. Acad. Sci. USA. 2007;104:18049–18054. doi: 10.1073/pnas.0706566104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doster W., Busch S., Scheer H. Dynamical transition of protein-hydration water. Phys. Rev. Lett. 2010;104:098101. doi: 10.1103/PhysRevLett.104.098101. [DOI] [PubMed] [Google Scholar]
- 26.Achterhold K., Ostermann A., Parak F.G. Dynamical properties of the hydration shell of fully deuterated myoglobin. Phys. Rev. E. 2011;84:041930. doi: 10.1103/PhysRevE.84.041930. [DOI] [PubMed] [Google Scholar]
- 27.Badger J., Caspar D.L. Water structure in cubic insulin crystals. Proc. Natl. Acad. Sci. USA. 1991;88:622–626. doi: 10.1073/pnas.88.2.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nucci N.V., Pometun M.S., Wand A.J. Site-resolved measurement of water-protein interactions by solution NMR. Nat. Struct. Mol. Biol. 2011;18:245–249. doi: 10.1038/nsmb.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang L., Wang L., Zhong D. Mapping hydration dynamics around a protein surface. Proc. Natl. Acad. Sci. USA. 2007;104:18461–18466. doi: 10.1073/pnas.0707647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rupley J.A., Careri G. Protein hydration and function. Adv. Protein Chem. 1991;41:37–172. doi: 10.1016/s0065-3233(08)60197-7. [DOI] [PubMed] [Google Scholar]
- 31.Lehnert U., Réat V., Pfister C. Thermal motions in bacteriorhodopsin at different hydration levels studied by neutron scattering: correlation with kinetics and light-induced conformational changes. Biophys. J. 1998;75:1945–1952. doi: 10.1016/S0006-3495(98)77635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clegg J.S., Drost-Hansen W. On the biochemistry and cell physiology of water. In: Hochachka P., Mommsen T.P., editors. Biochemistry and Molecular Biology of Fishes. Elsevier; Amsterdam: 1991. [Google Scholar]
- 33.Mattea C., Qvist J., Halle B. Dynamics at the protein-water interface from 17O spin relaxation in deeply supercooled solutions. Biophys. J. 2008;95:2951–2963. doi: 10.1529/biophysj.108.135194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Artero J.B., Hartlein M., Timmins P. A comparison of refined X-ray structures of hydrogenated and perdeuterated rat γE-crystallin in H2O and D2O. Acta Crystallogr. D Biol. Crystallogr. 2005;61:1541–1549. doi: 10.1107/S0907444905028532. [DOI] [PubMed] [Google Scholar]
- 35.Konarev P.V., Volkov V.V., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 36.Dolman M., Halling P.J., Waldron S. How dry are anhydrous enzymes? Measurement of residual and buried O-18-labeled water molecules using mass spectrometry. Biopolymers. 1997;41:313–321. [Google Scholar]
- 37.Frick B., Gonzalez M. Five years operation of the second generation backscattering spectrometer IN16–a retrospective, recent developments and plans. Physica B. 2001;301:8–19. [Google Scholar]
- 38.Wood K., Caronna C., Zaccai G. A benchmark for protein dynamics: ribonuclease A measured by neutron scattering in a large wavevector-energy transfer range. Chem. Phys. 2008;345:305–314. [Google Scholar]
- 39.Gabel F. Protein dynamics in solution and powder measured by incoherent elastic neutron scattering: the influence of Q-range and energy resolution. Eur. Biophys. J. 2005;34:1–12. doi: 10.1007/s00249-004-0433-0. [DOI] [PubMed] [Google Scholar]
- 40.Combet J., Frick B., Guerard B. Simultaneous diffraction and inelastic scattering on the backscattering instrument IN16. Physica B. 2000;283:380–385. [Google Scholar]
- 41.Mylonas E., Hascher A., Svergun D.I. Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry. 2008;47:10345–10353. doi: 10.1021/bi800900d. [DOI] [PubMed] [Google Scholar]
- 42.Bernado P., Blackledge M., Sancho J. Sequence-specific solvent accessibilities of protein residues in unfolded protein ensembles. Biophys. J. 2006;91:4536–4543. doi: 10.1529/biophysj.106.087528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernado P., Blanchard L., Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wuttke, J., A. Budwig, …, D. Richter. 2012. SPHERES, Jülich's high-flux neutron backscattering spectrometer at FRM II. Rev. Sci. Instrum.http://arxiv.org/abs/1204.3415. [DOI] [PubMed]
- 45.Schiró G., Caronna C., Cupane A. Direct evidence of the amino acid side chain and backbone contributions to protein anharmonicity. J. Am. Chem. Soc. 2010;132:1371–1376. doi: 10.1021/ja908611p. [DOI] [PubMed] [Google Scholar]
- 46.Gaspar A.M., Appavou M.S., Busch S., Unruh T., Doster W. Dynamics of well-folded and natively disordered proteins in solution: a time-of-flight neutron scattering study. Eur. Biophys. J. 2008;37:573–582. doi: 10.1007/s00249-008-0266-3. [DOI] [PubMed] [Google Scholar]
- 47.Nakagawa H., Kamikubo H., Kataoka M. Effect of conformational states on protein dynamical transition. Biochim. Biophys. Acta. 2010;1804:27–33. doi: 10.1016/j.bbapap.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 48.Mamontov E., O'Neill H., Zhang Q. Mean-squared atomic displacements in hydrated lysozyme, native and denatured. J. Biol. Phys. 2010;36:291–297. doi: 10.1007/s10867-009-9184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roh J.H., Tyagi M., Sokolov A.P. The dynamics of unfolded versus folded tRNA: the role of electrostatic interactions. J. Am. Chem. Soc. 2011;133:16406–16409. doi: 10.1021/ja207667u. [DOI] [PubMed] [Google Scholar]
- 50.Barre P., Eliezer D. Folding of the repeat domain of tau upon binding to lipid surfaces. J. Mol. Biol. 2006;362:312–326. doi: 10.1016/j.jmb.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 51.Choi U.B., McCann J.J., Bowen M.E. Beyond the random coil: stochastic conformational switching in intrinsically disordered proteins. Structure. 2011;19:566–576. doi: 10.1016/j.str.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qvist J., Ortega G., Halle B. Hydration dynamics of a halophilic protein in folded and unfolded states. J. Phys. Chem. B. 2012;116:3436–3444. doi: 10.1021/jp3000569. [DOI] [PubMed] [Google Scholar]
- 53.Modig K., Kurian E., Halle B. Water and urea interactions with the native and unfolded forms of a β-barrel protein. Protein Sci. 2003;12:2768–2781. doi: 10.1110/ps.03262603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bokor M., Csizmók V., Tompa K. NMR relaxation studies on the hydrate layer of intrinsically unstructured proteins. Biophys. J. 2005;88:2030–2037. doi: 10.1529/biophysj.104.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Awile O., Krisko A., Zagrovic B. Intrinsically disordered regions may lower the hydration free energy in proteins: a case study of nudix hydrolase in the bacterium Deinococcus radiodurans. PLOS Comput. Biol. 2010;6:e1000854. doi: 10.1371/journal.pcbi.1000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frolich A., Gabel F., Zaccai G. From shell to cell: neutron scattering studies of biological water dynamics and coupling to activity. Faraday Discuss. 2009;141:117–130. doi: 10.1039/b805506h. discussion 175–207. [DOI] [PubMed] [Google Scholar]
- 57.Khodadadi S., Roh J.H., Sokolov A.P. Dynamics of biological macromolecules: not a simple slaving by hydration water. Biophys. J. 2010;98:1321–1326. doi: 10.1016/j.bpj.2009.12.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jha A.K., Colubri A., Sosnick T.R. Statistical coil model of the unfolded state: resolving the reconciliation problem. Proc. Natl. Acad. Sci. USA. 2005;102:13099–13104. doi: 10.1073/pnas.0506078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.